

Abstract
Many subcellular structures assemble via liquid-liquid phase separation (LLPS) to form compartments without membranes. Though it has been shown that RNA is a central driver and modulator of LLPS, it is not yet known how these liquid droplets establish and maintain individual identities. Here we examine how mRNAs are recruited to or excluded from liquid compartments based on their sequence and ability to self-associate. We find that the specific secondary structure of a cyclin mRNA is required for it to assemble into distinct droplets and be excluded from other droplets containing functionally-unrelated mRNAs. This molecular mechanism explains how sequence-encoded shape information in RNA promotes the coexistence of the diverse array of RNA-rich liquid compartments found in a single cell.
One Sentence Summary Identity in cellular, phase-separated compartments emerges from RNA-RNA complexes encoded by mRNA secondary structures.
The formation of non-membrane bound organelles through the condensation of macromolecules is a newly appreciated mechanism of intracellular organization. These condensates often display liquid-like properties and form through liquid-liquid phase separation (LLPS) (1, 2). The growing list of LLPS-assembled compartments includes the nucleolus, RNA granules, cell signaling hubs, the spindle matrix, chromatin, the synaptonemal complex, and many pathological neuronal granules (3–12). Despite the growing appreciation of the variety of liquid-like assemblies employed in diverse cellular processes, a fundamental unsolved problem is how liquid droplets recruit distinct constituents and retain independent identities, rather than fusing into a singular compartment. This is especially remarkable given the fact that many of the constituents are highly disordered proteins and the droplets they form display such a propensity to fuse (2). RNA has been shown to be a driver of LLPS and can modulate the material properties of droplets (13–20), yet there is little known about how RNA can impact the identity and maintenance of coexisting liquid compartments. Here we show that mRNA secondary structure is required for droplet identity and likely acts through higher-order interactions between mRNAs and RNA-binding proteins. This illustrates how molecular scale interactions can encode the identity and emergent properties of micron-scale liquid compartments in cells.
Whi3 is a polyQ-containing RNA-binding protein identified in Saccharomyces cerevisiae through its role in cell size control (21). Whi3 is known for its roles in morphogenesis, memory of mating, and stress responses, where the protein forms aggregates and associates with RNA-processing bodies (22–26). The homolog in the filamentous fungus Ashbya gossypii has an expanded polyQ tract, a sequence known to promote self-assembly and forms liquid-like droplets that function to locally regulate nuclear cycling and hyphal morphogenesis (27, 28). In vitro, Whi3 polyQ-dependent LLPS is driven by the presence of specific RNAs encoding regulators of either the cell cycle (e.g. the cyclin CLN3) or actin (e.g. the formin BNI1) (14). We see two distinct types of Whi3 droplets in Ashbya cells: droplets clustered around nuclei where CLN3 mRNAs are positioned and droplets at growing cell tips where BNI1 mRNAs are localized (Fig. 1A, movie S1, (27, 28)). These different droplets have different amounts of Whi3 (Fig. 1B) and different rates of Whi3 incorporation based on accumulation of Whi3-Halo, visualized after a pulse of dye (Fig. 1C). These differences suggest Whi3 droplets around nuclei and at tips are somehow structurally distinct.

A. Above, Representative fluorescent image shows Whi3-tomato condenses into droplets around nuclei and at tips in Ashbya gossypii. Green arrows denote a tip droplet and pink arrows denote nuclear associated droplets. Scale bar 5 um. Below, nuclear associated Whi3 droplets (pink arrow) accumulate and fuse over time.
B. Mean intensity of Whi3-tomato is higher in tip droplets (green) than nuclei droplets (pink).N= 70 nuclei and 30 tips. Black bars indicate the medians.
C. Incorporation of JF649 Halo dye into Whi3-Halo droplets at tips is greater than into Whi3-Halo droplets around nuclei over time in N= 40 Nuclei and 15 tips. Black bars indicate the medians.
D. BNI1 mRNAs (green) and CLN3 mRNAs (pink) do not co-localize in Ashbya cells. In contrast, BNI1 mRNAs (green) co-localize with SPA2 mRNAs (pink). The green arrow marks where the two RNAs overlap at the growing tip. Nuclei are labeled in blue. Images are representative of N≥20 cells. Scale bar 5 um. E. Quantification of fraction of mRNA co-localized between BNI1 and CLN3 or BNI1 and SPA2 at either tips or around nuclei. N= 40 tips and nuclei for 20 cells.
