

Abstract
Redox biochemistry plays a key role in the transduction of chemical energy in all living systems. Observed redox reactions in metabolic networks represent only a minuscule fraction of the space of all possible redox reactions. Here we ask what distinguishes observed, natural redox biochemistry from the space of all possible redox reactions between natural and non-natural compounds. We generate the set of all possible biochemical redox reactions involving linear chain molecules with a fixed numbers of carbon atoms. Using cheminformatics and quantum chemistry tools we analyze the physicochemical and thermodynamic properties of natural and non-natural compounds and reactions. We find that among all compounds, aldose sugars are the ones with the highest possible number of connections (reductions and oxidations) to other molecules. Natural metabolites are significantly enriched in carboxylic acid functional groups and depleted in carbonyls, and have significantly higher solubilities than non-natural compounds. Upon constructing a thermodynamic landscape for the full set of reactions as a function of pH and of steady-state redox cofactor potential, we find that, over this whole range of conditions, natural metabolites have significantly lower energies than the non-natural compounds. For the set of 4-carbon compounds, we generate a Pourbaix phase diagram to determine which metabolites are local energetic minima in the landscape as a function of pH and redox potential. Our results suggest that, across a set of conditions, succinate and butyrate are local minima and would thus tend to accumulate at equilibrium. Our work suggests that metabolic compounds could have been selected for thermodynamic stability, and yields insight into thermodynamic and design principles governing nature’s metabolic redox reactions.
Introduction
Redox reaction networks are at the center of energy exchange in biochemical processes. The two main biogeochemical carbon-based transformations - respiration and photosynthesis - are at heart oxidative and reductive electron transport processes. In addition to these central respiratory and photosynthetic electron-transport chains, a very large fraction of catalogued enzymatic reactions (∼40%) are oxidoreductive in nature 1,2.
Thermodynamic and other physicochemical properties impose constraints on the evolution of metabolism in general and redox biochemistry in particular. A classic case of this is the expansion of metabolic pathways during the Great Oxygenation Event 3. The rise in atmospheric oxygen levels resulted in a standard redox potential of ∼1.1 eV available from oxidizing the universal redox cofactor NAD(P)H with molecular oxygen. For instance, as Bloch and Woodward pointed out, cholesterol biosynthesis - with six of its steps requiring molecular oxygen - emerged only after the increase in available oxygen 4. Other physicochemical properties such as hydrophobicity and charge at physiological pH also act as constraints that shape the evolution of metabolite concentrations 5.
Previous work has uncovered thermodynamic principles and constraints governing carbon redox biochemistry 6,7. This line of work has focused on the three main types of redox reactions that change the oxidation state of carbon: reductions of carboxylic acids (-COO) to carbonyls (-C=O); reduction of carbonyls to alcohols (C-O), and reduction of alcohols to hydrocarbons (C-C). A first principle is the “rich-get-richer” principle: the more reduced a carbon functional group is, the higher its redox potential. Thus, alcohol reduction is more favorable than carbonyl reduction, which in turn is more favorable than carboxylic acid reduction. This explains the set of reactions where ATP is invested in carbon fixation pathways, with all ATP used to drive reductions of carboxylic acid functional groups. Another principle involves the optimally tuned redox potential of NAD(P). With a value ranging from -370 to -250 mV, it is well matched to reversibly reduce/oxidize the vast of majority of central metabolic redox half-reactions. In addition, its standard potential is lower than that of the average carbonyl compound, thus effectively reducing the steady-state concentrations of damage-causing carbonyls in the cell.
In this work our goal is to contribute to the understanding of thermodynamic and physicochemical principles governing carbon redox biochemistry. We focus on a question that to our knowledge has not been systematically or quantitatively addressed in the literature: What distinguishes the set of observed, natural carbon redox reactions from the null space of all possible redox reactions? In order to address this, we use a three-pronged approach involving cheminformatics, quantum chemistry, and pathway thermodynamic modelling techniques. We generate the networks of all possible redox reactions involving n-carbon linear-chain molecules and partition these into natural and non-natural reactions and compounds. We find that natural compounds are significantly enriched for carboxylic acid functional groups and depleted for carbonyls. Using cheminformatics, we estimate and compare physicochemical properties of natural and non-natural compounds in the networks. Natural compounds have significantly higher solubility and lower lipophilicity (as measured by their octanol-water partition coefficients). Next, using a calibrated quantum chemistry approach, we estimate the transformed standard reduction potentials (E°’) of all reactions in the networks. We find that, across a range of pH and steady state redox cofactor potentials, E(cofactor), thermodynamics favors the accumulation of natural metabolites over non-natural compounds, with some interesting exceptions. We generate a pH/E(cofactor) phase diagram (a Pourbaix diagram 8) to highlight the molecules with minimal energy as a function of phase space parameters. Our work provides concrete evidence for the argument that thermodynamic stability guides the natural selection of metabolic compounds, and yields insight into thermodynamic and design principles governing nature’s metabolic redox reactions.
