

Abstract
The symbiotic relationship between cnidarians and dinoflagellates is the cornerstone of coral reef ecosystems. Although research is focusing on the molecular mechanisms underlying this symbiosis, the role of epigenetic mechanisms, which have been implicated in transcriptional regulation and acclimation to environmental change, is unknown. To assess the role of DNA methylation in the cnidarian-dinoflagellate symbiosis, we analyzed genome-wide CpG methylation, histone associations, and transcriptomic states of symbiotic and aposymbiotic anemones in the model system Aiptasia. We find methylated genes are marked by histone H3K36me3 and show significant reduction of spurious transcription and transcriptional noise, revealing a role of DNA methylation in the maintenance of transcriptional homeostasis. Changes in DNA methylation and expression show enrichment for symbiosis-related processes such as immunity, apoptosis, phagocytosis recognition and phagosome formation, and unveil intricate interactions between the underlying pathways. Our results demonstrate that DNA methylation provides an epigenetic mechanism of transcriptional homeostasis during symbiosis.
Introduction
Coral reefs are ecologically important marine ecosystems, which cover less than 0.2% of our oceans but sustain an estimated ~25% of the world's marine species and 32 of 33 animal phyla 1, 2, 3. Coral reefs are also economically important by providing food and livelihood opportunities to at least 500 million people; worldwide, they have a net present value of almost USD 800 billion, and they generate USD 30 billion in net economic benefits annually 3.
Unfortunately, these ecosystems are under severe threat from anthropogenic stressors including global warming and water pollution, among others, which can cause coral bleaching (loss of intracellular endosymbionts from coral) and overall coral reef decline. Despite increasing efforts on studying the mechanisms underlying the regulation and environmental stress related breakdown of this symbiotic association 4, 5, we still lack knowledge on basic molecular processes, for instance whether epigenetic mechanisms are involved in symbiosis regulation and could potentially contribute to increased resilience in response to environmental stress as reported in other organisms 6, 7.
DNA methylation plays an important role in many biological processes of plants and animals 8, 9, 10, 11. It has been proposed as a mechanism for organisms to adjust their phenotype in response to their environment in order to optimize organismal response to changing environmental conditions 7, 12. For instance, recent findings in mice show an important function for DNA methylation in inhibiting spurious transcription along the gene body, allowing for reduction of nonsense transcripts from highly expressed loci 13. However, its role and function in cnidarians is, at present, unknown 14. The sea anemone Aiptasia is an emerging model to study the cnidarian-dinoflagellate symbiosis. Like corals, it establishes a stable but temperature sensitive symbiosis with dinoflagellates of the genus Symbiodinium but, unlike corals, can also be naturally maintained in an aposymbiotic state. Its ease of culture and facultative symbiosis provides a tractable system to study the molecular mechanism underlying symbiosis without the impeding stress responses associated with coral bleaching stress 15, 16.
Using the model system Aiptasia (strain CC7, sensu ExAiptasia pallida), we obtained whole-genome CpG DNA methylation, ChIP-Seq and RNA-Seq data from aposymbiotic (Apo) and symbiotic (Sym) individuals to study the function of DNA methylation in transcriptional regulation and its role in the cnidarian-dinoflagellate symbiosis.
Results
Aiptasia DNA Methylation patterns change with symbiotic states
To assess changes in DNA methylation in response to symbiosis, we performed whole- genome bisulfite sequencing with an average coverage of 53× per individual on 12 anemones, providing 6 biological replicates per treatment (symbiotic vs. aposymbiotic). Methylation calling using the combined dataset identified 710,768 CpGs (6.37% of all CpGs in Aiptasia genome), i.e. methylated sites in the Aiptasia genome. Notably, the percentage of CpGs is much lower than in mammals (60–90%) 17, but comparable to the coral Stylophora pistillata (7%) 18. We identified 10,822 genes (37% of all 29,269 gene models identified in the Aiptasia genome) with at least 5 methylated positions that were subsequently defined as methylated genes. On average, these genes had 18.4% CpGs methylated, 3-fold higher than the average methylation density across the entire genome (Chi-squared test p value < 2.2 × 10−16) and 167-fold higher than the methylation levels in non-coding regions. These findings indicate that the distribution of CpG methylation is non-random and mainly located in gene bodies, similar to corals 18, 19 and other invertebrate species 20, 21.
