

Summary
Staphylococcus aureus needs to control the position and timing of cell division and cell wall synthesis to maintain its spherical shape. We identified two membrane proteins, named CozEa and CozEb, which together are important for proper cell division in S. aureus. CozEa and CozEb are homologs of the cell elongation regulator CozESpn of Streptococcus pneumoniae. While cozEa and cozEb were not essential individually, the ΔcozEaΔcozEb double mutant was lethal. To study the functions of cozEa and cozEb, we constructed a CRISPR interference (CRISPRi) system for S. aureus, allowing transcriptional knockdown of essential genes. CRISPRi knockdown of cozEa in the ΔcozEb strain (and vice versa) causes cell morphological defects and aberrant nucleoid staining, showing that cozEa and cozEb have overlapping functions and are important for normal cell division. We found that CozEa and CozEb interact with the cell division protein EzrA, and that EzrA-GFP mislocalizes in the absence of CozEa and CozEb. Furthermore, the CozE-EzrA interaction is conserved in S. pneumoniae, and cell division is mislocalized in cozESpn-depleted S. pneumoniae cells. Together, our results show that CozE proteins mediate control of cell division in S. aureus and S. pneumoniae, likely via interactions with key cell division proteins such as EzrA.
Introduction
Bacterial cell division initiates when the tubulin-like protein FtsZ polymerizes into a ring structure (Z-ring) located at the future division site. The Z-ring then serves as a scaffold for recruitment of cell division and cell wall synthesis proteins, forming the multiprotein complex known as the divisome. Several of the proteins constituting the divisome are widely conserved in most bacteria, while others are specific for bacterial subgroups or have diverged significantly (Pinho et al., 2013). Positioning and timing of Z-ring assembly and cell wall synthesis are dependent on the shape of the bacterium; and there are large variations between coccal, ovococcal and rod-shaped bacteria.
Staphylococcus aureus often serves as the model organism for cell division studies in spherical bacteria. S. aureus is an opportunistic pathogen, which persistently colonizes around 20 % of the human population (Grice & Segre, 2011), causing both superficial infections on the skin and invasive, life-threatening sepsis as well as endocarditis in humans (Rasigade & Vandenesch, 2014, Foster et al., 2014). Furthermore, S. aureus is an important pathogen among livestock (causing mastitis and other infections) and is a problematic food pathogen. Treatment of S. aureus infections with antibiotics is increasingly challenging due to the rise of antibiotic resistant strains, including MRSA (methicillin-resistant S. aureus which are resistant to β-lactam antibiotics) and VRSA (vancomycin-resistant S. aureus).
Cell division in spherical S. aureus occurs in three consecutive planes, where every new round of division is orthogonal to the previous division plane (Pinho et al., 2013). Many key cell division proteins known from other model bacteria are conserved in S. aureus, including FtsZ, FtsA, EzrA, GpsB, DivIB, DivIC, FtsL, MurJ, DivIVA, MreC and MreD (Pinho & Errington, 2003, Pinho & Errington, 2004, Steele et al., 2011, Pinho et al., 2013, Bottomley et al., 2014, Monteiro et al., 2018). These proteins are in different ways involved in formation of the division ring and for ensuring proper cell wall synthesis and cell division. For example, EzrA is a key early cell division protein linking division ring formation and the cell wall synthesis machinery (Jorge et al., 2011, Steele et al., 2011). Synthesis of new cell wall in staphylococci mainly occurs at midcell. The recruitment of cell wall synthesis proteins (i.e. transpeptidases PBP1, PBP3 and PBP4 and the bi-functional transpeptidase/transglycosylase PBP2 responsible for synthesizing the peptidoglycan sacculus) to midcell was recently shown to be driven by the putative lipid II flippase MurJ (Monteiro et al., 2018). The current models of the S. aureus cell cycle suggest an initial, gradual increase in cell volume by slight elongation, followed by recruitment of MurJ and PBPs to the septum, which drives cross-wall synthesis and cell constriction. To split the daughter cells, hydrolases perforate the cell wall before the actual splitting/popping of daughter cells occurs on a timescale of milliseconds (Monteiro et al., 2015, Zhou et al., 2015, Lund et al., 2018, Monteiro et al., 2018).
During each cell cycle, peptidoglycan synthesis and cell division need to be coordinated with DNA replication and chromosome segregation. This is to ensure correct cell size homeostasis and that the two daughter cells each get one copy of the chromosome in time before the cell splits. Misregulation would result in daughter cells of variable sizes without DNA or guillotining of the chromosome by the septal cross-wall. How the timing and localization of Z-ring assembly and cell wall synthesis are regulated in S. aureus, given the geometry of cell division in three consecutive perpendicular planes, is still an unanswered question. One protein involved in this coordination is probably Noc (nucleoid occlusion protein) which both controls DNA replication (Pang et al., 2017) and inhibits Z-ring formation across the nucleoid (Veiga et al., 2011). Recent results also predict the protein DivIVA to have an important role in linking chromosome segregation with cell division (Bottomley et al., 2017).
