

Abstract
A localized transcriptome at the synapse facilitates synapse-, stimulus-, and transcript-specific synthesis of the local proteome in response to neuronal activity. While enzyme-mediated mRNA modifications have been shown to regulate cellular mRNA turnover and translation, the role of these modifications in regulating synaptic RNA has not been studied. We established low-input m6A-seq of synaptosomal RNA to determine the chemically modified local transcriptome in healthy adult mouse forebrain and identified 4,329 selectively enriched m6A RNA peaks in 2,987 genes, which we refer to as the synaptic m6A epitranscriptome (SME). SME is functionally enriched in synthesis and modulation of tripartite synapses, and in pathways implicated in neurodevelopmental and neuropsychiatric diseases. Interrupting m6A-mediated regulation via knockdown of reader YTHDF1 in hippocampal neurons alters expression of SME member Apc, and causes synaptic malfunctions manifesting immature spine morphology and dampened excitatory synaptic transmission concomitant with decreased PSD-95 clustering and GluA1 surface expression. Our findings indicate that chemical modifications of synaptic mRNAs critically contribute to synaptic function.
Introduction
Synapses connect billions of neurons into functional neuronal and neuroglial circuits that underlie information processing and drive behavior. Remarkably, synaptic connections are highly dynamic, undergoing continuous synaptogenesis and synaptic elimination throughout development and adulthood,1,2. Synaptic plasticity, the ability of synapses to change at the level of their molecular components, structure, and transmission efficiency in response to neuronal activity, is a fundamental feature of neural network architecture and brain function3.
At the center of activity-induced synaptic alteration, local protein synthesis plays a key role in consolidating neuronal activity into persistent structural and functional change4–13. Aberrant translation at synapses has been linked to autism, fragile X syndrome and other intellectual disabilities, identifying dysregulated local translation as a core pathophysiological mechanism in neurodevelopmental and neuropsychiatric disease14–17. In addition to ribosomes, thousands of transcripts are localized to synaptic compartments encoding proteins involved in diverse functions, including synapse- and neuron-specific molecules, as well as components of the protein synthesis and degradation machinery18.
Recent advances in understanding specific cell-type contributions to synapse refinement has revealed that glial cells play active roles in signaling and facilitating synapse formation, function, plasticity, and elimination, giving rise to the concept of a “tripartite” synapse that comprises presynaptic and postsynaptic terminals as well as glial cells19–21. The mechanisms of mRNA trafficking and local translation extend to astrocytes, where selectively enriched transcripts and translational machinery can be detected in perisynaptic processes22. These new findings implicate multicellular contributions to input-specific, synapse-restricted protein synthesis.
Given the significant contribution of local translation to synaptic function both under physiological and disease conditions, an area of active investigation concerns the post-transcriptional regulatory mechanisms that support the rapid, spatio-temporally coordinated alteration of local proteome composition underlying synaptic plasticity. Current models suggest that signatures carried by individual RNAs, such as sequence motifs, secondary and tertiary structures affect specific interactions with RNA binding proteins and microRNA-mediated regulatory networks23–28. Recent studies of the cellular transcriptome have focused attention on epitranscriptional regulation via enzymatic modification as a mechanism that can dynamically regulate the fate of individual RNA transcripts29,30.
Between 0.1 and 0.4% of all adenosines in mammals are modified by the addition of a methyl group to the N6 position of the adenine to form N6-methyladenosine (m6A), the most abundant mRNA internal modification. m6A is reversibly regulated by methyltransferases (“writers”) and demethylases (“erasers”), and has been shown to affect the splicing, trafficking, stability and translation of individual RNAs31. In contrast to A-to-I editing, which alters the coding sequence of AMPA or 5HT receptors to regulate signaling, m6A alters the local structure and protein binding properties of the modified RNA32. The brain contains the highest level of m6A expression among the major organs33, and many neuronal genes, including genes involved in locomotion, circadian clock regulation, dopaminergic midbrain circuitry, and consolidation of cued fear memory, contain specific modification sites34–38. Although evidence for m6A as a key post-transcriptional regulator continues to grow, few studies have been performed in neurons and there has been, to our knowledge, no investigation of its presence in the synaptic compartment. We therefore sought to address whether m6A could localize to synapses and regulate the local transcriptome.
Here we report an m6A epitranscriptome, which we term “SME”, in the synaptic compartment of the adult mouse forebrain. We report that m6A is present in the synaptic transcriptome, selectively modifying diverse transcripts in all cellular domains of tripartite synapses. Although elevated methylation has a negative impact on synaptic distribution and abundance of the methylated mRNA in a general way, select mRNAs are hypermethylated at synapses. Localized hypermethylated mRNAs showed strong functional enrichment in synaptic pathways, in sharp contrast to hypomethylated transcripts. We report nuclear and somatodendritic distribution of m6A readers, writers and erasers in cortical pyramidal neurons in vivo as well as in cultured primary hippocampal neurons. To interrupt m6A-mediated regulation in cultured hippocampal neurons, we knocked down dendritically localized reader YTHDF1. YTHDF1-deficient neurons had aberrant spine morphology, with decreased spine volume and dampened spontaneous excitatory synaptic transmission concomitant with decreased PSD-95 clustering and surface expression of AMPAR subunit GluA1. Translation of at least one dendritically localized, highly methylated SME member mRNA encoding APC, a microtubule plus-end tracking protein, was inhibited by YTHDF1 deficiency, further demonstrating m6A-mediated regulation of synaptic gene expression and function.
Results
m6A RNA modification in adult mouse forebrain synaptosomes
We initially crossed two m6A-databases with 12 lists of mRNAs localized to axons, dendrites, and neuropils. This comparison revealed substantial overlap (14.0%~56.7%, Supplementary Table S1), strongly suggesting synaptic m6A localization. Next, we isolated a synaptosome (SYN) fraction from the forebrains of adult ICR mice (Supplementary Fig. S1a). SYN was enriched for synaptic proteins (Synaptophysin and PSD-95) and dendritically targeted mRNA of Camk2a, and depleted of soma-restricted markers (Supplementary Fig. S1b and S1c). An integrity check confirmed intact rRNA bands in all samples and enriched mitochondrial rRNA bands in SYN fractions (Supplementary Fig. S1d); m6A dot-blot detected strong m6A immunoreactivity in poly(A) RNA isolated from homogenized tissue (HOM) and pooled F3 and F4 (SYN), but not in an in vitro transcribed unmodified RNA (Fig. 1a, Supplementary Fig. S1e). Densitometry measured that m6A abundance in SYN was ~50% that of HOM (Fig. 1b, Supplementary Fig. S1e).