To analyze the relationship between methylation density (percentage of CpGs) and gene density (the number of genes per 10,000 bp), we ran a sliding window (window size: 40 kb, step: 30 kb) and visualized the results in a Circos plot (Fig. S1) 22. The correlation of CpG content and distribution of methylation showed a negative correlation (Pearson correlation coefficient: r = −0.31, p value < 2.2 × 10−16) suggesting that methylation tends to preferentially occur in CpG- poor regions (Fig. S2). Gene density had a positive correlation with methylation density (r = 0.21, p value < 2.2 × 10−16) consistent with the finding that methylation is predominantly located in gene bodies (Fig. 1). We also observed that within gene bodies, introns showed significantly higher methylation densities than exons (Fig. 1B).

(A) Distribution of CpG across intergenic (18%), genic (82%), intronic (42%) and exonic (40%) regions in the Aiptasia sp. genome. (B) Normalized percentage of methylated CpGs in different regions. Chi-squared test shows significant differences between intergenic and genic regions, and between exons and intron (****p<0.0001). (C) Relative frequencies of methylated positions across a normalized gene model.
Methylated genes are marked by H3K36me3
Analysis of methylation patterns (see above) within gene bodies showed rapidly increasing methylation levels after the transcription start site (TSS) that are maintained before slowly decreasing towards the transcription termination site (TTS) (Fig. S3A). Interestingly, we found that gene body methylation in Aiptasia is positively correlated with expression (Fig. 2A), suggesting that DNA methylation either increases the expression of genes or that DNA methylation is established as a consequence of transcription whereby increased expression results in increasing methylation levels. The latter interpretation would be in line with recent findings in mouse embryonic stem cells 13, which demonstrated that gene body methylation is established and maintained as a result of active transcription by RNA polymerase II (Pol II) and recruitment of the histone modifying protein SetD2 that trimethylates histone H3 at lysine 36 (H3K36me3). This histone mark is specifically bound via the PWWP domain present in the DNA methyltransferase Dnmt3b, which in turn methylates the surrounding DNA accordingly, resulting in the inhibition of transcription initiation from cryptic promoters within the gene body and thus a significant reduction of spurious transcription.

(A) Gene expression is positively correlated with median methylation levels, t-test p values are 7.65 × 10−21, 3.75 × 10−14 and 1.75 × 10−13 for the first quartile (Q1) and the second quartile (Q2) of methylation levels, Q2 and Q3, and Q3 and Q4, respectively. (B) ChIP-Seq analysis of H3K36me3 signals show significant enrichment in methylated genes (t-test p values: 2.48 × 10−20 for highly methylated genes (HM) and all methylated genes (allM), and 1.06 × 10−72 for unmethylated genes (UM) and allM). Highly methylated genes show the strongest enrichment with H3K36me3 followed by all methylated genes. In contrast unmethylated genes show only weak enrichment of H3K36me3 over input controls. (C) Distribution of H3K36me3 enrichment and DNA methylation levels across two exemplary gene models. H3K36me3 and DNA methylation show coinciding distribution patterns over genes.
Analysis of the Aiptasia gene set identified a DNMT3 gene (AIPGENE24404) that also encodes a PWWP domain as reported for the mouse homolog. In order to test if the previously described mechanism is conserved in Aiptasia, we performed a ChIP-Seq experiment using a validated antibody against H3K36me3 (Fig. S4). As predicted, our analysis confirmed a significantly higher association of H3K36me3 with methylated genes (p = 2.48 × 10−20 for highly methylated genes and all methylated genes, Fig. 2B and C). We then analyzed if methylated genes also exhibited significantly lower levels of spurious transcription in Aiptasia. Analysis of transcriptional profiles of methylated and unmethylated genes indeed showed significantly lower levels of spurious transcription along the gene body of methylated genes (p < 2 × 10−6, Fig. 3A).

(A) Spurious transcription in gene bodies is significantly lower in methylated and highly methylated genes. The y-axis shows the natural logarithm of the coverage fold change of exons 1–6 vs. exon 1. *: p < 0.05; **: p < 0.01; ***: p < 0.001; ****: p < 0.0001. (B) There is a linear relationship between the inverse of transcriptional noise (CV−1) and log expression level (log10fpkm). Given same expression level, methylated genes always show lower levels of transcriptional noise. For methylated genes, n = 8,561, r2 = 0.46, p < 2.2 × 10−16, for unmethylated genes, n = 2,491, r2 = 0.52, p < 2.2 × 10−16.