The distinct droplet properties in cells may depend on extrinsic features of the local cytosolic microenvironment near nuclei and at tips or may arise due to the presence of different constituents within the droplets. CLN3 and BNI1 mRNAs did not co-localize in the cytoplasm as assessed by smRNA F.I.S.H., although they were co-expressed by the same nuclei ~20% of the time, shown by bright signals within the same nucleus (Fig. 1, D and F). This suggests that there are intrinsic, compositional differences between droplets and that the components are mutually exclusive. In contrast, SPA2 mRNA, another Whi3-interacting partner that encodes a different cell polarity regulator, did frequently co-localize with BNI1 mRNAs especially at growth sites (Fig. 1, D and F) indicating that Whi3 droplets can contain mRNAs with different sequences. Thus, mRNAs encoding functionally-related proteins co-localize while functionally unrelated mRNAs bound by Whi3 do not. How can distinct Whi3-binding mRNAs segregate to different droplets in a common cytoplasm?
To address this question, we tested if the mRNA sequence alone was sufficient to generate individuality in droplets in a minimal reconstitution system of pure Whi3 protein and cyclin and formin mRNAs. In vitro, similarly as seen in cells, Whi3 droplets made out of BNI1 mRNA were more enriched in Whi3 protein than droplets made with CLN3 mRNA and accumulated more protein with time (Fig. S1A). Remarkably, when CLN3 mRNA is added to Whi3 droplets made with BNI1 mRNA (Fig. 2A), CLN3 mRNA preferentially assembled into new, distinct droplets that did not fuse with the droplets containing BNI1 mRNA (Fig. 2, B and C). In contrast, labeled BNI1 mRNA readily incorporated into the droplets made of BNI1 mRNA (Fig. 2, B and C). Notably, labeled SPA2 mRNA also incorporated into BNI1 droplets (Fig. 2, B and C). CLN3 mRNAs also preferentially assembled new droplets instead of being incorporated into SPA2 droplets (Fig. S1B). Similar results were obtained when droplets were assembled in concentrated fungal cell extract (Fig. S1C). Thus, as seen in cells, CLN3 and BN11/SPA2 mRNAs assemble into distinct droplets in vitro, suggesting that droplet identity is encoded by the mRNA.

A. Experimental schematic of in vitro droplet recruitment assay.
B. Representative images show cy3-labeled CLN3 mRNA (pink) is not efficiently recruited into Whi3-BNI1 droplets (green). In contrast, cy3-labeled BNI1 or SPA2 mRNA (pink) is recruited into Whi3-BNI1 droplets (green).
C. Measurement of how much cy3-labeled mRNA is recruited into Whi3-BNI droplets from three independent experiments. NS, not significant, p > 0.05; **, p < 0.01; ***, p < 0.001 by unpaired t test.
D. Simplified schematic describing in vitro RNA-only, droplet assay where CLN3, BNI1, or SPA2 are incubated with positively charged spermine tetrahydrochloride to induce electrostatic mediated phase separation into liquid or gel-like droplets. Representative images show RNA only assemblies.
E. Representative images showing CLN3 droplets (pink) are excluded from BNI1 droplets (green) when the RNAs are mixed in the presence of spermine. In contrast, BNI1 droplets (green) overlap with SPA2 droplets (pink).
F. Pearson’s Correlation Coefficient in RNA only experiments between BNI1 and either CLN3 (left) or SPA2 (right). BNI1 and SPA2 fluorescent signals are significantly more correlated to each other than BNI1/CLN3 and CLN3/SPA2 signals (far left), indicating CLN3 is still excluded from functionally unrelated assemblies in the absence of Whi3.
The mRNA sequences could control the identity of droplets by favoring homotypic or specific heterotypic interactions between RNA molecules. To test for specific RNA-RNA interactions, we incubated RNAs with polyvalent cations in the absence of Whi3 to induce electrostatic mediated phase transition of the mRNAs (29). The mRNAs by themselves were all are capable of homotypic assembly into either liquid or gel-like droplets (Fig. 2D). Strikingly, CLN3 mRNAs had only minimal co-localization with BNI1 or SPA2 mRNAs, whereas BNI1 and SPA2 mRNAs had significantly more co-localization with each other than CLN3 had with either mRNA associated with cell polarity (Fig. 2E). Thus, sequence-based features of the mRNA can underpin the assembly of distinct, immiscible structures.