The important human pathogen S. pneumoniae is an ovococcal bacterium, in which both septal (division) and peripheral (elongation) cell wall synthesis occur in the mid-cell area (Ducret & Grangeasse, 2017). In these cells, positioning of the Z-ring at mid-cell has been shown to depend on several factors, including the chromosomal origin of replication (van Raaphorst et al., 2017) and the peptidoglycan binding protein MapZ (Fleurie et al., 2014a, Holeckova et al., 2014). Most likely, the septal and peripheral cell wall growth in pneumococcal cells are mediated by separate protein machineries, whose actions are tuned by different regulatory proteins such as StkP, MreCD, GpsB, DivIVA or EloR (Ducret & Grangeasse, 2017, Fleurie et al., 2014b, Rued et al., 2017, Straume et al., 2017, Stamsås et al., 2017, Beilharz et al., 2012, Zheng et al., 2017). Another protein involved in regulation of cell wall synthesis in pneumococci, named CozE (for coordinator of zonal elongation, SPD_0768 in strain D39 and Spr0777 in strain R6), was recently identified (Fenton et al., 2016, Straume et al., 2017). CozE, a multi-transmembrane spanning protein, was found to be essential for normal growth, however, its essentiality was abolished in the absence of the bifunctional penicillin-binding protein PBP1a or the cell wall elongation proteins MreC and MreD (Fenton et al., 2016). In protein-protein interaction assays, CozE was found to be associated with the same proteins (PBP1a, MreC, MreD) as well as DivIVA and PBP2b (Fenton et al., 2016, Straume et al., 2017). CozE was thus proposed to be a key regulator of cell elongation in S. pneumoniae by positioning PBP1a via interactions with MreC and MreD (Fenton et al., 2016, Ducret & Grangeasse, 2017). CozE proteins are widespread among different bacteria (Fenton et al., 2016). Here we studied the two homologs of CozE in spherical S. aureus cells. We show that the CozE proteins are involved in coordinating cell division in S. aureus and that this function is conserved also in S. pneumoniae.
As a means to study the functionality of essential genes, we also develop a CRISPR interference (CRISPRi) system for S. aureus. With CRISPRi, the CRISPR/Cas9-system is harnessed to knock down gene expression of any gene of interest (Bikard et al., 2013, Qi et al., 2013). Transcriptional knockdown is achieved by two components: a catalytically inactive Cas9 protein (dCas9) and a single guide RNA (sgRNA). Unlike the Cas9 nuclease, dCas9 does not cleave DNA, but the DNA-binding capability is still intact. A single guide RNA (sgRNA), containing a gene-specific base-pairing region and a structured region for interaction with dCas9, is designed to target the gene of interest. Upon co-expression, the dCas9-sgRNA complex will bind DNA and serve as a transcriptional roadblock for the RNA polymerase, thereby downregulating transcription.
Results
The two CozE homologs of S. aureus
The protein CozE (for coordinator of zonal elongation) was recently identified as an essential cell division protein in oval shaped pneumococcal cells, where it has been shown to be involved in regulation of proper cell elongation (Fenton et al., 2016, Straume et al., 2017). Spherical S. aureus does not elongate to the same extent as S. pneumoniae and other rod-or oval-shaped bacteria, although a short elongation phase has been observed during the cell cycle (Pinho et al., 2013, Monteiro et al., 2015). Nevertheless, homology searches showed that S. aureus encodes two proteins homologous to CozE, and we therefore set out to unravel the function of CozE in these spherical cells. Sequence comparison of the pneumococcal CozE (hereafter CozESpn) with the two CozE-homologs of S. aureus SH1000, SAOUHSC_00948 (hereafter CozEa) and SAOUHSC_01358 (hereafter CozEb), shows that they are 31 % and 30 % identical to CozESpn, respectively (Fig. S1). When compared with each other, CozEa and CozEb are 30 % identical. Topology predictions suggest that these proteins have 8 or 9 transmembrane segments (Fig. S1). The cozEa gene is predicted to be monocistronic, while cozEb is located as the last open reading frame on a three-gene operon which also encodes a transcription antiterminator (glcT) and a small, putative membrane spanning protein (SAOUCHSC_01357) (Fig. 1A).

A.Genetic organization of the cozEa (SAOUHSC_00948) and cozEb (SAOUHSC_01358) genetic loci.
B.Growth curves SH1000 wild-type, SAMK24 (ΔcozEa) and SAMK21 (ΔcozEb) in BHI medium at 37°C.
C.Micrographs of SH1000, SAMK21 and SAMK24. Phase contrast (PC) images and staining with fluorescent vancomycin (VanFL) and DAPI are shown as well as an overlay of the two latter. The scale bars are 2 μm
D.Histogram of the cell diameters of SH1000, SAMK24 and SAMK21 (>250 cells analyzed per sample) as measured using MicrobeJ (Ducret et al., 2016). Both SAMK21 and SAMK24 were significantly smaller than SH1000 (P < 0.05, Kolmogorov-Smirnov test).
Using the temperature sensitive vector pMAD (Arnaud et al., 2004), cozEa and cozEb were deleted individually in S. aureus SH1000 by allelic exchange with a spectinomycin resistance cassette. The deletion mutants SAMK24 (ΔcozEa∷spc) and SAMK21 (ΔcozEb∷spc) did not exhibit any growth defect compared to wild-type (Fig. 1B). Analysis of cell sizes showed that the cell diameter of both mutants, on average, are slightly smaller compared to the wild-type (Fig. 1C and D). No obvious differences in cell wall labelling (using fluorescent vancomycin, VanFL) or nucleoid staining patterns (using DAPI) were observed between the mutants and wild-type (Fig. 1C).
In order to see whether the two mutant strains, SAMK21 and SAMK24, had acquired any suppressor mutations elsewhere in the genome, we resequenced their genomes and compared it to the SH1000 wild-type genome. SAMK24 did not contain any additional mutations. In SAMK21, a single conservative SNP was found in the gene thiI (SAOUHSC_01824) encoding a probable tRNA sulphurtransferase. This SNP (A970T) resulted in a conservative substitution of isoleucine with a phenylalanine (I324F). Our later experiments (see below) show that this mutation is not important for the functionality of cozEb (or cozEa) and we therefore conclude that neither CozEa nor CozEb are essential for normal growth and cell division in S. aureus SH1000.
Single deletions of cozEa or cozEb both cause a small reduction in cell size. To investigate the effects of a double deletion, another pMAD deletion vector (pMAD-cozEa∷cam) was constructed to delete cozEa in the ΔcozEb∷spc background. However, despite multiple attempts, we were unable to obtain the double deletion strain. This suggests that cozEa and cozEb may have complementary and essential functions.