A dampening effect of DNA methylation on transcription was also observed with regard to transcriptional noise similar to findings in the coral Stylophora pistillata 18. Regression analysis of median methylation levels and the coefficient of transcriptional variation of genes showed that, given the same expression level, methylated genes always exhibited lower levels of transcriptional variation (Fig. 3B).
DNA methylation regulates transcriptional homeostasis during symbiosis
Based on our previous findings, we investigated if DNA methylation is also involved in the regulation of symbiosis by identifying differentially methylated genes (DMGs) between symbiotic and aposymbiotic Aiptasia. Comparison of DNA methylation patterns using Principal Component Analysis (PCA) clearly separated symbiotic and aposymbiotic individuals by the first principal component, which accounted for ~18% of the variance (Fig. 4A). This analysis echoed the findings from a PCA analysis on gene expression where symbiosis state was separated by the second principal component accounting for ~21% of the variance (Fig. 4B) 23 and highlighted that specific changes in DNA methylation patterns occurred in response to symbiosis.
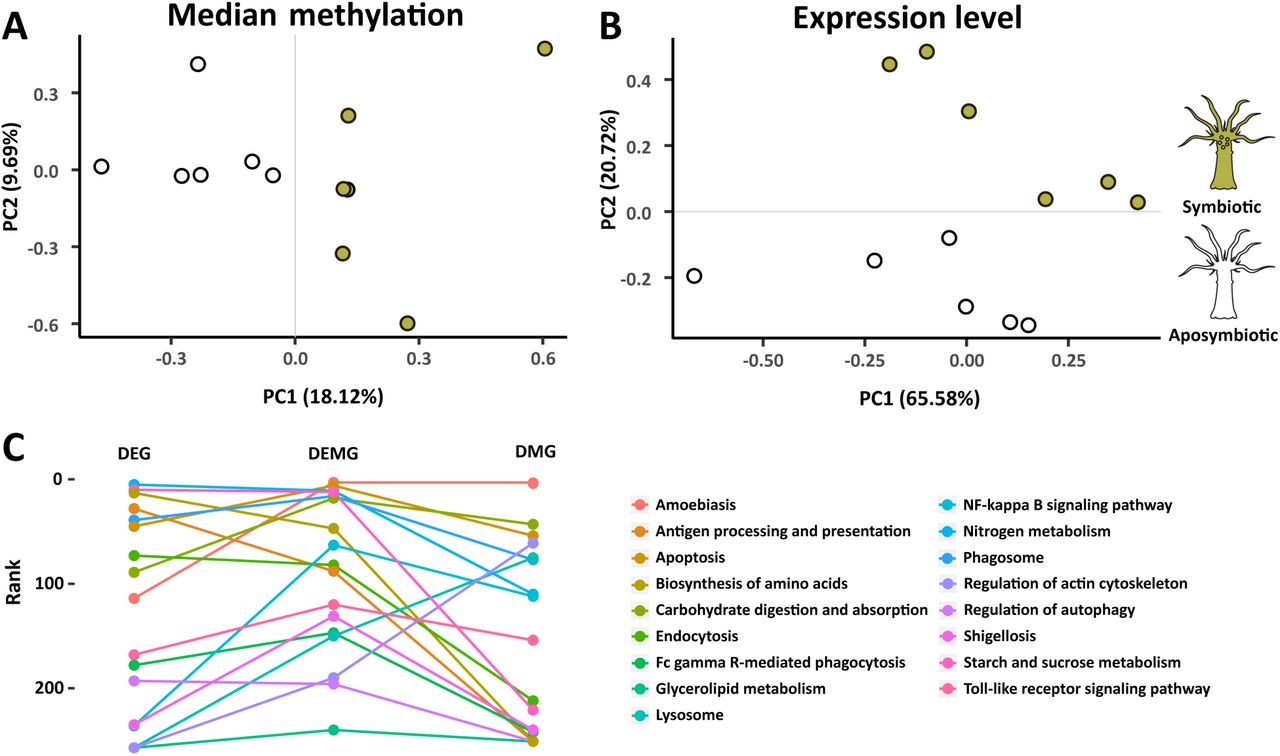
(A, B) PCA (Principal Component Analysis) of gene expression and median methylation levels of Aiptasia genes. Both gene expression and DNA methylation separate samples by symbiosis state. (C) KEGG pathway enrichment analysis. The combined sets of differentially expressed and differentially methylated genes (DEMG) provides significant lower p values (front ranks) for symbiosis related pathways.