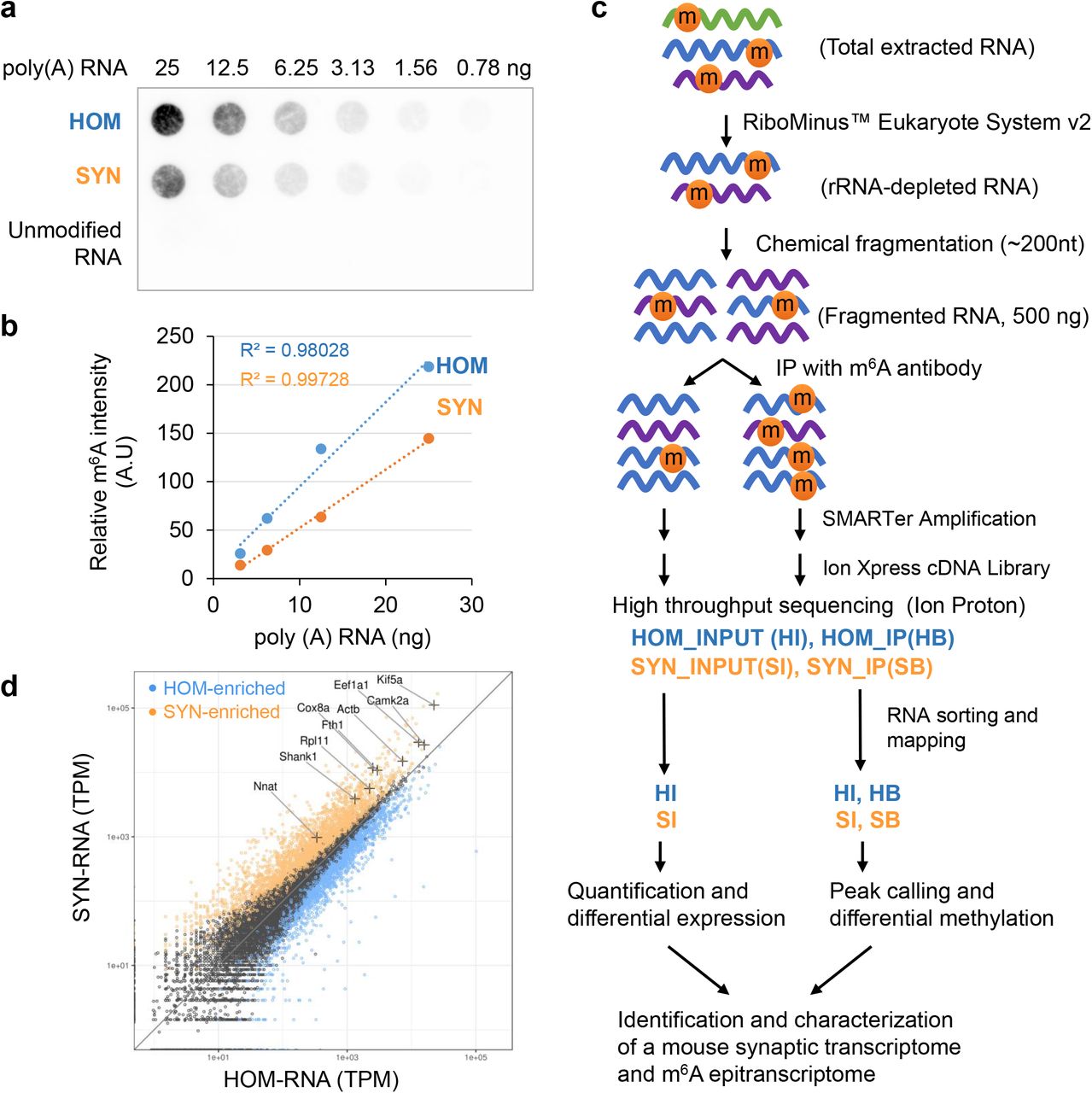
a. m6A dot-blot of poly(A) RNA purified from HOM (homogenate lysate) and SYN (synaptosomes) starting from 25ng in a twofold dilution series. An equal quantity of in vitro transcribed RNA using unmodified nucleotides was used as a negative control. b. Representative dot density plots for the first four dots show linear relationship to the quantity of RNA applied. Approximately 50% m6A immunoreactivity was observed in SYN compared to HOM. A.U.: Arbitrary Units. c. Workflow of low-input m6A-seq protocol. Four sequencing libraries HI, SI, HB, SB representing HOM input, SYN input, HOM IP, and SYN IP were constructed and characterized. d. Scatter plot comparing HOM and SYN gene expression demonstrates SYN-enriched (orange), SYN-depleted (blue), and genes that are equally represented between HOM and SYN (black). Known localized mRNA species (+) are found in the SYN-enriched gene group, supporting successful enrichment of localized transcripts. TPM: Transcripts per million.
Low-input m6A-seq protocol and synaptic enrichment of select RNA
We then sought to identify and characterize the full synaptic m6A-epitranscriptome using m6A-seq39. The limited yield of RNA from synaptosomes presented challenges using existing m6A-seq protocols. Total yield from a single forebrain was approximately 2μg, 1/125th that required by standard protocols39. We established a protocol as described in Online Methods to use submicrogram input, by enhancing sample quantification and optimizing IP, and conducting SMARTer amplification prior to cDNA library construction. Our protocol reliably produced INPUT and IP libraries using ribo(-) RNA from 8 mouse forebrains per replicate (Fig. 1c).
Four libraries (INPUT and IP for HOM and SYN) with biological replicates were sequenced (Supplementary Table S2; reproducibility was evaluated using Pearson’s r squared, Supplementary Fig. S2). To focus on cytoplasmic RNA species, we computationally removed abundant nucleus-restricted and mitochondrial genome-derived RNA using SortMeRNA (Supplementary Table S3). The remaining uniquely-mapped sequences were used for downstream analysis (Supplementary Table S4). Comparing INPUT libraries (HOM: 17,470 RNA; SYN: 15,767 RNA) revealed 3,067 significantly enriched genes at the synapse (p<0.05 FDR, log2FC>0.51) including many known localized mRNA, supporting successful enrichment of synaptic transcripts (Fig. 1d, Supplementary Table S5).
Identification and characterization of synaptic m6A peaks
We called exon-based m6A peaks in HOM and SYN, using recurrent peaks in biological replicates as high-confidence m6A sites. We identified 17,009 peaks annotated to 9,600 genes in HOM, and 16,539 peaks annotated to 9,323 genes in SYN (Fig. 2a, Supplementary Table S6). HOM peaks showed high and significant concurrence with previously published peaks in mouse brain tissue33,40,41 (Fig. 2b, statistical analysis in Supplementary Fig. S3a), indicating robust and reliable peak detection using the low-input protocol. HOM and SYN peaks showed extensive overlap (Fig. 2c), suggesting a common methylation site selection pathway used by both transcriptomes. The established “RRm6ACH” consensus motif (R: A or G; H: A or C or U) was detected in both HOM and SYN m6A peaks (Fig. 2d); the distance from peak summit to the discovered motifs was typically less than 100 bases, associating the motif with the antibody binding site (Fig. 2e for SYN peaks, Supplementary Fig. S3b for HOM peaks). In agreement with previous studies, the majority of peaks were detected in CDS and 3’UTR (Fig. 2f). The peak distribution along mRNA showed characteristic accumulation at the STOP codon (Fig. 2g, 33,42). Peaks associated with START and STOP codons were identified (Fig. 2h, Supplementary Table S7). Notably, genes with peaks associated with STOP codon, but not START codon, were functionally enriched in human phenotypes of “intellectual disability”, “microcephaly”, “muscular hypotonia”, “hypoplasia of the corpus callosum”, and “seizures,” highlighting their relevance to human brain development and cognitive functions (Fig. 2i and Supplementary Fig. S4; GO analyses using expressed genes in Supplementary Tables S8 and S9).