Construction of a two-plasmid CRISPR interference system for S. aureus
Since double deletions of cozEa and cozEb could not be obtained, we instead wanted to study the phenotypes of the cells when cozEa or cozEb gene expression was knocked down in ΔcozEb or ΔcozEa background, respectively. We therefore constructed a CRISPR/dCas9 knockdown system to allow inducible depletion of essential genes. The CRISPR interference systems developed for S. pneumoniae and Bacillus subtilis (Liu et al., 2017, Peters et al., 2016) were used as models. A dcas9 gene, encoding a catalytically inactive Cas9, was cloned downstream of an IPTG-inducible promoter in the low-copy number plasmid pLOW (pSK41 minireplicon, Fig. 2A) (Liew et al., 2011). A single guide RNA (sgRNA) construct, consisting of a 20 nt base-pairing region and a Cas9-handle region, was inserted downstream of a synthetic, constitutive promoter in the plasmid pCG248 (replicon T181, Fig. 2A) (Helle et al., 2011). Targeting of the gene of interest is accomplished by replacing the 20 nt sequence using inverse PCR as described in the Methods section. Notably, multi-sgRNA plasmids can be constructed by using the BglII and BamHI restriction sites located up- and downstream of the sgRNA construct, as outlined in Fig. S2. A schematic view of the resulting two-plasmid CRISPRi system is shown in Fig. 2A.

A.Schematic representation of the plasmids carrying dCas9 and sgRNA. The sgRNA is constitutively expressed, while the level of dCas9 is controlled by the inducible Plac promoter. Upon addition of IPTG, dCas9 will be expressed and the dCas9-sgRNA-DNA complex formation will lead to transcription block and knockdown of the target gene.
B-C. Knockdown of GFP expression in a strain constitutively expressing m(sf)gfp (SAMK56).
B.Fluorescence after induction with various IPTG concentrations. The fluorescence values are given relative to the fluorescence of a non-depleted strain. The experiment was repeated twice with similar results.
C.The temporal dynamics of GFP depletion after addition of 100 μM IPTG. The fluorescence at the time of IPTG addition was set to 1, and measured at different time points. The experiment was repeated twice with similar results.
D-F. Growth and phenotypic characterization of cells with depletion using CRISPRi. Growth curves in BHI medium at 37°C and micrographs are shown. The cultures were diluted to OD600 ∼ 0.01 prior to growth analysis. The scale bars are 2 μm.
D.Control cells carrying non-targeting sgRNA.
E.Depletion of pbp1. pbp1 depleted cells were significantly larger than wild-type cells (1.78 ± 0.38 μm, n = 126 for the pbp1 depletion versus 1.41 ± 0.34 μm, n = 250 for the control, P < 0.05, Kolmogorov-Smirnov test).
F.Depletion of dnaA, resulted in formation of anucleate cells (10.2 %, n = 234). The arrowhead points to an example of an anucleate cell.
To quantify the efficiency of our CRISPRi system, we created an RN4220-derivative strain with constitutive expression of a monomeric superfolder GFP (m(sf)GFP), SAMK56, and designed an sgRNA targeting the m(sf)gfp gene. As shown in Fig. 2B, GFP expression could be titrated by increasing the IPTG concentrations. Maximum depletion was obtained with ≥100 μM IPTG. To investigate how quick GFP expression was switched off after IPTG induction, SAMK56 was induced with 100 μM IPTG and samples were taken every 30th min for 3 hrs. The GFP fluorescence levels (Fig. 2C) decreased rapidly (signal reduced by ca. 90% within 60 min), suggesting that expression was switched off almost immediately. Specific transcriptional knockdown was also demonstrated using qPCR (Fig. S3, see below for details). Note that for some of our later experiments, we observed a faster depletion of cell division proteins when increasing the IPTG concentration to 400 μM. Furthermore, as a proof of the functionality of the CRISPRi system in targeting essential cell cycle genes, we created sgRNAs targeting the DNA replication initiator dnaA (encoded on an operon with dnaN) and pbp1 (monocistronic) encoding a penicillin-binding protein. The CRISPRi strains were analyzed by growth assays and microscopy (Fig. 2D-F), and the observed phenotypes were as expected, confirming the suitability of the CRISPRi system to study the function of essential genes; compared to the control strain (Fig. 2D), the pbp1 depletion resulted in clustered, larger cells with aberrant morphologies (Fig. 2E) (Pereira et al., 2007, Pereira et al., 2009) while dnaA depletion resulted in anucleate cells with variable sizes and nucleoid morphologies (Fig. 2F).
CozEa and CozEb have overlapping functions and are important for proper cell cycle progression in S. aureus
We made sgRNA constructs targeting cozEa and cozEb, and depleted expression of cozEa in the ΔcozEb background and vice versa. Note that cozEa is monocistronic, while cozEb is located as the last gene in the operon, and the knockdown will therefore have minimal polar effects (Peters et al., 2016, Liu et al., 2017). No growth reduction was observed upon knockdown of the individual genes in wild-type background (as expected from the deletion mutants) (Fig. 3A and B). We performed RT-qPCR on these CRISPRi-strains and verified that transcription of cozEa and cozEb was specifically knocked down (Fig. S3). After diluting the cells to OD600 of 0.05 with and without 150 μM IPTG and culturing for 2 hours, the expression of cozEa and cozEb was reduced 23-fold and 13-fold in the respective strains when comparing induced and non-induced conditions.

A.Strains carrying sgRNA targeting cozEb in wild-type background (circles, strain SAMK57) compared to the ΔcozEa background (triangles, strain GS1167). Growth curves with (red) or without (black) induction of dCas9 expression with 150 μM IPTG are shown.
B.Strains carrying sgRNA targeting cozEa in wild-type background (circles, strain SAMK60) compared to the ΔcozEb strain (triangles, strain GS1163). Growth curves with (red) or without (black) induction of dCas9 expression with 150 μM IPTG are shown.