Subsequently we analyzed changes in DNA methylation and gene expression between symbiotic and aposymbiotic Aiptasia to assess their correlation on potential biological functions in symbiosis. We determined differentially methylated genes using a generalized linear model from Foret et al. 24 that was modified to allow for replicate-aware analysis. This approach identified 2,133 DMGs (FDR ≤ 0.05, Supplement Table S1) that specifically changed their methylation status in response to symbiosis. To verify these results, we sequenced a subset of 14 DMGs using bisulfite PCRs. The results show a strong correlation (r2 = 0.815 and p = 1×10−5 for Apo, r2 = 0.922 and p = 5.2 × 10−8 for Sym) to our WGBS and confirm the observed methylation changes within these loci (Fig. S5).
Analysis of gene expression changes in the same 12 samples (i.e., 6 symbiotic and 6 aposymbiotic anemones) identified 1,278 differentially expressed genes (DEGs, FDR ≤ 0.05, Supplement Table S2), of which 14 genes were subsequently confirmed via qPCR (Fig. S6). However, analysis of the overlap between DMGs and DEGs showed only 103 genes that were shared, suggesting that differentially expressed genes are not necessarily the same cohort of genes that are differentially methylated. Functional enrichment analyses based on Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of all DMGs and DEGs identified several symbiosis relevant functions and pathways in both groups (Supplement Table S3-S10).
Based on the finding that gene body DNA methylation is likely a consequence of active transcription, we hypothesized that changes in DNA methylation patterns might also provide a record of transcriptional activity over longer periods of time. We therefore tested if differential methylation and acute transcriptional changes, obtained from our RNA-Seq analysis, provide a complementary view of the processes underlying symbiosis. For this we compared enrichment of symbiosis-specific pathways across the sets of 2,133 DMGs, 1,278 DEGs, and the combined set of both DMGs and DEGs (3,308 DEMGs). Interestingly, we observed that the combined data set (DEMGs) provided significantly lower p-values for previously identified symbiosis-related pathways, including