Results
In the full linear chain n-carbon molecule redox networks, aldose sugars have the maximal number of connections
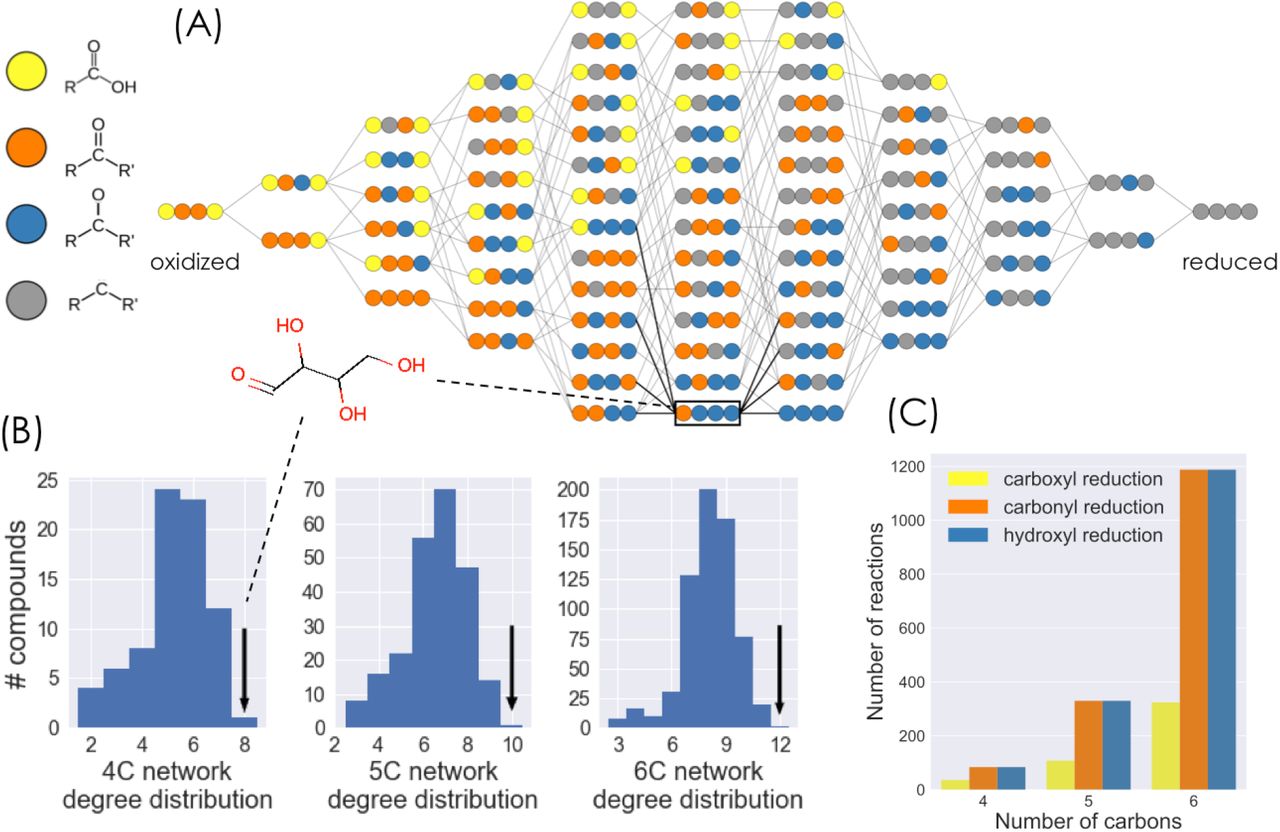
We generate and study the properties of redox networks of linear chain molecules with n-carbon atoms (Figure 1), focusing the majority of the analysis on the 4-carbon network. For a given molecule in a redox network, each of its n carbon atom can be in one of four different oxidation states: carboxylic acid, carbonyl (ketone or aldehyde), alcohol, and hydrocarbon (Figure 1a). Molecules in the network are connected by three different types of reduction (or the reverse oxidation) reactions that change the oxidation state of a single carbon atom: reduction of a carboxylic acid to a carbonyl; reduction of a carbonyl (either an aldehyde or a ketone) to an alcohol; and reduction of an alcohol to a hydrocarbon. In order to make the network model tractable to analysis, we decrease its complexity by not including carbon-carbon bond cleavage or formation reactions (e.g. carboxylations or decarboxylations), keto-enol tautomerizations, double-bond formation, intramolecular redox reactions, or different stereoisomers for a fixed molecular oxidation state. In what follows, we focus the results and analysis on the properties of the 4-carbon linear chain molecule redox network.

a) The network of all possible 4-carbon linear-chain molecules obtained from the three different two-electron redox reactions considered: reduction of carboxylic acid to carbonyl, reduction of carbonyl to alcohol, and reduction of alcohol to hydrocarbon (and corresponding oxidations). Compounds within each column have the same molecular oxidation state. Carbon atoms are represented as colored circles, with color corresponding to oxidation state. b) The degree distributions for the 4-, 5-, and 6-carbon linear chain molecules. Aldose sugars are the only compound with the maximal number of possible reductions and oxidations c) Number of reactions of each of the three redox categories considered in the 4-, 5-, and 6-carbon redox networks.