a. Summary of peaks identified in this study. Numbers in () are counts after joining peaks spanning introns. Fold change represents differential coverage density range between IP and INPUT samples. b. Pairwise comparison of m6A peaks identified in HOM to three published studies using mouse brain RNA. The number of peaks identified in each study (grey box), number of overlapping peaks in groups to the left (darker blue), and number of overlapping peaks in groups on the top (light blue). c. Venn diagram of peaks identified in HOM and SYN reveals extensive overlap. () indicates co-occurrence in SYN PEAKS. d. De novo motif discovery identified m6A consensus motif “RRACU” in both HOM and SYN peaks with variation in adjacent nucleotides. e. Frequency plot of motif per 5nt per peak (y axis) against distance from peak summit (x axis) in SYN. f. Percentage of peaks in HOM and SYN associated with annotated transcript features. TTS: transcription termination site; UTR: untranslated region; TSS: transcription start site. g. Metagene profiles depicting relative frequency of peak coverage along transcripts in HOM and SYN. h. Number of peaks associated with START and STOP codons in HOM and SYN. i. Enriched human phenotypes among genes with stop codon-associated SYN peaks (ToppGene). j. IGV view of peaks in representative synaptically hypermethylated and hypomethylated genes. Ckap5: cytoskeleton associated protein 5; Lysmd4: LysM domain containing 4.
We identified peaks specifically associated with SYN or HOM. Visualization with IGV, shown in Fig. 2j and Supplementary Fig. S5, consistently confirmed differential methylation of RNAs at synapses.
Methylation negatively impacts synaptic location and abundance of transcripts
We next investigated the relationship between methylation and synaptic RNA concentration, observing weak but significant negative correlation between transcript abundance and methylation (Supplementary Fig. S6a). Furthermore, the concentration of transcripts at synapses relative to whole cells was also negatively correlated with methylation (Supplementary Fig. S6b). This result is consistent with the overall decreased m6A level in synaptosomal mRNA (Fig. 1a), suggesting there may be a combinatorial result of local degradation, and retention of m6A-mRNA in the soma. Interestingly, methylation in the 5’UTR was not significantly correlated with the synaptic location of transcripts (slope −0.02, Pearson’s r −0.02, p value 0.073). This position-specific feature may explain the elevated peak frequency in 5’ region of synaptosomal mRNAs (Fig. 2g).
A synaptic m6A epitranscriptome (SME)
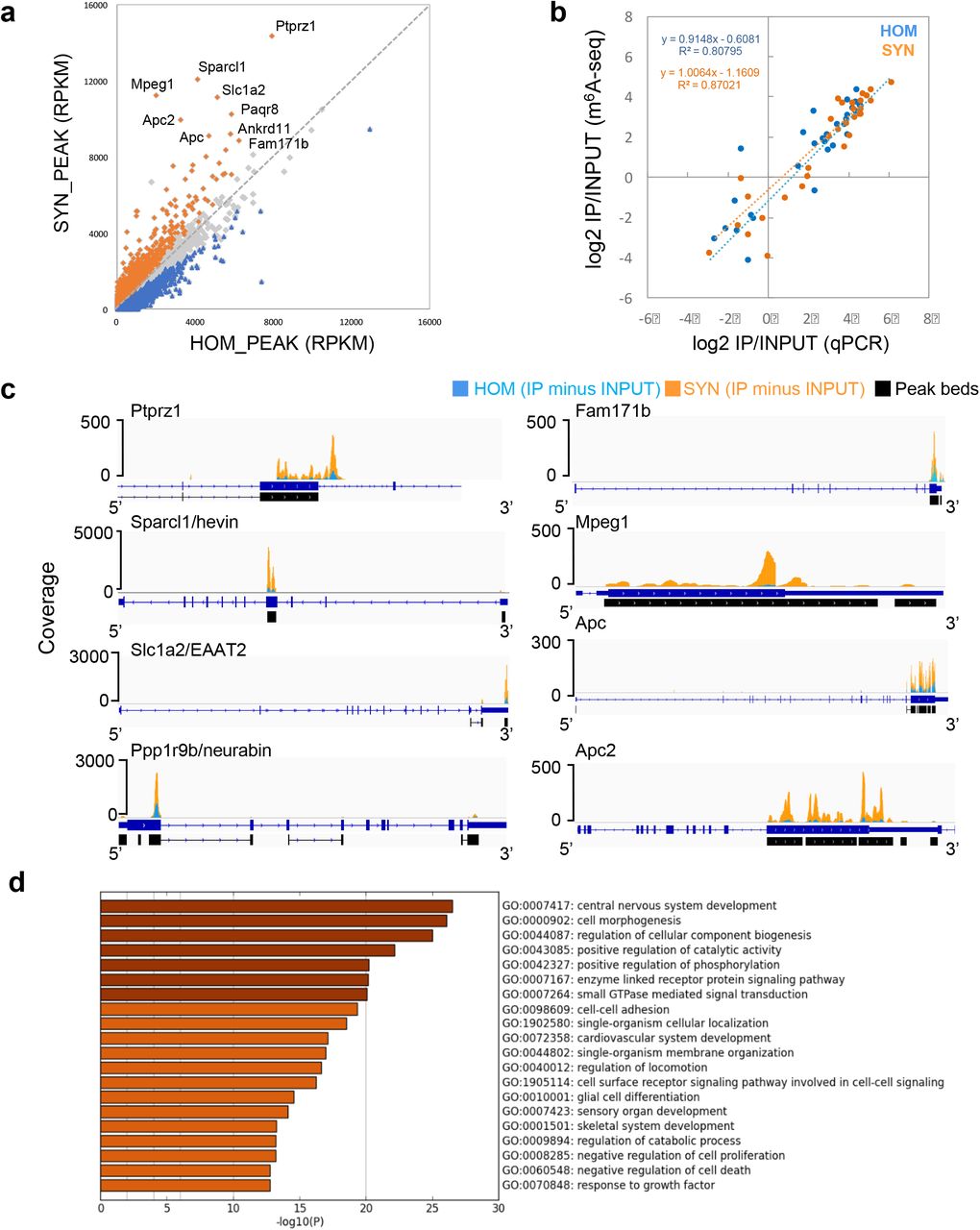
Defining that synaptic m6A RNA should not only be detected in SYN, but also enriched in SYN, we next aimed to identify the full synaptic m6A epitranscriptome. We merged all m6A peaks in HOM and SYN and scored the coverage in the merged peak regions. After normalization to transcript length, 4,329 significantly enriched peaks in SYN in 2,987 genes were identified (p<0.05 FDR, log2FC>0.14, Fig 3a, Supplementary Table S10). We refer to these methylated transcripts as the synaptic m6A epitranscriptome (SME).