We next investigated what features of the mRNA sequence generate specificity. An mRNA with a scrambled CLN3 coding sequence (cln3 scr) but identical nucleotide composition and Whi3 binding sites formed Whi3 droplets (Fig. S2A) but no longer showed any droplet specificity (Fig. 3, A and B). Given that the charge, length, nucleotide composition and number of Whi3 binding site number (valency) were identical in this construct, we hypothesized that the secondary structure of the mRNA was responsible for droplet specificity. When CLN3 mRNA was heated to 95°C to completely disrupt secondary structure, and then immediately added to Whi3-BNI1 droplets, this “melted” mRNA readily incorporated into BNI1 droplets (Fig. 3, A and B). When melted CLN3 mRNA was slowly refolded using step-wise temperature changes, the “refolded” CLN3 mRNA showed significantly less mixing than melted CLN3 mRNA, but more mixing than native CLN3 mRNA (Fig. 3B). Thus, specificity information in CLN3 mRNA can be eliminated by scrambling the sequence outside of Whi3 binding sites or by disrupting secondary structure without introducing any changes to the RNA sequence.

A. Representative images showing the recruitment of scrambled (cln3 scr), melted, and refolded (CLN3 refold) CLN3 mRNA (pink) into Whi3-BNI1 droplets (green) (8 uM Whi3, 5 nM RNA).
B. The recruitment coefficients from A from three independent experiments. Outliers are indicated with (●) marks. *, p<0.05; ***, p < 0.001 as determined by unpaired t test.
C. Base-pairing probability compared among CLN3, cln3 scr, and CLN3 refold. Arcs represent base pairs and are color-coded by probability.
D. Secondary structure models for the first 400 nucleotides of CLN3, CLN3 refold, and cln3 scr. Whi3 binding sites are denoted in orange.
What exact features of CLN3 mRNA secondary structure promote specificity? We performed SHAPE-MaP, a method that uses a small electrophilic molecule to modify nucleotides in regions that are highly flexible, followed by reverse transcription to detect modified nucleotides and sequence changes, and deep sequencing to identify structured RNA regions. (30, 31). SHAPE-MaP was carried out on untreated CLN3, refolded CLN3, and cln3 scrambled mRNA to evaluate differences in secondary structure (Fig. 3C, S2, B and C). Many of the first 400 nucleotides in the CLN3 sequence have low SHAPE reactivity and low Shannon entropy (Fig. S2D, purple shaded regions), suggesting a high frequency of paired nucleotides and a highly-folded, secondary structure. This region was one of the most changed in the refolded CLN3 sequence, with a statistically significant increase in SHAPE reactivity compared to the native CLN3 sequence (Wilcoxon rank sum test, p <0.001), as many nucleotides that were originally less reactive became more reactive (Fig. 3C, S2, B and D), suggesting that this region transitioned to a more unstructured state during the refolding process. These data suggest that melting and refolding allows the RNA to sample conformations that are significantly different from the structures formed during transcription. The cln3 scrambled sequence showed an almost completely different SHAPE profile with dramatically altered secondary structure throughout the sequence (Fig. 3, C and D, Fig. S2C). Thus, secondary structure perturbations accompany the loss of droplet specificity.
One hypothesis to explain how RNA secondary structure might impact droplet specificity is that stem-loop elements might selectively display or mask sequences capable of base-pairing with other RNAs. The CLN3 sequence contains five regions capable of heterotypic base-pairing with BNI1 sequences (Fig. S3A). Most of these regions were located in areas of low SHAPE reactivity and thus more structured regions (Fig. S3B), suggesting that in the native CLN3 RNA these complementary regions are relatively inaccessible for base pairing with BNI1. However, when the CLN3 mRNA is melted, these regions might become available to base-pair with complementary sequences on BNI1, providing a possible explanation for the structure-dependent loss of droplet specificity.
A striking difference in the structures between melted, scrambled, and untreated CLN3 RNAs was the position of Whi3 binding sites relative to topological features such as stem-loops (Fig. 3D). In the untreated CLN3, three of the four Whi3 binding sites occurred in or overlapped single-stranded loops, whereas after melting and refolding, these sites were no longer in the same accessible context but rather found in stems, which may be less accessible to Whi3-binding. In cln3-scr, the Whi3 sites were also rearranged and instead incorporated into a single, extended run of stem-loops (Fig. 3D). Thus, in addition to biasing specific RNA-RNA interactions, RNA secondary structure may also influence how Whi3 interacts with target RNAs. This idea prompted us to examine roles for Whi3-binding to different RNAs in maintaining droplet specificity.