The 4-carbon network consists of 78 molecules connected by 204 reactions. The molecules span 11 different molecular oxidation states, from fully oxidized 2,3-dioxosuccinic acid (a dicarboxylic acid and di-carbonyl) to the fully reduced alkane, butane (Figure 1a). 84 reactions reduce carbonyls to alcohols and the same number reduce alcohols to hydrocarbons. Since carboxylic acids are restricted to the carbon atoms at the edge of a molecule, only 36 reactions reduce carboxylic acids to aldehydes (Figure 1c).
The number of reactions that connect a single molecule to its oxidized or reduced products - the degree of a molecule - ranges from 2 to 8 (Figure 1b). Only a single molecule in the network, the aldose sugar erythrose (and its stereoisomers, e.g threose), has the maximal degree value of 2n=8, where n is the number of carbon atoms. This holds true for networks with different number of carbon atoms: only the corresponding aldose sugars in the 2, 3, 5, and 6 carbon networks have the maximum value in the degree distribution (2n). This can be explained by the fact that aldose sugars satisfy the two constraints required to have a maximal number of connections in an n-carbon linear chain redox network: 1) Each atom must be in an intermediate oxidation state that can be both oxidized and reduced. Therefore all “inner” carbon in the molecule atoms must be in the alcohol oxidation state, while carbon atoms in the edge can be either in the carbonyl (aldehyde) or alcohol oxidation state; 2) The molecule must not be symmetric under a 180 degree rotation along its center; thus the two edge atoms must be in different oxidation states. This leads to the aldose sugar oxidation state configuration.
Natural compounds are significantly enriched for carboxylic acids, depleted for carbonyls, and have higher solubilities
We then asked: what distinguishes molecules in the 4-carbon redox network that are found in cellular metabolism from the set of non-natural compounds? To explore this question, we divided the 78 molecules in the network into natural and non-natural categories by identifying compounds that match metabolites in the KEGG database1,2. We find that 30 molecules in the network match natural compounds while 48 are non-natural (Methods, Figure 2a). Compounds that appear in KEGG with the same oxidation state as a network molecule but with substituted functional groups were considered a match (see Methods for further details). For example, the natural metabolites oxaloacetate and aspartate, which have the same oxidation state at each carbon atom but differ by the substitution of an alcohol into an amine, are both considered a match to the corresponding molecule in our network. Similarly, we consider metabolites with phosphate groups instead of alcohols, and those with activated carboxylic acids (with thioesters or phosphate groups) as matches to network molecules.

a) Molecules that match natural metabolites in the KEGG database. Compounds that only match KEGG metabolites with one or more alcohol groups substituted by an amine or phosphate are marked in black and red squares, respectively. b) Enrichment and depletion of functional groups in the natural compounds. Gray squares correspond to the analytically-derived null distributions for randomly sampled sets of molecules from the network. See Figure 4.3 for statistical analysis of 2-mer and 3-mer functional group patterns. c) Comparison of predicted aqueous solubility log(S) at pH=7 for natural and non-natural compounds in the 4-carbon linear-chain redox network. Natural compounds have significantly higher solubilities than the non-natural set (p < 0.005).
We next analyzed the enrichment or depletion of functional groups (oxidation states) in the natural and nonnatural sets. To do this, we compared the observed number of times that each functional group appears in the natural metabolites, in comparison to what would be expected by chance (Methods). Natural metabolites are significantly enriched in carboxylic acids (p<0.001) while being significantly depleted for ketones (p<0.001) (Figure 2b). After properly normalizing for the observed single functional group statistics (Methods) we computed the expected of 2-carbon and 3-carbon atom functional group patterns (Figure S1), and find that only the 2-gram pattern [alcohol-hydrocarbon] is depleted in the natural set, albeit not significantly (p=0.05). All other 2-mer patterns, including the low observed number of nearest neighbor dicarbonyls, can be explained by the single carbon atom functional group statistics.
We also explored whether the observed enrichments and depletions for carboxylic acids and carbonyls result in different physicochemical properties of the natural and nonnatural sets. To explore this, we used cheminformatic tools to estimate the values of solubility (logS) and lipophilicity (as measured by the octanol-water partition coefficient, logP) at pH=7 (Methods). The enrichment of natural metabolites for carboxylic acid functional groups results in natural metabolites having significantly higher solubilities at pH = 7 (p<0.005) (Figure 2c). We also find that the natural metabolites have a significantly lower predicted octanol-water partition coefficient at pH=7 (p<0.01) (Figure S2).
The thermodynamic landscape of carbon redox biochemistry
We apply a calibrated quantum chemistry strategy to predict the thermodynamics of all redox reactions in the networks. Briefly (see Methods section for further details), we use density functional theory (DFT) with a double-hybrid functional9,10 to compute the electronic energy of several geometry-optimized conformations of the fully protonated state of every compound in the network. We estimate the standard redox potential E° (for the fully protonated species) as the difference in electronic energies of the products and substrates, ΔEelectronic. Using cheminformatic pKa estimates (Marvin 17.7.0, 2017, ChemAxon) and the Alberty Legendre transform 11,12, we convert the standard redox potentials to a transformed standard redox potentials E’°(pH), which is a function of pH. Finally, in order to correct for systematic errors in the quantum chemistry model and cheminformatic pKa’s, we calibrate the transformed standard redox potentials E’°(pH) against a dataset of available experimental data using linear regression.