a. Scatter plot of normalized reads in HOM peaks (x axis) and SYN peaks (y axis) delineates SYN-enriched peaks (orange), SYN-depleted peaks (blue), and peaks expressed at equal levels in HOM and SYN (grey). Genes carrying the most abundant and enriched peaks at synapses are labeled. b. Peak quantification was validated by qPCR of cDNA libraries. Scatter plot of log2 fold change (PCR, x axis) and log2 fold change (RPKM, y axis) reveals high concordance. c. IGV views of selected abundant and synaptically enriched peaks. Peaks are represented as subtracted read densities (IP minus Input) in SYN (orange) and HOM (blue). d. Top 20 clusters with their representative enriched terms in SME.
High concordance between relative expression and (de)enrichment of methylation between m6A-seq and qPCR measurement was detected (R2=0.80795 for HOM and 0.87021 for SYN, Fig. 3b, Supplementary Fig. S7), validating the results of bioinformatic analysis. Inspection of individual peaks showed more prominent m6A peaks in SYN (Fig. 3c). Gene ontology (GO) analysis identified the most significant ontology cluster associated with SME as “central nervous system development” (Fig. 3d), demonstrating the strong functional specificity of the localized m6A epitranscriptome. A subset of functional clusters was converted into a network layout to illustrate their functional relationships (Supplementary Fig. S8).
Known functions of top candidates based on peak abundance in SYN were examined using manual annotation and data mining in Pubmed. Genes with the top differentially methylated peaks were associated to regulatory mechanisms of synapse formation, function, and elimination in response to local environment, mediated by neuron, astrocytes, and microglia. A recurring theme was neurological disorders such as schizophrenia, autism, anxiety, and intellectual disability (Table 1).
Top methylated transcripts in synaptic m6A epitranscriptome30
Functional partitioning of synaptic transcriptome by m6A modification
To investigate the functional distinction between hypermethylated and hypomethylated transcripts at synapses, we compared SME to the synaptic transcriptome (ST, Fig. 1d). 50% of SME genes overlapped with ST (1,505 overlapping genes), while 1,562 non-overlapping ST-specific genes represent a hypomethylated population, and 1,482 SME-specific genes represent a synaptically hypermethylated population of transcripts (Fig. 4a and Supplementary Table S11). GO analysis revealed that the synaptically hypomethylated genes are functionally polarized to cellular metabolic processes such as “formation of a pool of free 40S subunits”, “oxidation-reduction process”, “metabolism of lipid and lipoproteins”, and “lysosome”. Transcripts enriched both by expression and methylation had greater functional applicability to CNS, such as “regulation of cell migration”, “cell morphogenesis”, and “response to growth factor”. The synaptically hypermethylated genes, however, had a striking functional delineation toward synapses, exemplified by the terms “central nervous system development”, “synapse organization”, and “anterograde trans-synaptic signaling”, all within the top 10 functional clusters with significant q-values (Fig. 4b). Furthermore, significance heat-map showed a compelling distinction between hypo- and hyper-methylated genes associated with the terms in cellular metabolism and synaptic function (Fig. 4c). These results suggest that m6A methylation predicts functional partitioning of transcripts at the synapse, and could thereby potentially affect the synthesis of critical synapse-specific components that alter synaptic organization and transmission (GO analyses using expressed genes as background in Supplementary Tables 12 and 13).