While no effect on growth was observed by depleting cozEa or cozEb expression in wild-type background, knockdown of the other gene in the respective deletion backgrounds caused dramatic reduction in growth (Fig. 3A and B). The initial doubling time after CRISPRi-induction is more affected in GS1167 (ΔcozEa, sgRNA(cozEb), td induced = 52 min and td non-induced = 34 min) than in GS1163 (ΔcozEb, sgRNA(cozEa), tdinduced= 38 min and tdnon-induced= 35 min). However, after approximately 300 min of dCas9-induction, the growth is dramatically reduced for both GS1167 and GS1163 (Fig. 3). From this we conclude that cozEa and cozEb have overlapping functions in S. aureus.
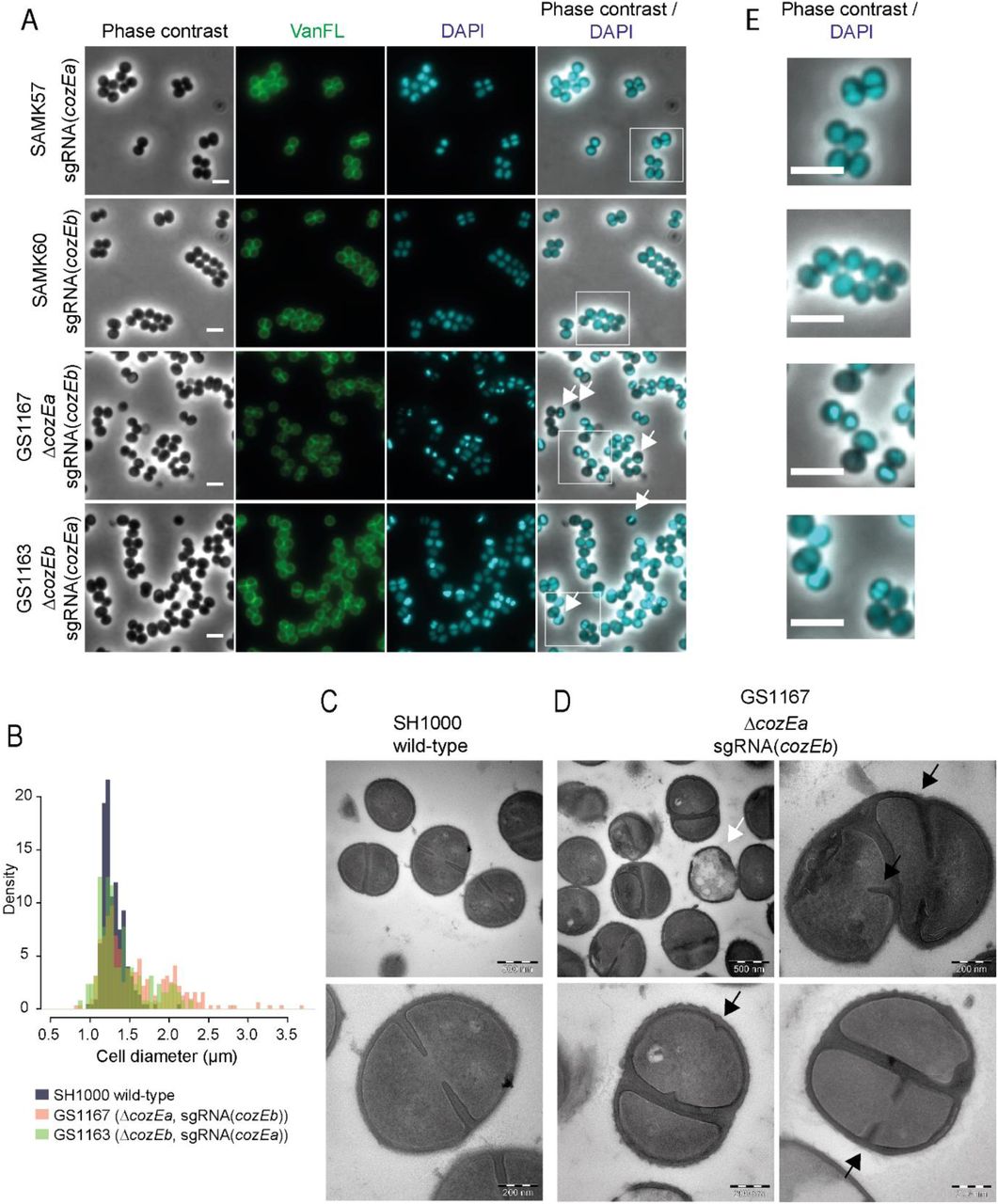
The phenotypes of the GS1167 (ΔcozEa∷spc, depleted cozEb) and GS1163 (ΔcozEb∷spc, depleted cozEa) strains were then further investigated by microscopy. Phase contrast micrographs revealed severely perturbed cell morphologies when the CRISPRi system is induced, displaying both variable cell shapes and sizes as well as increased clustering of cells (Fig. 4A). Measurements of the cell diameter of CRISPRi-induced GS1167 and GS1163 cells show that they have a very wide distribution compared to the wild type (Fig. 4B). We also made a double sgRNA strain allowing knockdown of both cozEa and cozEb simultaneously with the CRISPRi system (strain SAMK75), and as expected this strain displayed similar phenotype as the GS1167 and GS1163 strains (Fig. S4).