Our approach achieves significantly better accuracy than the group contribution method (GCM), the most commonly used approach to estimate thermodynamics of biochemical reactions (Figure 3) 13–16. The improvement in accuracy depends on the redox reaction category, and is most apparent for the set of carbonyl to alcohol reductions. By considering only the difference in group energies, GCM effectively ignores the molecular environment surrounding the reduced/oxidized carbon atom and its influence on thermodynamics. Thus GCM collapses the redox potentials of all carbonyl reductions to two values, corresponding to average linear-chain aldehyde and ketone standard reduction potentials (Figure 3a).

Experimental data was obtained from the NIST database for Thermodynamics of Enzyme-Catalyzed Reactions (TECRDB) 17a) Group contribution method prediction accuracies for reduction potentials of carboxylic acids, carbonyls, and alcohol functional groups in linear chain compounds. b) Calibrated quantum chemistry prediction accuracies for reduction potentials of carboxylic acids, carbonyls, and alcohol functional groups in linear chain compounds. Quantum chemistry calculations were performed using density functional theory with a double hybrid functional (B2PLYP)9,10, and calibrated against experimental data using linear regression.
Having estimated the potentials of all reactions, we study the thermodynamic landscape of the network. We assume that each redox reaction in the network is coupled to a redox cofactor with a fixed, steady-state, potential, E(cofactor). For instance, such a cofactor potential could correspond to the potential set by a steady state ratio of NAD/NADH inside the cell 18 or to that set by a certain concentration of molecular hydrogen in the context of an alkaline hydrothermal vent 19. With this cofactor potential value, we convert the standard redox potentials of each reaction into standard Gibbs reaction energies. (Figure 3a, Methods). This allows us to study the thermodynamic properties of the network across a wide range of cofactor potentials and pH values.
We find that across a range of cofactor potentials, natural metabolites have on average significantly lower relative Gibbs energies than the non natural compounds (Figure 4, Figure 5a, Figure S3). An important exception to the relatively lower energies of the natural compounds is the aldose sugar erythrose, the ketose sugar erythrulose, and the sugar alcohol threitol (and all corresponding stereoisomers), which have a high relative Gibbs energy across a large range of cofactor potentials.

Gibbs energies are normalized relative to the metabolite with the lowest energy (butane). Thus the cumulative Gibbs energies of a metabolite is obtained by summing up the Gibbs reaction energies of all reactions leading to it from the reference metabolite. Compounds within a column (i.e. with the same molecular oxidation state) are sorted according to their energies. The three compounds - butane, butanoic acid, and succinate - which are local minima in the thermodynamic landscape are shown. These local minima have lower energy than any of their neighboring molecules which are accessible by either a reduction or an oxidation.
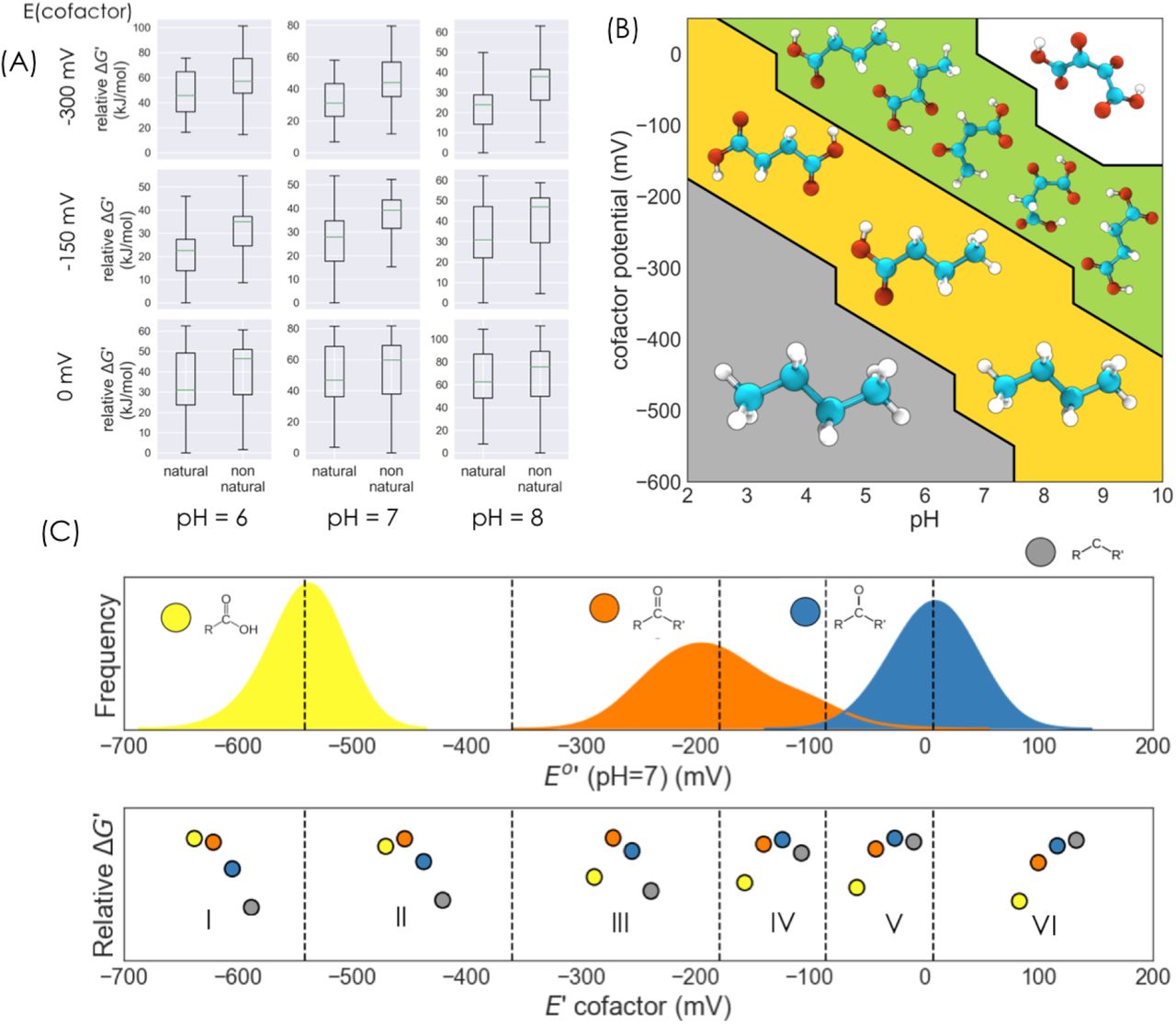
a) Relative Gibbs energies of natural and non-natural metabolites for a range of pH and E(cofactor) values. At each value of pH and E(cofactor), Gibbs energies are normalized relative to the compound with the lowest energy. b) The Pourbaix phase diagram of the 4-carbon linear chain redox network. Local minima in the energy landscape at each region of the pH, E(cofactor) phase space are shown. For instance, at low pH and E(cofactor) values, only butane is a local (global) minimum, while at high pH and E(cofactor) values, only the fully oxidized 4-carbon molecule is a local (global) minimum. Some non-natural metabolites which are also local minima in the yellow and green regions are not shown. c) The top panel shows the standard redox potentials of the three reducible functional groups at pH=7. The bottom panel shows a schematic representation of the relative functional group Gibbs energies at regions corresponding to different cofactor potential values.