apoptosis, phagosome, nitrogen metabolism, and arginine biosynthesis, among others (paired t-test: DEMG vs. DEG p = 0.015; DEMG vs. DMG p = 0.009) (Fig. 4C and Supplement Table S11). This suggested that changes in methylation and transcription indeed provide complementary information with regard to transcriptional adjustments in response to symbiosis.
DMGs and DEGs are involved in all stages of symbiosis
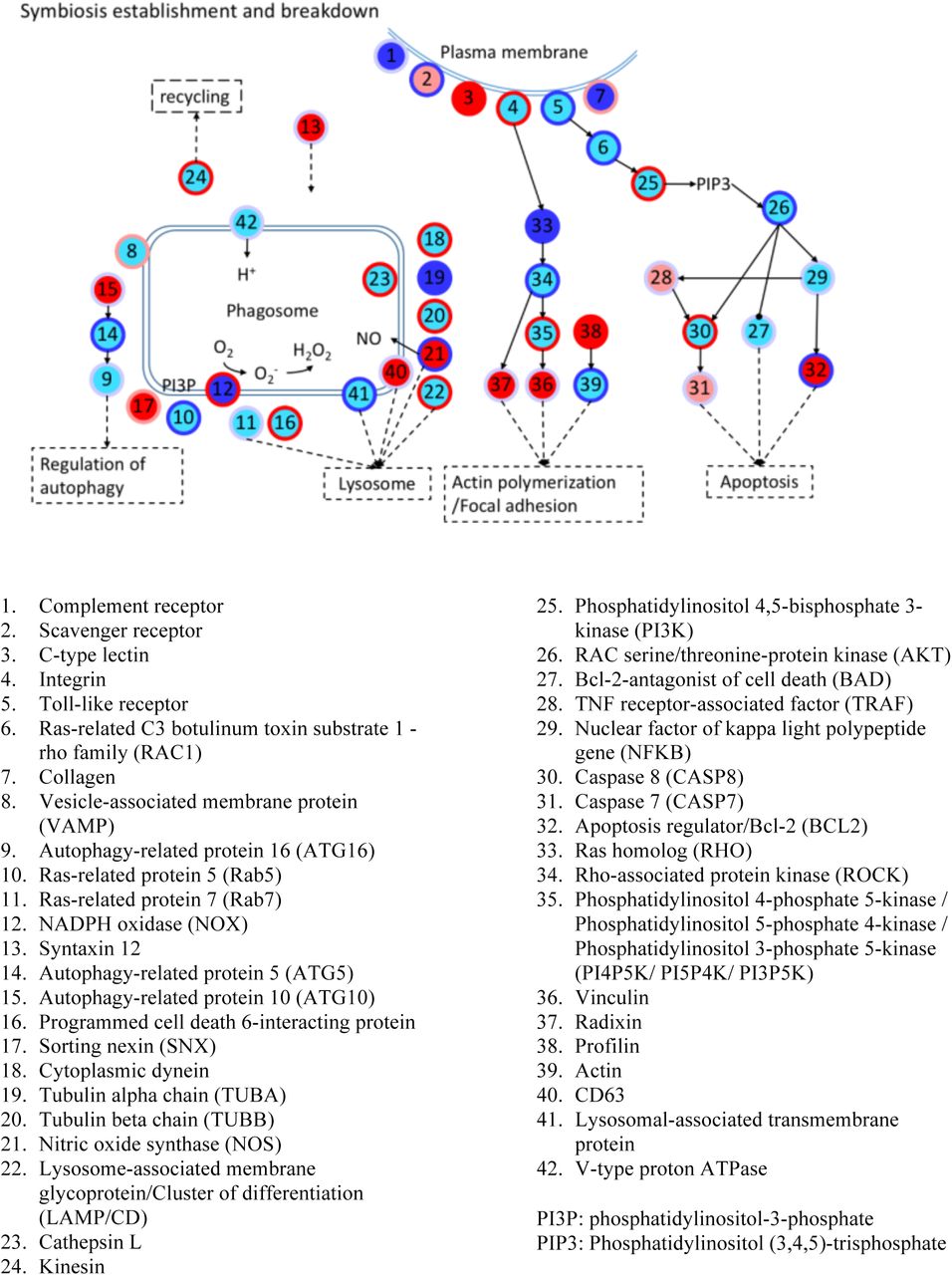
Analysis of the combined DMG and DEG gene set showed significant enrichment of genes involved in the distinct phases of symbiosis, that is symbiosis establishment, maintenance, and breakdown 4. Using an integrated pathway analysis based on known molecular interactions between proteins we found that these processes are linked through several DMGs and/or DEGs (Fig. S7 and Fig. S8, and see Supplement Table S11-S12 and Supplementary discussion).
For instance, we found numerous symbiosis-related receptors to respond to symbiosis on a transcriptional and/or methylation level (Fig. S7), including C-type lectins (Fig. S7.3), Toll-like receptors (Fig. S7.5), and the scavenger receptor SRB1 (Fig. S7.2) that has previously been implicated in symbiont recognition in the sea anemone Anthopleura elegantissima 25. Following symbiont recognition, we also found several known engulfment and sorting-related genes to change in methylation and/or expression such as Rab5 (Fig. S7.10), sorting nexin (Fig. S7.17), Rac1 (Fig. S7.6), the lysosomal-associated membrane protein 1/2 (Fig. S7.22), and many genes related to the cytoskeleton and movement (Fig. S7.33-39).
As expected in a metabolic symbiosis 4, 26 we also identified a large number of genes involved in nutrient exchange. These included genes involved in the provision of inorganic carbon in the form of CO2 or bicarbonate (HCO3−) to fuel symbiont driven photosynthesis 27 (Fig. S8.1) as well as genes involved in the exchange of fixed carbon in the form of lipids (Fig. S8.11), sugars and amino acids (Fig. S8.10, S8.4) 28. Concordantly, we also found that genes involved in nitrogen acquisition, such as ammonium transporter (Fig. S8.2) and genes involved in glutamate metabolism (Fig. S8.5-7), respond to symbiosis.