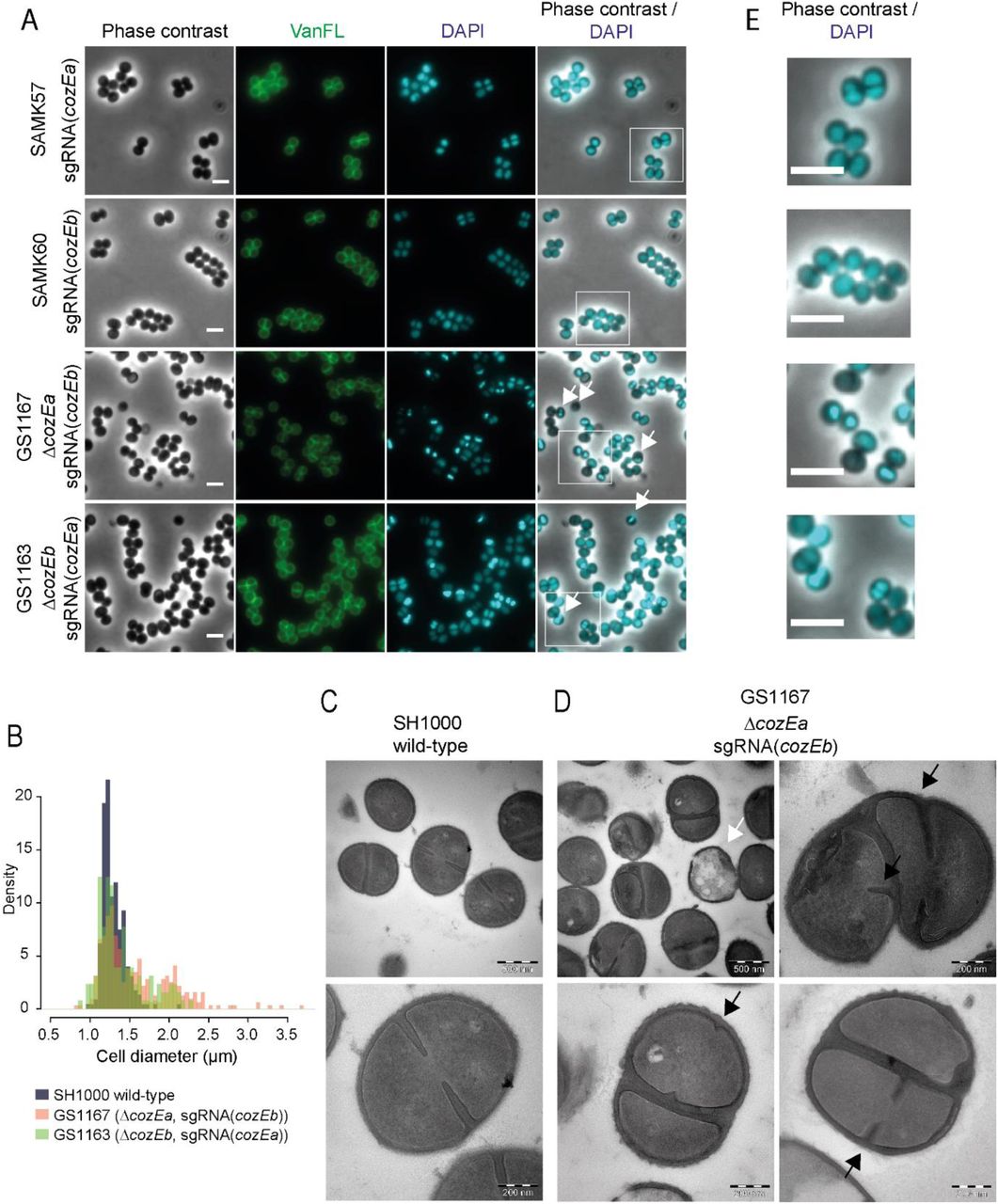
A.Phase contrast micrographs are shown to the left. The cells were also stained with fluorescent vancomycin and DAPI to visualize cell wall and the nucleoid, respectively. Overlay of DAPI and phase contrast images are shown. White arrows point to cell with aberrant nucleoids. The scale bars are 2 μm. The white squares indicate the area magnified in Fig. 4E.
B.Histogram of cell diameter distribution for wild-type cells (grey) and ΔcozEa∷spc with depleted cozEb (GS1167, orange) cells and ΔcozEb∷spc with depleted cozEa (GS1163, green) induced with 150 μM IPTG. Both GS1167 and GS1163 are different from wild-type (P < 0.05, Kolmogorov-Smirnov test), with high proportion of the cells with diameters larger than 1.5 μm (7.9 % for wild-type compared to 44.2% for GS1167 and 27.9 % for GS1163, n >150 for all strains). C-D. Transmission electron micrographs of wild-type cells (C, SH1000) and ΔcozEa∷spc with depleted cozEb (D, GS1167) cells. The white arrow points to a lysed cell. Black arrows point to septum initiation in GS1167 cells. Two different magnifications are shown, as indicated by the scale bars. E. Magnified insets from Fig. 4A with overlays of DAPI and phase contrast images, demonstrating the variation in nucleoid staining between the strains.
Perturbed morphologies in cells depleted of both cozEa and cozEb prompted us to further analyze cell division placement by transmission electron microscopy (TEM) (Fig. 4C - D and S4 Fig.). In GS1167 cells (ΔcozEa∷spc with depleted cozEb) depleted for 4 hours, cells could initiate septum formation in only one of the daughter cells prior to cell splitting (Fig. 4D, lower panels) and non-perpendicular septum formation resulting in misshaped cells was also observed (Fig. 4D, top right panel). Spatial and temporal coordination of cell division thus seem compromised in cells lacking CozEa and CozEb. Empty, lysed cells were also observed (Fig. 4D). Furthermore, the cell wall also appeared to be thicker in the mutant cells, and comparison of septum thickness based on the TEM images show that the GS1167 on average has thicker septal cross-wall compared to wild-type cells (Fig. S6). In mildly depleted cells (depletion for 1 hour), the phenotype is less severe, however, uncoordinated initiation of septum formation and aberrant septa were also observed here (Fig. S5). Electron micrographs of the individual deletions showed that ΔcozEa mutants display both lysed cells (Fig. S5) and have thicker septa than wild-type (Fig. S6). These phenotypes were not observed in ΔcozEb cells (Fig. S5 and S6).
Notably, nucleoid staining of the cells depleted of both cozEa and cozEb using DAPI was also abnormal, displaying non-homogeneous staining patterns. A large fraction of the cells appeared to have high intensity or highly condensed DAPI signals (47.1 % for GS1167, 22.9 % for GS1163, n > 250) compared to the wild type, and some cells were also anucleate under these conditions (4.1 % for GS1167 and 2.0 % for GS1163, n > 250) (Fig. 4A and zoomed images in Fig. 4E). The chromosome biology of the cells thus also seems to be perturbed when CozEa and CozEb are lacking.