a. Venn diagram relating synaptic transcriptome (ST) and synaptic m6A epitranscriptome (SME). b. Top 10 clusters with their representative enriched terms in synaptically hypomethylated and hypermethylated gene lists. c. Significance heat-map (by p value, FDR) showing enrichment of each term category in synaptically hypo- and hypermethylated gene lists, significance cutoff, 0.05.
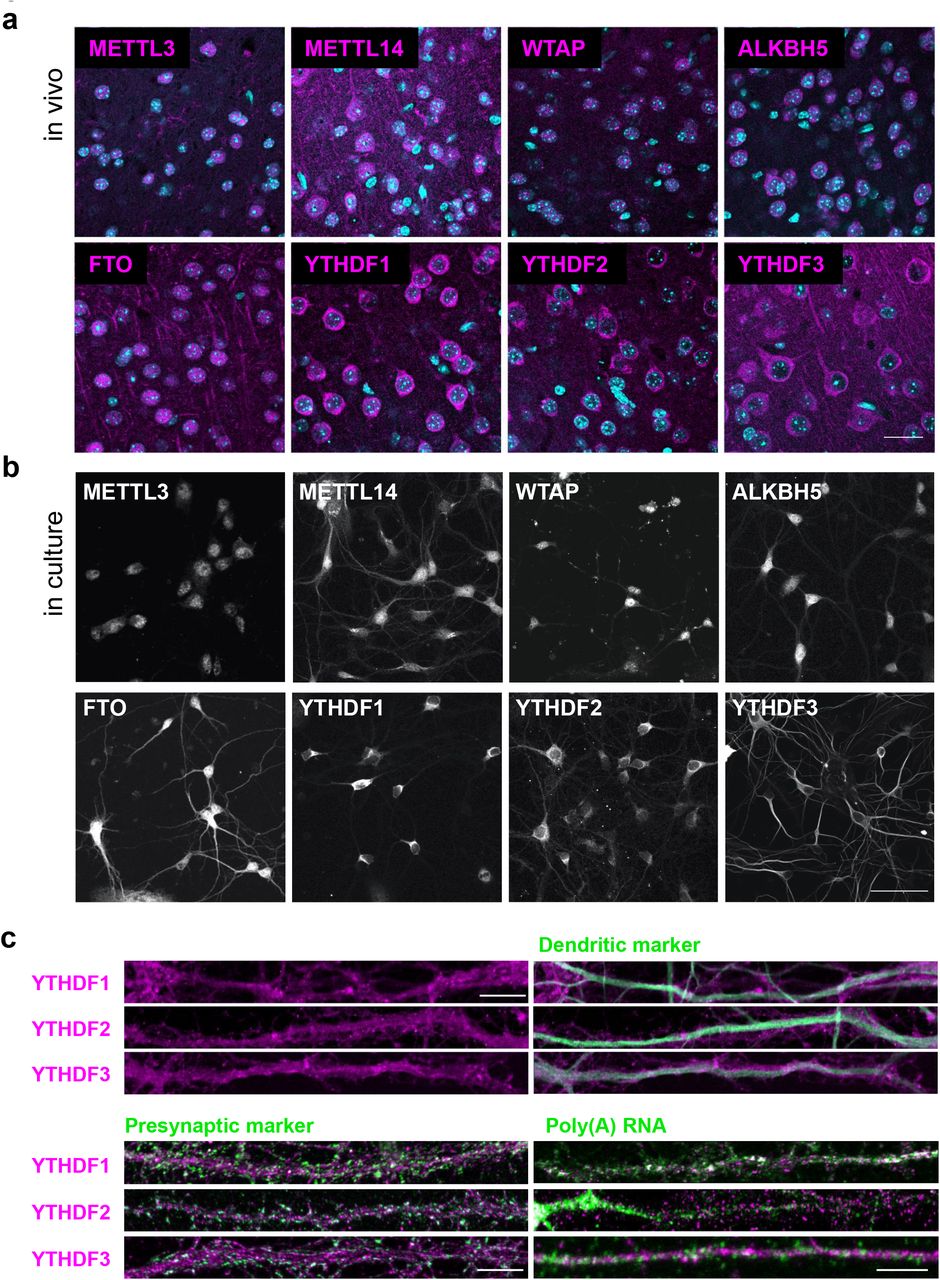
Dendritic localization of m6A regulatory proteins
Selective enrichment of methylated transcripts at synapses raises the possibility of synapse autonomous regulatory mechanisms. We next asked whether m6A regulators could be detected near synapses. Using immunocytochemistry, we found wide distribution of m6A writers, erasers and readers in adult mouse brain slices in the cortex, thalamus, hippocampus and other brain regions. In cortical pyramidal neurons, METTL14, FTO, and YTHDF1, YTHDF2, and YTHDF3 were readily detectable in extra-somatic regions (Fig. 5a). These in vivo localization patterns were replicated in dissociated hippocampal neuronal cultures (Fig. 5b). Co-staining with phalloidin detected METTL14, FTO, and all three YTHDF readers in dendritic shafts adjacent to F-actin-rich spines (Supplementary Fig. S9). Particularly, all three m6A readers YTHDF1, YTHDF2, and YTHDF3 were localized to dendrites in a non-diffuse pattern that partially overlapped with MAP2-positive microtubules (Fig. 5c). Co-staining with presynaptic markers (synapsin I or synaptophysin) demonstrated that all three readers were localized to the vicinity of synapses. Among the three readers, poly(A) RNA colocalized the best with YTHDF1, then YTHDF3, and the least with YTHDF2 in dendrites (Fig. 5c). These results suggest that YTHDF1 may bind to m6A-mRNA in dendrites near synapses, thus may regulate their metabolism locally. In perfused brain slices, YTHDF1 was detected in the MAP2 positive layers, albeit the densely packed cortical environment did not resolve clear colocalization (Supplementary Fig. S10).

a. Confocal images of m6A writers, erasers, and readers in pyramidal cell layers of perfusion-fixed mouse cortical slices. DAPI: cyan; Immunoreactivity: magenta; Scale bar, 20μm. b. Replicate of a in cultured hippocampal neurons. Scale bar, 40μm. c. Confocal images of dendritic processes of dissociated hippocampal neuronal cultures immunostained with m6A readers YTHDF1–3, co-stained with dendritic marker MAP2, presynaptic marker synapsin I or synaptophysin, and poly(A) RNA. Scale bar, 3 μm.
Reduced expression of m6A reader YTHDF1 causes synaptic dysfunction
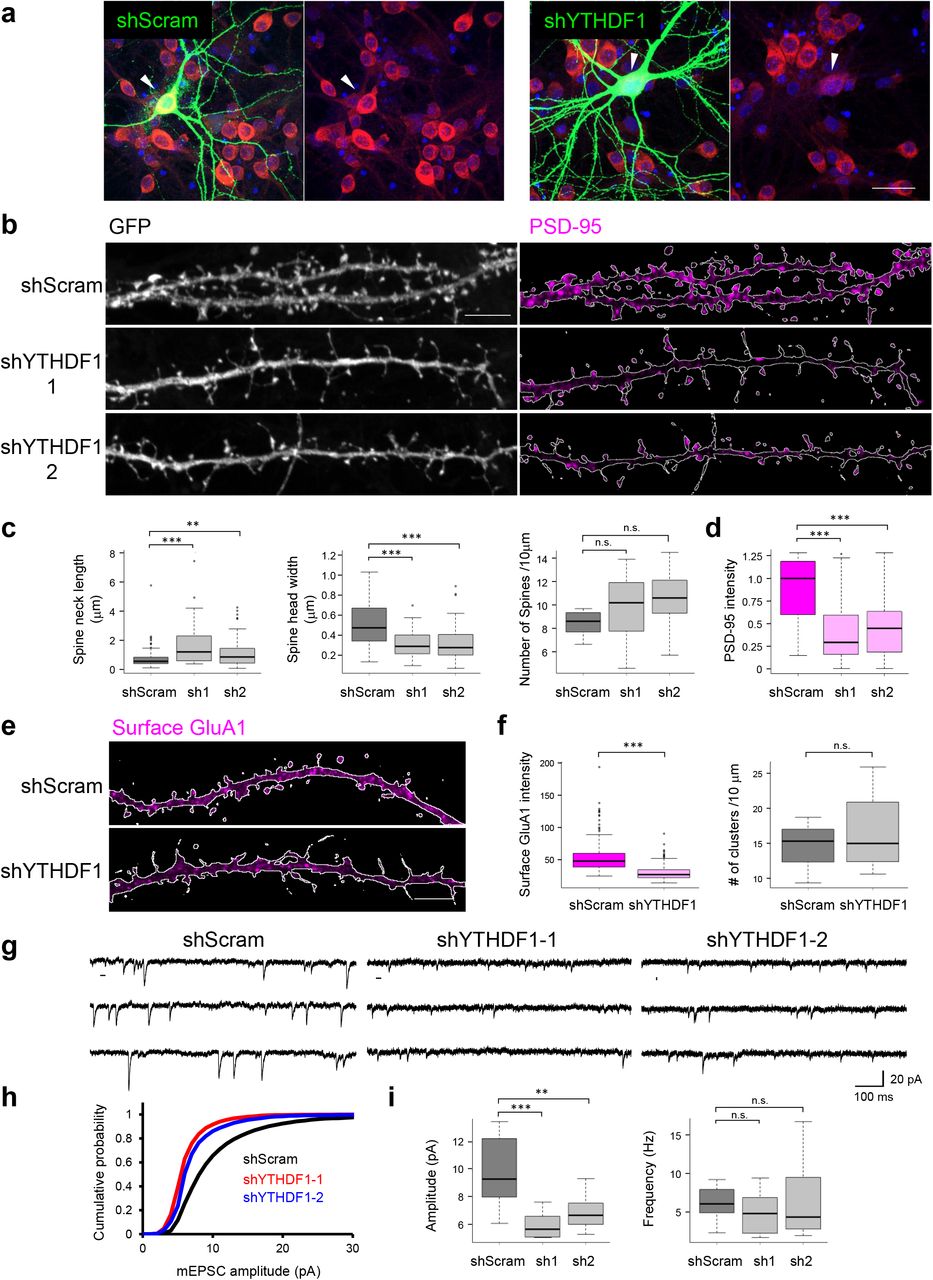
We first attempted to reduce m6A activity by knocking down methyltransferase METTL3 in cultured neurons and observed massive cell death, which prohibited further functional analysis (Supplementary Fig. S11). We next sought to manipulate m6A-mediated regulatory pathways by expressing YTHDF1 shRNAs in dissociated hippocampal neurons (Fig. 6a, Supplementary Fig. S12). In YTHDF1-KD neurons, altered spine morphology, characterized by increased spine neck length (1.69±0.17 in shYTHDF1–1, 1.10±0.09μm in shYTHDF1–2, 0.73±0.06μm in shScramble) and decreased spine head width (0.32±0.01 in shYTHDF1–1, 0.31 ±0.02 μm in shYTHDF-21, 0.51 ±0.02 μm in shScramble) was detected, as well as significantly reduced PSD-95 cluster intensity (1.9-to 2.2-fold decrease in shYTHDF1–1 and shYTHDF1–2, respectively, Fig. 6b-d). Although spine density was not significantly affected, the percentage of spines containing visible PSD-95 clusters decreased (36.8% and 58.6% in shYTHDF1–1 and shYTHDF1–2, 95.0% in shScramble). In parallel experiments, expressing CRISPR/Cas9 targeting YTHDF1 in hippocampal neurons recapitulated the abnormal spine morphology (Supplementary Fig. S13), supporting specific association of the phenotypes with reduced YTHDF1 expression. These data suggest that m6A recognition/binding protein YTHDF1 is required for normal spine development and post-synaptic density formation.