The thermodynamic landscape reveals that a few metabolites are local energetic minima of the network as a function of pH and E(cofactor). A molecule is a local minima at a value of pH and E(cofactor) if its Gibbs energy is lower than all of its neighbors with whom it’s connected through a reductive or oxidative transformation. Figure 5b shows a Pourbaix phase diagram representation 8 of these local minima. In a Pourbaix diagram, the lowest energy phase or state of an electrochemical system is displayed as a function of pH and potential.
At the lower left corner of the diagram, in the region corresponding to more acidic pH and lower cofactor potentials, the fully reduced 4-carbon alkane butane is the only local (and the global) minimum in the thermodynamic landscape. This illustrates the fact that more negative values of the cofactor potential and more acidic pH drive the redox network to an overall reduced state.
Notably, succinate and the 4-carbon short-chain fatty acid (SCFT) butyrate - two natural compounds - emerge as additional local minima at relatively more oxidative regions of the phase diagram (the region containing the values E(cofactor) ∼ -300 mV and pH = 7). Both of these natural compounds chemically consist of “inner” carbon atoms that are in the hydrocarbon oxidation state, and “edge” carbon atoms in either the fully reduced hydrocarbon or the fully oxidized carboxylic acid state. Thus, at cofactor potentials around -550 mV and 0 mV, thermodynamics drives carbon atoms to either the most oxidized (carboxylic acid for edge atoms or carbonyl for inner atoms) or the most reduced (hydrocarbon) oxidation state (Figure 5c).
Further increases in either pH or the cofactor potential (Figure 5b) result in the emergence of additional compounds, both natural and non-natural, as local minima in the energy landscape. In regions of phase space with increasing values of pH and E(cofactor), the natural metabolites oxaloacetate, acetoacetate, and alpha-ketobutyrate emerge as local energetic minima. Finally, in the region of alkaline pH (pH∼8) and high cofactor potential (E(cofactor) ∼ -50 mV), the fully oxidized four carbon compound 2,3-dioxosuccinic acid becomes the sole local (global) minimum in the energy landscape.
Discussion
We generated the set of all possible redox reactions involving linear chain n-carbon compounds. Analyzing the structure of the resulting redox networks, we found that aldose sugars -glyceraldehyde (n=3), erythrose (n=4), ribose (n=5), and glucose (n=6), and their corresponding stereoisomers - are unique in that they have the highest possible number of oxidative and reductive connections (2n) to neighboring compounds. Thus our analysis reveals an aspect of the redox network topology that makes these sugar molecules unique. Whether this maximal number of connections played a role in the emergence of aldose sugars as key players in metabolism remains to be explored.
Comparing the properties of natural and non natural compounds in the network, we find that the natural set is significantly enriched for carboxylic acids. This enrichment correlates with a significantly higher value of solubility (logS) at pH=7. Other benefits of carboxylates for cellular metabolism potentially include decreasing the permeability across cell membranes, which is captured by our predicted octanol-water partition coefficients, logD(pH=7), as well as potentially enhancing enzyme recognition.