CozEa and CozEb do not affect cell wall composition, but interact with key cell division proteins
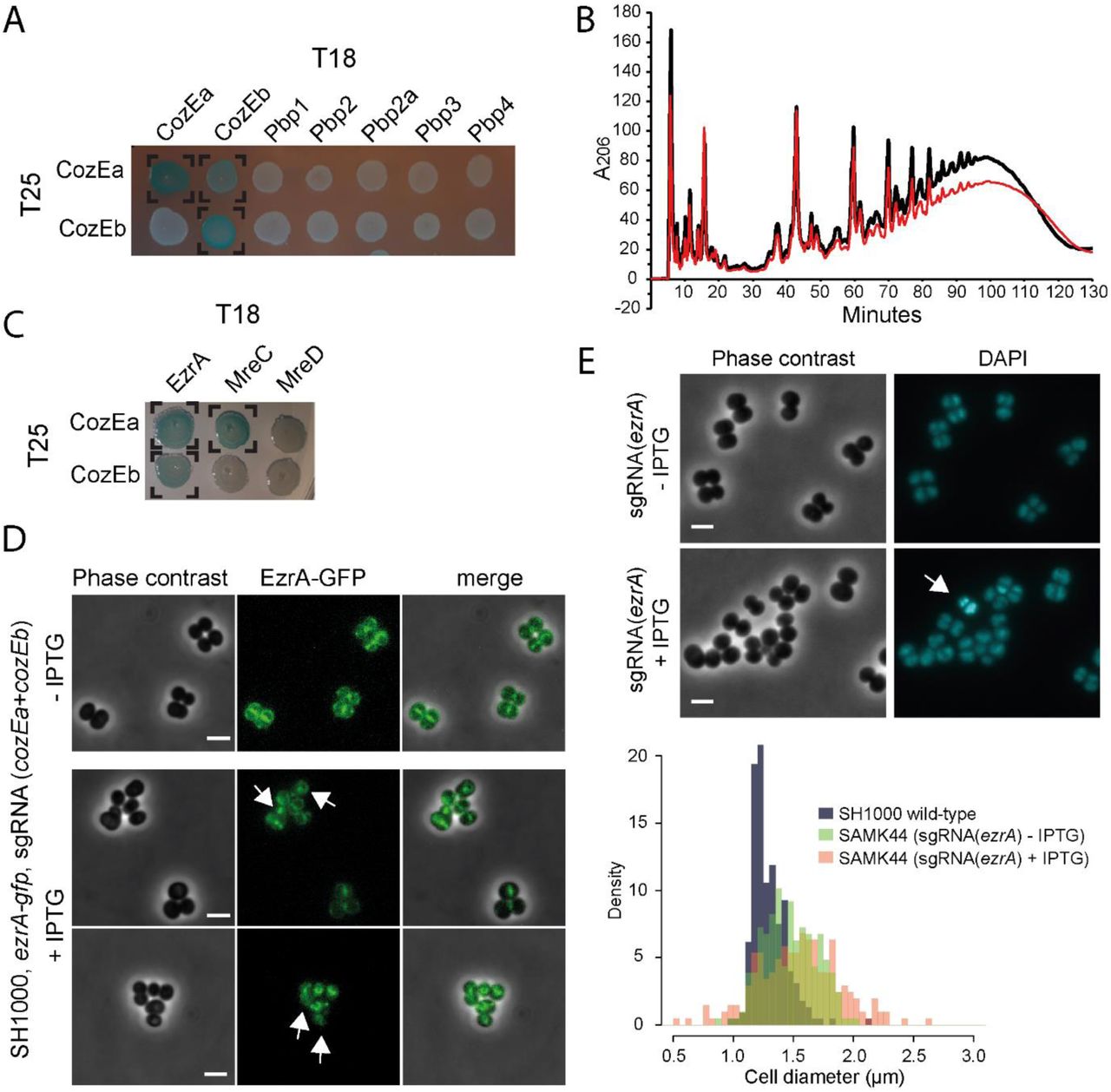
TEM images showed that the septal cell wall appeared different between wild-type and the cozE-deficient cells; coordination of cell wall synthesis seems to be compromised (Fig. 4) and the septal cell wall is thicker in cells lacking cozEa (Fig. S5.). In S. pneumoniae, CozESpn has been shown to interact with the bi-functional penicillin binding protein PBP1a (Fenton et al., 2016). To get insight into whether CozEa and CozEb could influence cell wall synthesis, we first investigated whether these proteins could interact with any of the four PBPs of S. aureus (PBP1, PBP2, PBP3 and PBP4) using bacterial two-hybrid assays (see Material and Methods for detailed description) (Karimova et al., 2005). While CozEa and CozEb both self-interacted and interacted with each other, no interaction was found with any of the PBPs of S. aureus (Fig. 5A, Fig. S7). We also tested the methicillin-resistant PBP2a (MecA) from S. aureus COL, but we could not find any interactions with the CozE proteins (Fig. 5A). Next, we analyzed the muropeptide composition of peptidoglycan derived from strain the GS1167 (ΔcozEa with depleted cozEb), to see whether the cell wall architecture was altered in this mutant. However, the muropeptide composition of GS1167 was similar to the wild-type (Fig. 5B). This suggests that CozEa and CozEb affect positioning and timing of cell division and cross-wall synthesis, but that the cell wall synthesis pathway is unaltered.

A. Bacterial two-hybrid analyses of interactions between CozEa and CozEb fused to the T25 domain with proteins fused to the T18. Positive interactions are observed as blue colonies and marked with brackets. See Fig. S7 for control experiments. All interaction results were repeated at least five times.
B. Cell wall muropeptide composition of SAMK15 control cells (black) and GS1167 depletion cells (red) induced with 150 mM IPTG for 4 hours as analyzed with UHPLC. See Fig. S7 for control experiments.