a. Confocal images of DIV19 dissociated hippocampal neurons expressing shScramble-GFP (left panel) or shYTHDF1-GFP (right panel). DAPI (blue), GFP (green), YTHDF1 (red). Arrowheads point to GFP(+) shScram or shYTHDF1 transfected neurons. Scale bar, 10 μm. b. Confocal images of dendritic processes of DIV19 dissociated hippocampal neurons expressing shScramble-GFP or shYTHDF1-GFP (1 or 2). Left, GFP labels morphology of dendritic shaft and spines; Right, PSD-95 staining in the same samples to label post-synaptic density. Contour of affected dendrites was traced as the white lines through GFP expression. Fluorescence PSD-95 signals outside of the contour were masked to restrict quantification to the affected neurons. Scale bar, 5 μm. c. Group quantification of spine neck length, spine head diameter, and spine density; d. Group quantification of PSD-95 cluster staining intensity in spines. e. Confocal images of surface GluA1 staining in DIV20 dissociated hippocampal neurons expressing shScramble-GFP or shYTHDF1-GFP. Contour of affected dendrites is indicated by white line. Scale bar, 5 μm. f. Group quantification of fluorescence intensity of surface GluA1 cluster and GluA1 cluster density along 10μm dendrites. g. Representative mEPSC traces recorded from neurons transfected with shScramble or shYTHDF1 (sh1 or sh2). h. Cumulative plots of mEPSC amplitude distribution. i. Group quantification of amplitudes and frequencies of mEPSCs (n = 12 cells per condition).
Lack of PSD-95 clustering in spines indicated dysregulation of postsynaptic signaling complex assembly and synaptic transmission at the excitatory synapses. We therefore measured surface expression of AMPA-type glutamate receptors, which mediate fast excitatory transmission. Using an antibody recognizing extracellular N-terminus of GluA1 subunit under membrane non-permeable staining conditions, we detected significantly reduced surface expression of GluA1 (fluorescence intensity 53.4±23.7 in shScram vs 29.8± 11.0 in shYTHDF1) but unaltered cluster density (14.5±4.73 in shScram vs 16.7±6.55 clusters per 10 μm in shYTHDF1), indicating impaired trafficking of GluA1 to the synaptic membrane and/or anchoring (Fig. 6e and 6f). Such deficits can result in compromised synaptic transmission. We therefore proceeded to record miniature excitatory post-synaptic currents (mEPSCs) and found a significant reduction of amplitude (9.88±0.70 pA in shScram vs 5.92±0.27 pA and 6.86±0.35 pA in shYTHDF1–1 and shYTHDF1–2, respectively) but not of frequency (6.19±0.57 Hz in shScram vs 4.93±0.78 Hz and 6.25±1.34 Hz in shYTHDF1–1 and shYTHDF1–2, respectively, Fig. 6g-i). These results collectively demonstrate that reduced expression level of m6A reader YTHDF1 in neurons causes both structural and functional deficits of excitatory synapses.
SME member APC expression is altered in YTHDF1-KD neurons
The functional deficits of synapses may be a product of altered SME expression in YTHDF1-KD neurons. YTHDF1 has been previously shown to facilitate translation of m6A mRNAs43. Toward this end, we surveyed potential candidates by crossing SME to a previously published YTHDF1 CLIP/IP dataset43. Among 345 common mRNA, an interesting target was APC (adenomatous polyposis coli), whose mRNA was stabilized but translational efficiency was decreased in siYTHDF1 cells43. Given the critical role of APC in regulating microtubule dynamics, we suspected that its altered expression in YTHDF1-KD neurons may have contributed to the synapse malfunction.
To test this possibility, we performed single-molecule staining of Apc mRNA simultaneously with YTHDF1 protein in cultured hippocampal neurons. GFP expression was used for single neuron morphology labeling. Apc mRNA was detected in dendrites including distal sites, and partially colocalized with YTHDF1 protein (Fig. 7a). In YTHDF1-KD neurons, a remarkable cell-wide decrease in APC immunoreactivity was detected, including depletion from dendrites near synapses; in contrast, APC2, another top SME candidate and APC homologue, showed unaltered expression (APC: 168.9±31.5 vs 25.4±11.3 A.U., APC2: 129.5±16.4 vs 130.6±14.5 A.U. Fig. 7b and 7c, Supplementary Fig. S14). Importantly, Apc mRNA level was comparable between YTHDF1-KD and control cells (10.3±9.90 vs 7.00±7.15 fluorescence puncta per cell, Fig. 7d and 7e). These data suggest that expression of localized SME members is modulated by YTHDF1 expression, exemplified by Apc mRNA, whose translation, but not stability or dendritic targeting, is dependent on m6A reader YTHDF1.

a. Confocal images of DIV7 dissociated hippocampal neurons transfected with pCAG-EGFP. Left, merged images of GFP (white), YTHDF1 immunostaining (magenta) and Apc mRNA (green); Right, dual-color images of YTHDF1 (magenta) and Apc mRNA (green) in insets a1 and a2; Contour of GFP(+) dendrites is indicated by white lines. Scale bar, 10 μm and 5 μm. b. Confocal images of DIV6 neurons with merged GFP (green) and APC (top, magenta) or APC2 (bottom, magenta). Scale bar, 10 μm. c. Group quantification of APC and APC2 immunostaining intensity in GFP(+) neurons. d. Confocal images of DIV7 dissociated hippocampal neurons transfected with shScramble-GFP or shYTHDF1-GFP (sh1) neurons with merged DAPI (blue), GFP (white) and Apc mRNA FISH (green). Scale bar, 10 μm. e. Quantification of Apc mRNA staining clusters per cell in GFP(+) neurons.