Finally, our analysis also highlighted genes putatively involved in the expulsion or degradation of symbionts in response to environmental stress or as a means to control symbiont densities. Autophagy is of interest in this regard because it links to other membrane trafficking pathways and to apoptosis, and evidence suggests that autophagy also plays a role in removal of symbionts during bleaching 29, 30. Intracellular degradation of the symbiont is a result of reengagement of the phagosomal maturation process or autophagic digestion of the symbiont by the host cell 4, and we find both apoptosis- and autophagy-related genes to significantly change in their methylation and/or expression level. These include the apoptosis genes RAC serine/threonine-protein kinase (Fig. S7.25), Caspase 7 (Fig. S7.31), Caspase 8 (CASP8) (Fig. S7.30), Nitric oxide synthase (Fig. S7.21) and Bcl2 (Fig. S7.27), as well as the Autophagy proteins 5 and 10 (Fig. S7.14-15), among others.
Discussion
To assess the role of CpG methylation in the cnidarian-dinoflagellate symbiosis, we undertook a global analysis of changes in the DNA methylomes and transcriptomes of aposymbiotic and symbiotic Aiptasia. In contrast to their vertebrate counterparts, only 6.37% of the CpGs in the Aiptasia genome are methylated, but their distribution is highly non-random (p < 3 × 10−300) and that methylated CpGs are most highly localized in gene bodies (18.4% of CpGs). Analysis of the distribution of the histone modification H3K36me3 further showed significant enrichment of this epigenetic mark in methylated genes, echoing findings in mammals and invertebrates 31. More importantly, we find that methylated genes show significant reduction of spurious transcription and transcriptional noise (Fig. 2B), suggesting that both the underlying mechanism of epigenetic crosstalk as well as the biological function of DNA methylation is evolutionary conserved throughout metazoans. These results highlight a tight interaction of transcription and epigenetic mechanisms in optimizing gene expression in response to changing transcriptional needs 13. Further support for such a role is provided by the analysis of differentially methylated and differentially expressed genes, which, when combined, showed significant increase in enrichment of symbiosis relevant processes. This suggests that DNA methylation and transcriptome analyses provide complementary views of cellular responses to symbiosis whereby methylation changes provide a transcriptional record of longer-term transcriptional adjustments.
While our analysis identified several genes, processes, and pathways previously reported to be involved in symbiosis, it further highlights their intricate molecular interactions. Symbiosis recognition, sorting and breakdown are interconnected processes, which is reflected in the observed changes in methylation and expression. The molecular machinery involved in phagosome maturation is tightly linked to autophagy and apoptosis enabling the host to respond to potential pathogen invasion but also to degrade and remove dead or unsuitable symbionts.
This is strongly supported by immunofluorescence examinations of Aiptasia pulchella gastrodermal cell macerates, showing that Rab5 appears around healthy, newly ingested and already established Symbiodinium, but is replaced by Rab7 in heat-killed or DCMU-treated newly ingested Symbiodinium. Conversely, Rab7 is absent from untreated newly infected or already-established Symbiodinium 32, 33.
Rab5 is also required for the exosomal release of CD63 34, which mediates the endocytotic sorting process and transport to lysosomes 35. This process is further regulated by Rac1 36 in conjunction with sorting nexin and the GTPase Rho, all of which were also identified in our analyses. The sorting of phagocytosed Symbiodinium is critical to symbiosis establishment as Symbiodinium is phagocytosed at the apical end and transported to the base of the cell, where they are protected from digestion. In contrast, Symbiodinium staying at the apical end of the cell are degraded 37.
Similar to the processes of symbiosis initiation and breakdown, we also found significant enrichment of genes involved in nutrient exchange and many of these transporters have previously been implicated in symbiosis maintenance 4, 38. Notably, this also included genes involved in the transport and assimilation of ammonium. Nitrogen is a main limiting nutrient in coral reefs 39, 40, 41, and the coral-dinoflagellates symbiosis has been proposed to increase the efficiency of nitrogen utilization by both partners 42 whereby the underlying nature of this mechanism is currently debated 43, 44.
Conclusions
This study provides the first analysis of the function and role of DNA methylation in a symbiotic anthozoan. Our results show that the epigenetic crosstalk between the histone mark H3K36me3 and gene body methylation is conserved in cnidarians and reveal a role of gene body methylation in reducing of spurious transcription and transcriptional noise. Furthermore, we show that changes in DNA methylation patterns are specific to symbiosis and imply a functional in the establishment, maintenance, and breakdown of this important symbiotic association. Our findings therefore provide evidence for a role of DNA methylation as epigenetic mechanism involved in the maintenance of transcriptional homeostasis during the cnidarian-dinoflagellate symbiosis. The premise that epigenetic mechanisms play a role in organismal acclimation warrants future experiments targeted to investigate if DNA methylation could also contribute to resilience through the epigenetic adjustment of transcription in response to environmental stress in Aiptasia and corals.