C. Bacterial two-hybrid analyses of interactions between CozEa and CozEb fused to the T25 domain with EzrA, MreC and MreD fused to the T18 domain. Positive interactions are observed as blue colonies and marked with brackets. See Fig. S7 for control experiments. All interaction results were repeated at least five times.
D. Localization of EzrA-GFP without and with induction of CozEa/CozEb-depletion. The upper panel shows uninduced cells and two lower panels show representative cells after induction of CRISPRi with 400 μM IPTG. The arrows point to cells with obvious mislocalization of EzrA-GFP. The scale bars are 2 μm.
E. Phenotype of ezrA knockdown. Phase contrast micrographs and DAPI signal are shown for SAMK44 (CRISPRi targeting ezrA) with or without induction with 300 μM IPTG. The arrows point to cells with aberrant nucleoid staining. The scale bars are 2 μm. The lower panel shows cell diameter histograms of wild-type SH1000 cells as well as induced and non-induced SAMK44 cells. Both induced and non-induced cells are significantly larger than wild-type cells (Kolmogorov-Smirnov test, P < 0.05), with high proportion of the cells with diameters larger than 1.5 μm (7.9 % for wild-type compared to 43.9% for non-induced and 55.1 % for induced, n >100 for all strains).
We further analyzed whether CozEa and CozEb could interact with a selection of other key cell cycle proteins using bacterial two-hybrid assays (Fig. 5C, S1 Table, Fig. S7). CozESpn has been shown to interact with MreCSpn, MreDSpn and DivIVASpn (Straume et al., 2017, Fenton et al., 2016). We detected an interaction between CozEa and MreC, however, this was not the case for CozEb. No interactions with MreD or DivIVA were observed for any of the combinations (Fig. 5C, S1 Table). The only protein we could identify that interacted with both CozEa and CozEb was the early cell division protein EzrA. Among the other proteins tested, we also found that CozEa and GpsB interacted, however, not CozEb and GpsB. A full overview of all tested bacterial two-hybrid interactions are given in S1 Table.
The positive two-hybrid interactions suggest that CozEa and CozEb may mediate cell division control via interactions with EzrA. CozEa and CozEb both displayed a membrane localization in S. aureus SH1000, with no apparent enrichment in the septal region (Fig. S9). Notably, however, depletion of cozEa and cozEb expression in cells expressing a chromosomal ezrA-gfp fusion (Lund et al., 2018), demonstrate that EzrA-GFP is mislocalized under these conditions (Fig. 5D). Furthermore, knockdown of ezrA using the CRISPRi system leads to similar phenotypes as cells lacking CozEa and CozEb (Fig. 5E) with variable cell sizes and nucleoid staining. This is fully in line with previous results of ezrA deletions and knockdown mutants in S. aureus (Steele et al., 2011, Jorge et al., 2011). Note that the cell size effect is observed also, but to a lesser extent, when no IPTG is added, reflecting leaky expression from the Plac promoter. Abnormal DAPI staining pattern was also observed in the cells after IPTG induction, although this phenotype appear to be less pronounced in the ezrA knockdown cells (5.5 % of cells, n = 200) compared to cells depleted of CozEa and CozEb. It should also be noted that the growth rate was not severely affected upon induction of ezrA knockdown (Fig. S6), suggesting that ezrA is not essential for normal growth under these conditions (Bottomley et al., 2014).
The division ring is mislocalized in S. pneumoniae cells depleted of CozESpn
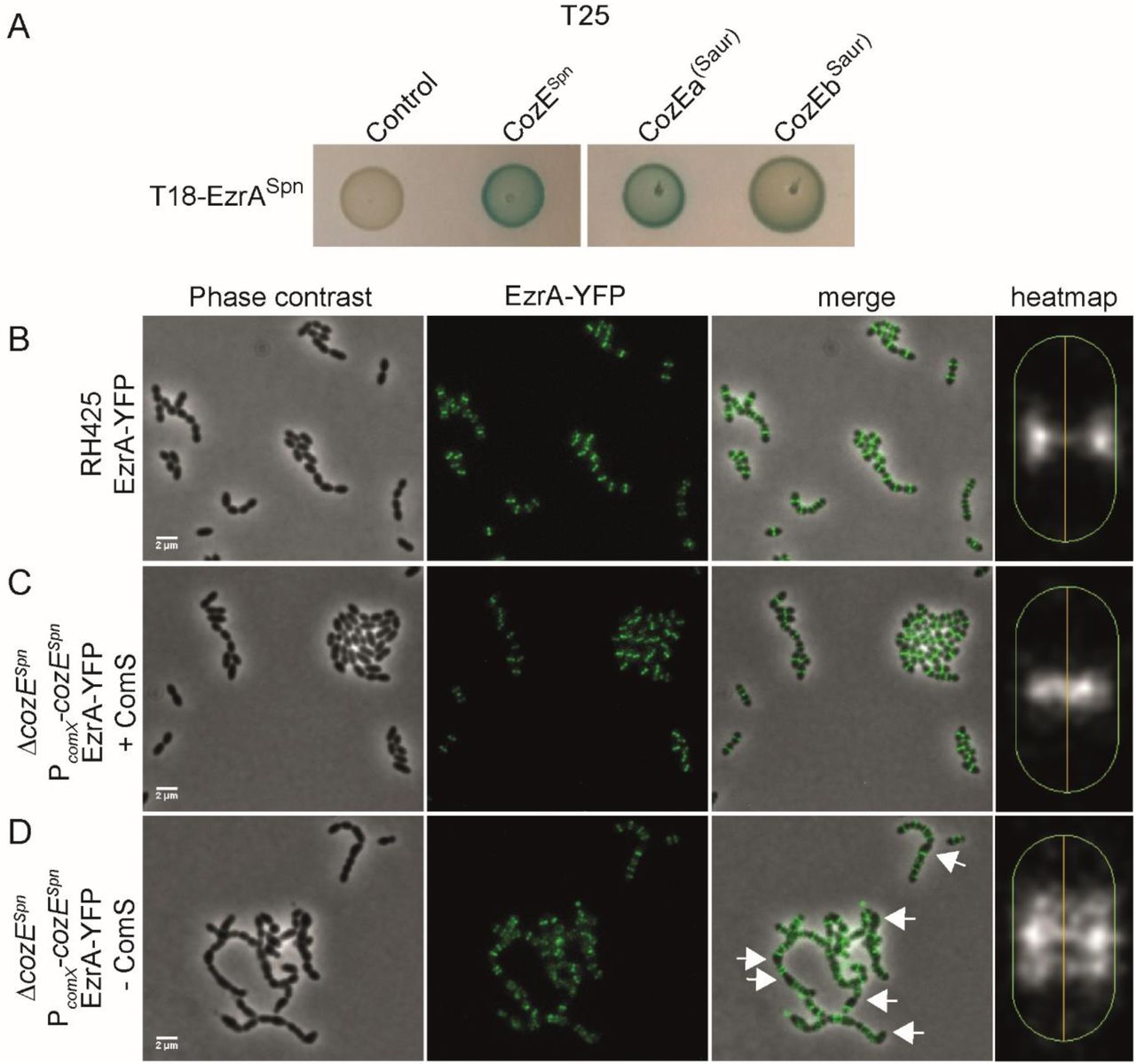
The results above demonstrate that CozEa and CozEb play functionally overlapping roles in controlling cell division in S. aureus, and both genes can be deleted individually. As mentioned above, a single protein CozESpn (SPD_0768 in strain D39 and Spr0777 in strain R6), is shown to be essential for growth and proper cell morphology in S. pneumoniae (Fenton et al., 2016, Straume et al., 2017). To investigate whether the EzrA-interactions detected here were specific for S. aureus or also relevant in S. pneumoniae, we used bacterial two-hybrid assays to test the interaction between CozESpn and EzrASpn. Just like the staphylococcal proteins, a strong interaction was found between the corresponding pneumococcal proteins (Fig. 6A). Strikingly, while EzrA localized to midcell in wild-type S. pneumoniae (Fig. 6B and C), the protein is clearly mislocalized in cells where cozESpn was depleted (Fig. 6D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A. Bacterial two-hybrid assay showing interactions between EzrASpn and different CozE-proteins.