We also find that natural compounds are significantly depleted for carbonyl functional groups. Carbonyls are significantly more reactive than carboxylic acids or alcohols, and can cause oxidative damage, spontaneously cross-link proteins, inactivate enzymes and mutagenize DNA 20. Only one natural compound in the 4-carbon network - diacetyl, which is part of the diacetyl/acetoin biosynthetic pathway in Lactococcus lactis bacteria 21 - contains a dicarbonyl. Selection against such dicarbonyls can again be explained by their very high reactivity as well as potential selectivity problems in the context of enzyme recognition stemming from having two identical active sites next to each other.
We used a calibrated quantum chemistry approach to predict the transformed standard redox potentials of all reactions in the network as a function of pH. Importantly, the quantum chemistry strategy results in significantly better accuracy than the commonly used alternative biochemical thermodynamics prediction tool, the group contribution method 13,14. In trying to predict the redox potentials of the type considered here, GCM collapses a wide range of values into one or two energies. In addition, the fact that the networks studied here contain numerous non-natural compounds implies that the reactant or component contribution methods 15,16, which make use of experimentally derived Gibbs formation energies of natural compounds to increase prediction accuracy, are of no use for many of our reactions.
The quantum chemical thermodynamic data shows that, across a range of pH and cofactor potential values, natural compounds have on average relatively lower Gibbs energies that the set of non-natural molecules. Although our reaction networks is a toy model of redox biochemistry - albeit a complex one - our analysis provides quantitative evidence that molecules observed in nature’s metabolic toolbox are on average thermodynamically favored over the non-natural compounds. Thus our analysis supports the reasonable yet “highly speculative” 22 argument put forward by Bloch that in the context of the origins of life “the thermodynamically most stable compounds had the best chance to accumulate and survive.”
The resulting thermodynamic landscapes also reveals which compounds - both natural and non-natural - are energetic local minima as a function of pH and steady state cofactor potential E(cofactor). We observe that at a relatively reductive range of pH and E(cofactor) values (e.g. pH = 7, -500 mV < E(cofactor) < -100 mV), the natural metabolites succinate and butyrate (the 4-carbon fatty acid), are local minima. Regarding the short-chain fatty acid (SCFA) butyrate (as well as equivalent fatty acids with higher number of carbon atoms), it is tempting to offer this as evidence that, under redox potentials existing in prominent origins-of-life scenarios 19,23 thermodynamics naturally leads to the accumulation of SCFAs. It is also of significant interest to note that SCFAs are a prominent bacterial fermentation product of dietary fibers by gut microbiota 24.
The fact that succinate - a key component of the TCA cycle - is a thermodynamic local minimum implies that it accumulates at these values of pH and cofactor potential. This is consistent with the behavior of the TCA cycle under anaerobic conditions 25–27. In such conditions, the cycle operates as a complete or incomplete fork, with a portion running in the reductive TCA modality (oxaloacetate sequentially reduced to malate, fumarate, and succinate), and leading to the accumulation succinate. In contrast, in oxidative conditions, the thermodynamic driving force is in the direction of succinate oxidation and oxaloacetate formation. These two different thermodynamic regimes are captured in our Pourbaix phase diagram representation, where oxaloacetate appears as a local minimum in oxidative pH, E(cofactor) regions of phase space.
We also note that succinate, in combination with glycine, is the starting building block for all porphyrins in one of the two known porphyrin biosynthetic pathways 28. In addition, acetoacetate appears at as a local minimum at slightly more oxidative conditions. Acetoacetate is the precursor for the immense set of isoprenoid compounds 29. Thus it is tempting to speculate that thermodynamics would naturally lead to the accumulation of these important precursor building-block metabolites.
Our analysis demonstrates that certain compounds that are not part of the KEGG database (i.e. non-natural) are also local energetic minima at certain pH and cofactor potential values. Notably, at reductive cofactor potential/pH values, the fully reduced alkane butane is the global minimum in the energy landscape. Alkane biosynthesis can occur in bacteria 30,31. However, such pathways involve the reduction of fatty acids to fatty aldehydes, followed by the decarbonylation of the terminal carbon, resulting in alkanes with one carbon shorter than the original fatty acid. Alternatively, enzymes referred to as fatty acid acyl-CoA reductases (FARs) can reduce the fatty aldehydes to fatty alcohols 32,33, and alkanes are known to be a component of the surface waxes of the alga K. flaccidum 34.
In future work, the network model could be expanded to include other types of biochemical transformations, such as keto-enol tautomerizations, double-bond formation, intramolecular redox reactions, or a detailed accounting of the different stereoisomers that correspond to a given molecular oxidation state. It would be particularly interesting to include carboxylation and decarboxylation reactions (both reductive/oxidative and non-reductive/non-oxidative), which would effectively connect the different n-carbon redox networks to each other. These would result in more realistic redox networks but in a more complex analysis. Having said so, we believe that the results presented in this paper will provide a general guidance for more general models of the redox reaction networks.
Materials and methods
Generation of full redox networks using RDKit
To generate the reactions, we used the RDKit cheminformatics software to design SMILES (simplified molecular-input line-entry system)35 reaction templates (reaction strings), which, when applied to a compound, will reduce it according to the functional groups detected. Reaction strings were created for the three redox categories of interest: reduction of carboxylic acids to aldehydes, reduction of ketones to alcohols, and reduction of alcohols to hydrocarbon.