Author contributions
M.A. conceived and coordinated the project. Y.L., G.C., M.J.C. and N.Z. performed experiments. M.A., C.R.V. and Y.J.L. provided tools and/or data. C.T.M. constructed libraries for whole genome bisulfite sequencing, ChIP-Seq and RNA-Seq. Y.L., Y.J.L. and M.A. analyzed expression, methylation and ChIP-Seq data. M.A. and Y.L. wrote the manuscript with input from Y.J.L. and C.R.V. All authors read and approved the final manuscript.
Supplementary Figures

(a) TE density. (b) Gene density. (c) Fraction of methylated CpGs in symbiotic treatment. (d) Fraction of methylated CpGs in aposymbiotic treatment. (e) CpG content.

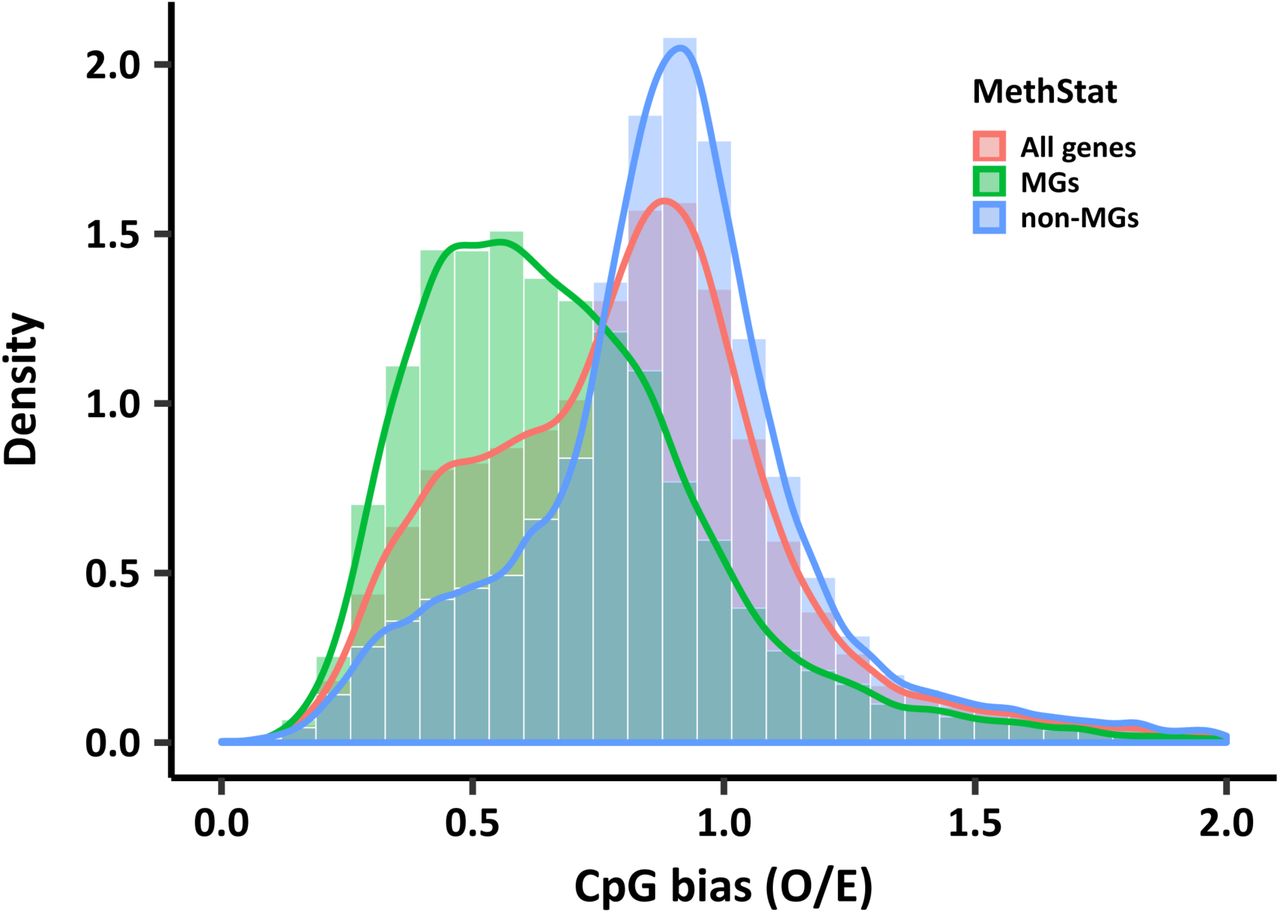
CpG distribution of methylated genes (represented by red curve) peaks at around 0.5, which is lower than in unmethylated genes (represented by green curve) peaking at around 0.9. mC to T conversion skews the CpG O/E distribution of all genes as expected (represented by blue curve), but methylated and unmethylated genes still show a large overlap of their CpG O/E distributions. These results indicate that gene body methylation cannot be accurately inferred from CpG O/E in Aiptasia.