We found that a mutant of the cln3 RNA (cln3 5m) in which the binding sites for Whi3 were altered no longer nucleated Whi3 droplets (Fig. S4A). Remarkably, however, cln3 5m nevertheless incorporated into Whi3-BNI1 droplets (Fig. 4A). Interestingly, this mutant RNA had an appreciably different secondary structure from wild-type CLN3 (Fig. S4, B and C), despite containing only five interspersed point mutations. To determine if Whi3 binding alters RNA conformation, we used single-molecule FRET (smFRET) to sample the conformational dynamics of CLN3 and BNI1 RNAs in the presence and absence of Whi3 (32). Fragments of CLN3 or BNI1 mRNA were labeled with donor fluorophores and hybridized to an 18 nt oligo with an acceptor fluorophore bound to a coverslip (Fig 4B). In the absence of protein, CLN3 RNA showed high FRET values indicative of a compacted state where donor and acceptor are in close proximity, while BNI1 RNA showed a broader distribution of and somewhat lower FRET values, indicating a less compact state (Fig. 4C, purple shaded regions). Upon addition of Whi3, CLN3 RNA FRET values decreased, indicating more extended RNA conformations were induced from the RNA-alone condition. In contrast, BNI1 RNA showed a more substantial broadening of FRET values when complexed with Whi3 than CLN3 (Fig. 4, C and D) suggesting that Whi3-BNI1 complex is more dynamic. Dwell-time analysis, which measures the time between minimum FRET values, revealed that the Whi3-induced dynamics are over three times faster for BNI1 than CLN3 (Fig. 4D). These differences in FRET behavior show that different mRNAs react differentially in their intramolecular fluctuations to the presence of Whi3 providing a second layer of distinction amongst the droplets made with different RNAs.
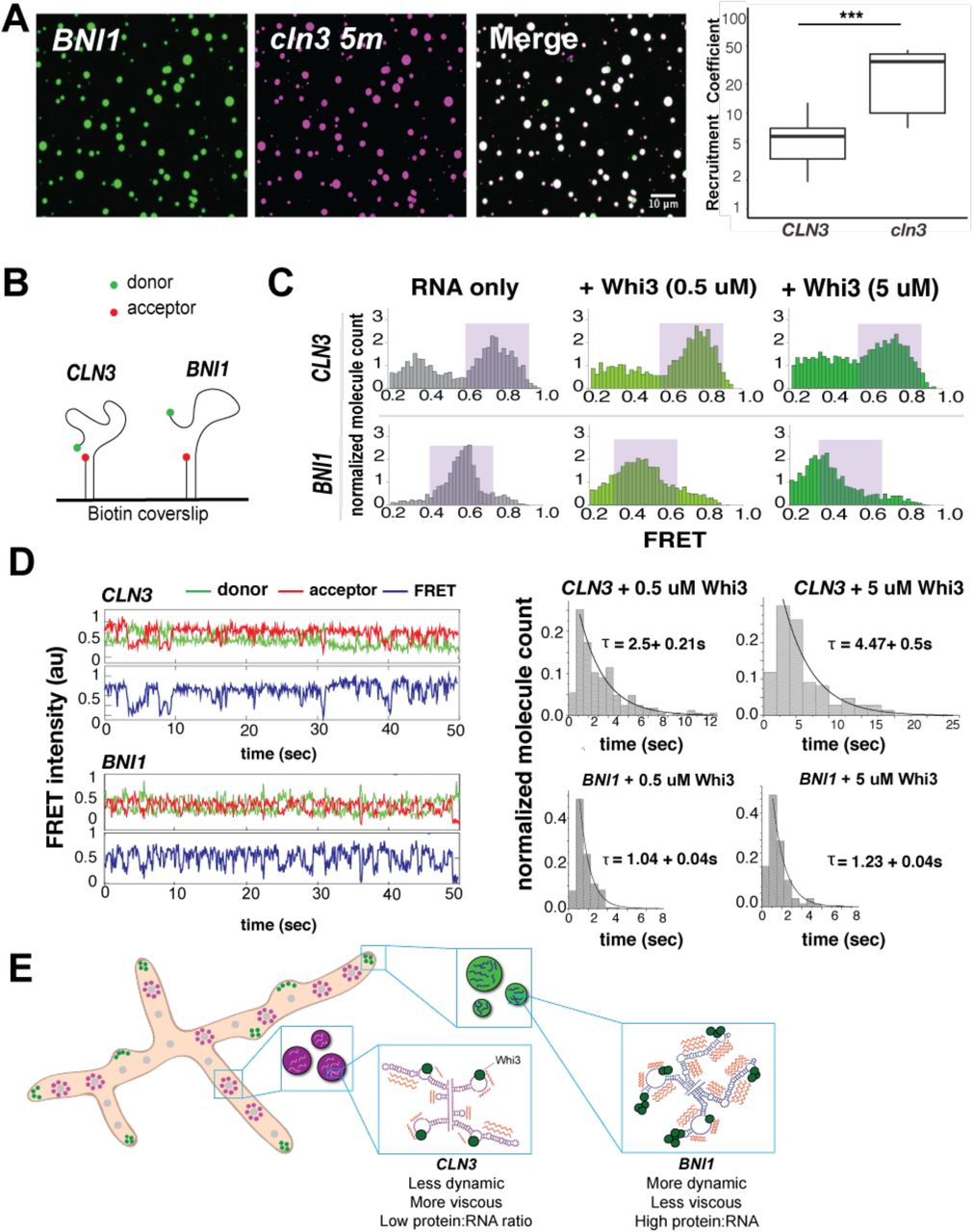
A. Representative images showing the recruitment of the mutant cln3-5m mRNA (pink) into Whi3-BNI1 droplets (green) (8 uM Whi3, 5 nM RNA). Recruitment coefficient of cln3-5m shows more efficient RNA recruitment compared to CLN3 from three independent experiments. *, p<0.05; ***, p < 0.001 as determined by unpaired t test.