SME highlights cell surface-associated intercellular signaling in neuroglia network
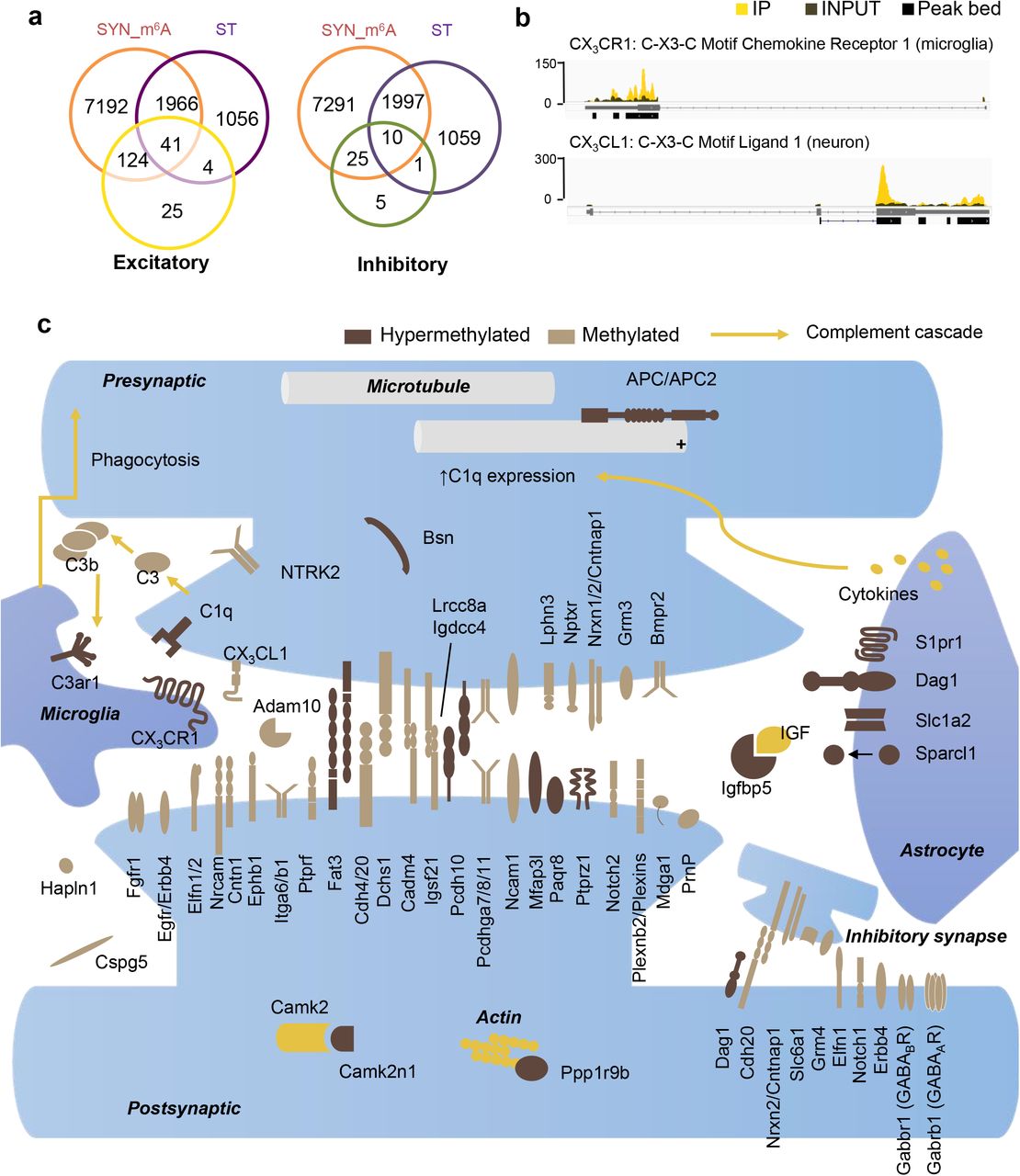
Within the SME-enriched functional terms “synapse” and “cell junction,” surface receptor pathways involved in cell-cell signaling were among the most abundant methylated transcripts. To further dissect SME function, we crossed our data set to a recently identified collection of synaptic cleft residents44, and found that 85% of synaptic cleft proteins at both excitatory and inhibitory synapses were encoded by methylated transcripts. Thus methylation-mediated regulation may have a wide impact on the biosynthesis of this functional class of proteins (Fig. 8a). In addition to highly abundant astrocytic methylated transcripts Sparcl1 and EAAT2, whose proteins promote synapse formation and remove glutamate from synapses, we identified complement cascades in microglia surveillance in SME. The mRNAs encoding CX3CL1, secreted by neurons potentially as a “find me” signal, and CX3CR1, the receptor for CX3CL1 expressed exclusively by resident microglia in enhancing synapse refinement, were both highly methylated (Fig. 8b, Supplementary Fig. S15). By further data mining and assigning the annotated SME genes to different cell-types, a preliminary yet exciting tripartite SME network emerged (Fig. 8c). Critical components in all synaptic domains such as pre-synaptic protein bassoon, post-synaptic CamK2n1, a large portfolio of pre- and post-synaptic adhesion proteins, secreted proteins and reciprocal receptors involved in neuron-astrocyte and neuron-microglia communication were encoded by methylated mRNAs at the synapse, of which many have been identified as playing critical roles in sculpting synaptic connections.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A. Venn diagram of methylated transcripts in SYN (SYN_m6A), transcripts enriched in SYN (ST), and excitatory synaptic cleft proteins (left) and inhibitory synaptic cleft proteins (right) recently identified by Loh et al., 2016. 165 of 194 (85%) excitatory, and 35 of 41 (85%) inhibitory, synaptic cleft proteins are encoded by methylated mRNA identified in SYN. B. IGV view of peak coverage of mRNAs encoding chemokine-mediated neuron-microglia communication pathway CX3CL1-CX3CR1. C. Curated assignment of proteins encoded by methylated mRNA encoding synaptic cleft proteins and by mRNAs carrying the most abundant methylation sites in SME at excitatory and inhibitory tripartite synapse.
Discussion
The protein composition of synapses is highly dynamic and regulated in an activity-dependent manner. mRNA trafficking and local protein synthesis at a synapse plays an essential role, supplying the initial changes in protein abundance required for maintaining and modifying connections18,45. A rich cellular toolbox of post-transcriptional regulatory mechanisms such as microRNA, nonsense mediated decay, RNA binding proteins, and post-translational modification of translation elongation factors has been shown to regulate activity-induced translation at synapses. The SME identified in the current study reveals another layer of complexity in the regulation of the synaptic proteome. This enzyme-mediated, reversible modification, and its impact on the metabolism and translation of individual mRNA, comprises a highly flexible regulatory mechanism, effective at the single copy level.