(A) DNA methylation is mainly located in the proximal part of gene bodies with slightly decreasing levels towards the end. (B) Methylation pattern over intergenic regions. (C) Methylation pattern around splice donor sites show increasing levels immediately after donor sites. (D) Methylation pattern around acceptor sites show decreasing levels immediately after splice acceptor sites. (E) Methylation pattern over initial exons show increasing methylation levels (3,147 exons with 35,885 methylation sites were used). (F) Methylation pattern over internal exons show decreasing methylation levels (7,977 exons with 139,009 methylation sites were used). (G) Methylation pattern over terminal exons show decreasing methylation levels (7,905 exons with 102,162 methylation sites were used). (H) Methylation pattern over introns from single-exon genes follow a similar trend as observed for multi exon genes with increasing methylation levels in the proximal and decreasing levels in the posterior part of the exon (298 exons with 4,735 methylation sites were used). (I) Methylation pattern over initial introns show increasing methylation levels (3,381 introns with 39,262 methylation sites were used). (J) Methylation pattern over internal introns maintain stable methylation levels (7,371 introns with 211,950 methylation sites were used). (K) Methylation levels over terminal introns decrease slightly (3,959 introns with 34,246 methylation sites were used). (L) Methylation levels over introns from one-intron genes change gently with initial increase followed by a decrease (1,055 introns with 10,709 methylation sites).

Western blot result for antibody affinity validation, target band is 15kDa in size as expected from molecular weight analysis.

Validation of methylation level using bisulfite PCR on selected genes. (A, B) validation of methylation level on genes (median methylation levels of methylated CpGs were used to represent genes). (C, D) validation of methylation level on locus (methylated CpGs). WGBS: whole genome bisulfite sequencing; Amplicon: MiSeq sequencing results of bisulfite PCR amplicons on selected genes.

Validation of gene expression changes using qPCR. Expression levels are shown as log10(fold change). All genes show similar expression changes as determined by RNA-seq and q-PCR.

Every node represents a category of genes, and generally has multiple corresponding genes. The inside colors of nodes represent the expression changes of corresponding genes, including non-DEGs (cyan), up-regulated (red), down-regulated (blue) and up- and down- regulated DEGs (light red). The colors of node edges represent the methylation level changes of corresponding genes, including non-DMGs (light blue), hypermethylated (red), hypomethylated (blue) and hyper- and hypo-methylated DMGs (light red). Numbers in circles denote genes/proteins as detailed below.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Every node represents a category of genes, and generally has multiple corresponding genes. The inside colors of nodes represent the expression changes of corresponding genes, including non-DEGs (cyan), up-regulated (red), down-regulated (blue) and up- and down-regulated DEGs (light red). The colors of node edges represent the methylation level changes of corresponding genes, including non-DMGs (light blue), hypermethylated (red), hypomethylated (blue) and hyper- and hypo-methylated DMGs (light red).
Acknowledgements
Research reported in this publication was supported by funding from King Abdullah University of Science and Technology (KAUST). We thank Prof. John Pringle for the provision of the initial Aiptasia CC7 and Symbiodinium SSB01 cultures. We thank Sebastian Baumgarten for his help with establishing the Aiptasia cultures and critical reading of the manuscript.
References and Notes
References