B. RNA substrates composed of truncated CLN3 or BNI1 sequences and a Cy5-Cy3 FRET pair are annealed to a Biotin labeled RNA and immobilized on a Biotin coverslip.
C. FRET histograms before (gray) and after (green) 0.5 or 5 uM Whi3 addition. Purple shaded regions denote high and mid FRET states for CLN3 and BNI1, respectively.
D. Averaged cy3 (green), cy5 (red) intensities, and representative FRET traces (blue) obtained from smFRET experiments of CLN3 and BNI1 in the presence of 5 uM Whi3. Dwell time analysis shows slower FRET fluctuations for CLN3 than BNI1 in the presence of Whi3 (0.5 uM and 5 uM).
E. Proposed model in which RNA secondary structures have been “tuned” to promote the selective uptake of distinct RNAs and protein constituents into droplets leading to diverse dynamics (orange zig-zags) of different droplet complexes
Our findings suggest that Whi3 binding alters the conformational dynamics of target RNAs. We speculate that this could lead to a form of kinetic sorting, that serves to maintain droplet identities. In this model, the slower fluctuations of CLN3 bound to Whi3 may be one source of exclusion from the more rapidly fluctuating BNI1-Whi3 complexes. Such altered dynamics might also drive the droplet-specific, emergent material properties (viscosity and surface tension) that we reported previously (14) and that may also serve as barriers to homogenization.
We show here that RNA secondary structure, derived from mRNA sequence and protein interactions, encodes a key level of specificity in the incorporation into liquid droplet compartments. We hypothesize that RNA secondary structures have been optimized to promote particular RNA-RNA interactions and the selective uptake of distinct RNAs and protein constituents into droplets. Furthermore, protein binding to different sequences can lead to varied dynamics of complexes (Fig. 4E). We hypothesize that these different RNA conformation dynamics are another feature promoting immiscibility of coexisting droplets. Our findings indicate how specificity in RNA recruitment can be achieved in intracellular biological condensates and show that mRNAs can encode this specificity in their secondary structure. Given the large number of distinct RNA-based liquid bodies in a single cell, these mechanisms are likely broadly relevant to explain how droplets achieve and retain individuality.
Author Contribution
EML designed and performed experiments, analyzed data, prepared figures and drafted manuscript; PB and AN designed and performed experiments, analyzed data, and edited manuscript.; GM performed experiments and analyzed data; CW performed experiments and analyzed data, KMW designed experiments, provided reagents, and edited manuscript. TG provided reagents and edited manuscript. CMT performed experiments. SM designed experiments and edited manuscript. ASG designed experiments, analyzed data, and drafted the manuscript.
Acknowledgments
Data presented are available upon request from EML or ASG; We would like to thank members of the Gladfelter Lab for critical discussions, Erik Griffin, Jamie Moseley, Danny Lew, Mark Peifer, and Henry Higgs for critically reading the manuscript, the HHMI HCIA at the Marine Biological Laboratory in Woods Hole for providing a community to incubate these ideas, Alain Laederach and Lab for pilot experiments and support in initiating RNA analysis and Timothy Straub for useful data analysis discussions; We would also like to thank Tom Christy and Patrick Irving of the Weeks Lab for SHAPE method and analysis help; This work was supported by NIH GM R01-GM081506 to ASG, the HHMI Faculty Scholars program to ASG, NIH R35 GM122532 to KMW, American Cancer Society Fellowship 130845-RSG-17-114-01-RMC to CW, and NIH 1DP2 GM105453, NIH R01 GM115631 to SM.





{kind=link}
{kind=link}
{kind=link}
{kind=link}