SME consists of 4,329 m6A peaks that are found selectively enriched at the synapse; half of the 2,987 genes carrying these peaks are not preferentially localized to synapse. This suggests the existence of mechanisms for differential methylation or, alternatively, selective mRNA trafficking which have yet to be identified. Functional enrichment analysis revealed that the 1,482 hypermethylated genes are enriched for synapse-related functions such as synapse maturation, organization, assembly, and modulation of synaptic transmission, in sharp contrast to the localized hypomethylated transcripts, which are primarily involved in cellular metabolic pathways. The functional impetus for this deliberate partitioning is not clear. An intriguing possibility is that m6A-mediated spatiotemporal control of gene expression is required for coordinating activity-dependent translation of a cohort of mRNA. In support of this, a rapid and site-specific surge in m6A level was shown in response to UV-triggered DNA damage, indicating that changes in modification status can occur in a subset of RNAs involved in DNA repair pathways46. Moreover, m6A can exert control over translation via direct recruitment of ribosomes47,48 and/or co-translational interaction dynamics between the protein products49. Together these observations support a localized, activity-dependent dynamic proteomic network regulated by m6A at the synapse.
We found writer METTL14 and eraser FTO distributed to the distal processes of pyramidal neurons and, although the methylation activity of standalone METTL14 is under debate31, their presence raises the possibility of local enzyme-mediated (de)methylation. In future studies, this possibility could be tested using in vitro measurement of RNA methylation after stimulating isolated synapses. We also detected all three cytoplasmic readers (YTHDFs) distributed distally, suggesting versatile downstream signaling potential. Previous studies have demonstrated that binding of YTHDF1–3 to m6A mRNAs enhances translation efficiency and RNA degradation43,47,48,50,51, and in general, m6A is associated with shorter RNA half-life51,52. These apparently conflicting roles of m6A has led to a working model of sequential binding of YTHDF1 and YTHDF2 to help methylated mRNA achieve steady-state levels rapidly, with YTHDF3 acting as an enhancer of both degradation and translation53,54, thereby decreasing response time between stimulus and phenotypic output43. This model may also help reduce expression “noise” by degrading excess mRNA and enhancing translation of scarce transcripts. Both mechanisms can benefit synaptic stability and plasticity in response to activity, and we predict the localized readers act synergistically with the local m6A epitranscriptome to achieve robust production of synaptic responses.
Knockdown of reader YTHDF1 in cultured primary neurons resulted in synaptic transmission deficits and immature morphology of spines. Such phenotypes can be associated with synaptic pathophysiology. We identified a downstream molecular target of YTHDF1, SME member Apc mRNA, whose abundance and dendritic localization were not affected, but expression decreased with YTHDF1 knockdown. Previously, the APC cKO mouse model has shown abnormality of central synapses (increased spine density, frequency of miniature excitatory synaptic currents, and long-term potentiation), leading to a multi-syndromic neurodevelopmental disorder55, suggesting that altered APC expression and subsequent cytoskeletal regulation might partially contribute to the synaptic dysfunction. Dysregulated m6A mRNA translation at synapses may underlie disrupted long-term synaptic plasticity, learning and memory, and neurodevelopmental disorders. In support of this, human genetic research has identified the m6A reader YTHDC2 as one of the risk factors for autism spectrum disorder56. The functional analysis of synapses in the current study therefore warrants further investigation in m6A-mediated local translation and its relation to neuropsychiatric disorders.
A fascinating aspect of SME is its invoking of the tripartite local translation model, implicating mRNA methylation-mediated local translation in multicellular interactions between neurons, astrocytes, microglia, and oligodendrocytes. Methylated mRNA encoded proteins that coordinate intercellular cross-talk, such as postsynaptic membrane receptors (Ptprz1), synaptogenic proteins secreted from peri-synaptic astrocyte processes (Sparcl1), neuronal regulators of synapse turnover (Paqr8 and Igfbp5), chemokines secreted by neurons to attract microglia (CX3CL1), and the reciprocal receptors on the microglial membrane (CX3CR1). Advanced understanding of molecular communication pathways is required to elaborate how local translation supports combined contributions of synaptic components to the activity and plasticity of tripartite synapses. SME provides an initial candidate list for future mechanistic dissections, highly enriched in specified synaptic functions. Interestingly, of 224 recently identified peri-synaptic astrocyte mRNAs, 180 (80%) overlapped with ST and 152 (68%) overlapped with SME, in contrast to only 23% overlap between the 116 soma-derived astrocyte mRNA and ST22. Thus, RNA metabolism in peri-synaptic astrocyte processes may be directly regulated by m6A. Future molecular dissection of m6A regulation in tripartite synaptic function requires site-directed manipulation of the methylated adenosine residue and/or adjacent residues to disrupt methylation dynamics on individual transcripts. A recent single nucleotide-resolution m6A-seq method may provide valuable information in this regard57.
Ultimately, m6A may be one of many covalent RNA modifications regulating synaptic epitranscriptome30,58. 5mC methylase NSun2 has been shown to colocalize with FMRP in dendrites, where it may exert control over synthesis of a subset of mRNA in an activity-dependent manner59. NSun2 mutations have been associated with intellectual disability as well as other neurodevelopmental deficits60,61. The diverse functional impact of modified transcripts suggests that RNA modification can greatly enhance customization of the local proteome. Such mechanisms are perhaps indispensable for generating context-dependent responses at synapses with spatiotemporal coordination; and aberrant modification events may therefore lead to the pathogenesis of neurodevelopmental and neuropsychiatric disorders.
METHODS
Methods and any associated references are available in the online version of the paper. Accession code PRJNA388019.
Author Contributions
Wang DO conceived and designed the project. Hong WT purified synaptosomes, performed RNA extractions and m6A dot-blot, data mining. Wang DO and Ohara T performed library construction; Merkurjev D, Iida K, Goldie BJ, Yamaguti H, Pellegrini M performed bioinformatics analysis; Ohara T performed immunostaining; Oomoto I constructed shRNA and performed KD in cultured hippocampal neurons. Kawaguchi S performed electrophysiology. Wang DO and Goldie BJ wrote the manuscript, Martin K and Hirano T supervised parts of the project. All authors participated in data analysis and interpretation, and made indispensible contributions.
Acknowledgements
We thank Yasunori Hayashi for critical reading of this manuscript. CeMI imaging center at iCeMS and supporting facility at medical school of Kyoto University for technical support. This work was supported by grants KAKENHI17H03546, KAKENHI26702038, KAKENHI26115515, Hirose foundation, Astellas foundation to DOW. BJG is supported by Japan Society for the Promotion of Science foreign researcher fellowship.
References (Harvard Style)