These templates are designed to be generic enough that they can be applied to any compound with the target functional group, but also with enough specificity to only generate a reaction belonging to the correct redox category.
As an illustrative example, we consider the reduction of pyruvate. Pyruvate contains two types of functional groups that can be reduced: a carboxylic acid and a ketone. The carboxylic acid can be reduced to an aldehyde, or the ketone can be reduced to a hydroxyl. To accomplish this we applied the appropriate SMILES reaction strings. The SMILES reaction string used for the ketone reduction of pyruvate to lactate is shown below. This reaction string can be visualized as a generic reduction of a ketone to a hydroxyl. The ReactionFromSmarts function in RDkit is used to generate a reaction object from the reaction string.

The molecular transformation encoded by the SMILES reaction string. The substrate and compound of each reaction are represented as strings and concatenated into a reaction string as follows: [#6:1][CX3:2](=O)[#6:3]>>[#6:1][CX4H1:2]([#6:3])[OX2H1]
This reaction object can be applied to any compound with a ketone functional group in order to reduce it to a hydroxyl. For cases in which the compound contains multiple target functional groups (e.g. dicarbonyls), every possible product will be generated. To generate the full network or redox reactions, these reaction strings were run iteratively, starting with the fully oxidized unbranched, carbon chain compounds of length 2 to 6 carbons. For example the seed compound for the full redox network of 6-carbon straight-chain molecules is shown below:

Once fully oxidized seed compound had been reduced one step at every possible carbon atom in the initial iteration, the function was repeatedly applied on the resulting products. This continued iteratively until, the fully reduced n-carbon hydrocarbon chain is obtained. Any duplicate reactions and products generated from this approach were eliminated during each iteration. Thus, a network of all possible redox reactions originating from the fully oxidized seed compound can be generated.
SMILES reaction strings
Computing the network degree distributions
The degree of a compound in the redox network is defined as the number of redox reactions - oxidations and reductions - that connect it to molecules with higher or lower oxidation state. We used the network analysis library NetworkX36 in Python to compute the degree distribution of compounds in the full redox networks.
Comparison against KEGG database
In order to classify compounds in the full redox networks as natural or non-natural, we looked for matches in the KEGG database of metabolic compounds. We did this in several steps.
In order to match natural compounds against the n-carbon network, we filtered out metabolites in KEGG containing n-carbon atoms. Then, using the RDKit toolbox, we matched molecules in the networks against KEGG metabolites using their canonicalized smiles string representation 37. In order to additionally capture KEGG compounds that have alcohol functional groups substituted by amine or a phosphate functional groups, we visually inspected all remaining n-carbon molecules in KEGG. Finally, to capture compounds with carboxylic acids activated by Coenzyme A, we generated a list of all KEGG compounds with n-carbon atoms plus a covalently attached Co-A molecule. Manual search of this list led to the final set of natural metabolites matching compounds in our full redox networks.
Computing the null distribution for the expected number of n-gram (single, pair and triplet) functional group patterns
Borrowing terminology from natural language processing, we call the set of all possible sequences of one, two, and three carbon functional groups the set of oxidation state n-grams. The goal is to count the number of times that each n-gram appears in the set of natural (or non natural) compounds (where N is the total number of natural compounds), and compare that against properly generated random sets of compounds (the null distribution).
The analytical null distribution for single functional group patterns (1-grams)
We first note that a given n-gram can appear more than once in a single molecule. For example, the metabolite succinate has the functional group sequence {carboxylic acid, hydrocarbon, hydrocarbon, carboxylic acid}. Thus it contains two instances of the {carboxylic acid, hydrocarbon} 2-gram. In general, a 4-carbon linear-chain compound can have up to 4 instances of a 1-gram, up to 3 instances of a 2-gram, and up to 2 instances of a 3-gram.
Let n(k; g) be the number of molecules in the full redox network with k instances of 1-gram g. For example, n(1; hydroxyl) is the total number of compounds in the network with a single hydroxyl functional group. Assume a set of N molecules are randomly sampled without replacement from the network. Let m(g) be the total number of instances of the 1-gram g in this random set. These m(g) instances can come from different sampling configurations of molecules, each with k instances of the 1-gram g. We call m(k; g) be the number of molecules in the random sample with k instances of the 1-gram g.
To give a concrete example, assume a random set of size N = 30 molecules contains 16 instances of the n-gram g; thus m(g) =16. One of the very many sampling configuration that can lead to this value of m(g) is sampling 17 molecules with zero instances of g, 10 molecules with 1 instance of g, and 3 molecule with two instances of g. Thus 
The total number of instances of the 1-gram in the sample is given by: 
Note that the following constraint is satisfied: 
In order to compute the probability of having m(g) instances of the 1-gram g, we need to account for all such possible sampling configurations that add up to m(g). The number of ways of sampling m(k;g) molecules with k instances of g is given by  . In general, given a sample size N and value of m(g) for n-gram g, the number of all possible sampling configurations that lead to that value of m(g) is given by:
. In general, given a sample size N and value of m(g) for n-gram g, the number of all possible sampling configurations that lead to that value of m(g) is given by: 
Where the summation is over terms that satisfy the following two constraints: 

Normalizing each value of P(m(g), N) over the sum of all values leads to the probability of observing m(g) instances of the 1-gram g in a sample of size N, p(m(g),N). We numerically obtain the value of n(k;g) for k = 0, 1, 2, 3, 4 and g = {carboxylic acid, carbonyl, hydroxyl, and hydrocarbon}. We then numerically compute the value of P(m(g), N) by obtaining all sampling configurations that satisfy the constraints. We take N to be equal to the number natural compounds in the full redox network.
The empirical null distributions for functional group pair and triplet patterns (2- and 3-grams)
Obtaining the proper null distribution for oxidation state pair and triplet patterns (2-grams and 3-grams) requires accounting for (or normalizing) for the observed single functional group statistics (1-grams). For example, the 2-gram pattern [carbonyl-carbonyl] seems to appear infrequently in the natural set of metabolites. Is this due to selection against this specific 2-gram pattern, or is it simply due to the general depletion of carbonyls (the 1-gram pattern) in the natural compounds?
In order to address this, one needs to generate random sets of N compounds that control for or conserve the 1-gram statistics of the natural set of compounds. We numerically generate random molecules that conserve 1-gram statistics. In the case of 4-carbon linear chain molecules, we randomly choose the identity of the functional group at positions n = (1, 2, 3, 4) by sampling from a discrete distribution 
Where gN is the number of instances of 1-gram g in the natural set, and N is the number of molecules in the natural set. Importantly, in order to avoid sampling carboxylic acids in the inner carbon atoms of a molecule (positions n = 2 and 3), we obtain separate functional group distributions for the inner and the outer carbon atom positions.
Cheminformatic prediction of solubility (logS)
We used the cheminformatics software ChemAxon (Marvin 17.7.0, 2017, ChemAxon) to predict the pH-dependent solubility, logS(pH), of natural and non-natural compounds in the full redox networks. Specifically, we use the calculator plugin cxcalc logs. The cxcalc solubility calculator is based on a parametrized fragment-based model (the atom-contribution approach) fit to sets of experimental logS data 38,39.
Predicting standard redox potentials with calibrated quantum chemistry approach
Our method relies on computing the electronic structure and energy of the fully protonated species of each metabolite. We obtain the smiles string for the fully protonated species and generate initial geometric conformation (with up to 10 initial conformers per metabolite) using ChemAxon (Marvin 17.7.0, 2017, ChemAxon).
All quantum chemistry calculations were performed using the Orca quantum chemistry software 40 version 3.0.3. We first perform a geometry optimization using density functional theory with the B3LYP functional 41, with Orca’s DefBas-2 basis set, COSMO implicit solvation42, and D3 dispersion correction43. We then perform an additional electronic single point energy (SPE) using the double-hybrid functional B2PLYP 9,10 (with the DefBas-5 Orca basis set, COSMO implicit solvation 42, and D3 dispersion correction 43). We note that the model chemistry selected - the combination of DFT functional, basis set, implicit solvent model, and dispersion correction for both the geometry optimization and the single point energy - was done based on a combinatorial exploration of different options.
We Boltzmann average the electronic energies of compounds, and obtain the difference in electronic energies of products and substrates for all redox reactions in the full redox networks. Every redox reaction (in the direction of reduction) was balanced by a hydrogen molecule H2 in the substrate side of the equation. Reductions of carboxylic acids to aldehydes and reductions of alcohols to hydrocarbons were balanced with a water molecule H2O in the product side of the equation.
The difference in product and substrate electronic energies is an estimate of the chemical redox potential for the fully protonated species, E°(fully protonated species). In order to convert this chemical potential to the biochemical potential at pH = 7, Eo’(pH=7), we use pKa estimates from Chemaxon (cxcalc, pKa) and the Alberty Legendre transform.
Our approach relies on several approximations, such as ignoring vibrational enthalpy and entropy contributions to the formation Gibbs energy of compounds. In order to correct for systematic in the quantum chemistry methodology and the empirical pKa estimates used, we calibrate predictions against available experimental data using linear regression.
Predicting standard redox potentials with the group contribution method
The group contribution method relies on a fragment-based decomposition of compounds into group, each of which is assigned a group energy based on available experimental data 13–16.
Reaction energy estimates are obtained by taking the difference of the group energy vectors of products and substrates. We used the group contribution method as implemented by Noor et al. 16 to estimate the redox potentials of the set of linear-chain carbon redox reactions with experimental values.
Determining statistical significance
For all tests of statistical significance (i.e. differences in solubilities, n-gram counts, octanol-water partition coefficients, Gibbs energies of natural vs. non-natural compounds) we performed Welch’s unequal variance t-test, which is an adaptation of Student’s t-test that does not assume equal variance.
Acknowledgements
We thank Arren Bar-Even for fruitful discussions and feedback, and Ron Milo, Manuel Razo-Mejia, and members of the Aspuru-Guzik lab for comments on the manuscript. The authors thank Harvard Research Computing for their support on using the Odyssey cluster. A.A.-G. and A.J. acknowledge support from SEAS^NVIDIA, Massively Parallel Programming and Computing (332986). J.E.G and D.S. were partially supported by grants from NASA, NSF and the Human Frontiers Science Program.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}