B-D. Localization of EzrA-YFP in S. pneumoniae. Phase contrast and fluorescence are shown individually and merged. The localization of EzrA-YFP in the cells are also shown as heatmaps, as generated using MicrobeJ. The heatmaps represent the localizations in >650 cells for each strain. EzrA-YFP localization was analyzed in RH425 wild-type (B) and a strain where the native cozESpn is deleted and instead expressed from the ComS-inducible promoter PcomX (Berg et al., 2011). The cozESpn-depletion strain (ΔcozESpn and PcomX-cozESpn) was grown with (C) or without (D) inducer peptide ComS. The arrows point to examples of cells with mislocalized EzrA-YFP. The scale bars are 2 μm.
S. aureus cozEa and cozEb can complement the ΔcozE Spn phenotype of S. pneumoniae In order to gain further insight into functional conservation of CozE proteins between S. aureus and S. pneumoniae, we tested whether CozEa or CozEb could functionally complement the essential CozESpn in S. pneumoniae. We created pneumococcal strains in which cozEa and cozEb were chromosomally integrated downstream of the ComRS-inducible promoter, PcomX. Induction of PcomX is achieved by addition of the peptide ComS to the growth medium; ComS is internalized where it activates the PcomX-binding transcriptional activator ComR (Berg et al., 2011). Next, we attempted to delete the native cozESpn by allelic exchange with the Janus cassette (Sung et al., 2001), with and without presence of the inducer ComS. A functional complementation with CozEa or CozEb in the pneumococcus would allow deletion of the cozESpn gene. Indeed, upon induction of cozEa or cozEb expression with 2 μM ComS, the native cozESpn could readily be deleted (Table 1). It should be noted that the CozEa and CozEb probably have a reduced functionality compared to CozESpn, as higher inducer concentrations were required to obtain correct transformants for the non-native CozE-proteins (Table 1). Additionally, CozEa seemed to function better than CozEb, since the number of transformants were higher for the former. Microscopy of the resulting strains further confirmed that the typical cozESpn-depletion phenotype in pneumococci, characterized by extensive chaining and slight rounding of cells (seen by reduced length and aspect ratio closer to 1) (Straume et al., 2017), could be complemented by both CozEa or CozEb (Fig. S10).
Complementation of ΔcozESpn in S. pneumoniae with CozEa and CozEb.
Finally, to get insights into how the staphylococcal proteins CozEa and CozEb could complement CozESpn, we analyzed by bacterial two-hybrid assays whether CozEa and CozEb could still interact with EzrASpn (Fig. 6A). Both CozEa and CozEb interact with EzrASpn in this assay. Thus, conservation of the interaction with EzrA could thus explain why CozEa and CozEb were functional in S. pneumoniae.
Discussion
The membrane protein CozESpn was recently identified as an essential regulator of cell elongation in oval shaped S. pneumoniae (Fenton et al., 2016, Straume et al., 2017). CozE proteins are widely conserved and present in the genome of bacteria from different phyla and of different morphologies (Fenton et al., 2016). We here show that the two CozE-homologs of S. aureus, which we named CozEa and CozEb, play overlapping roles to control proper cell cycle progression in these spherical cells.
While the deletion of either cozEa or cozEb has only minor effects, both genes cannot be deleted at the same time. To confirm the synthetic relationship between cozEa and cozEb, we developed a CRISPRi system for S. aureus to allow knockdown of expression of essential genes. Recent reports have already shown the suitability of using CRISPR/dCas9 for knockdown of genes in S. aureus (Dong et al., 2017, Zhao et al., 2017). The plasmid-based CRISPR/dCas9-derived system we developed here contains several unique features compared to the published ones (Dong et al., 2017, Zhao et al., 2017): (i) Knockdown is inducible by addition of IPTG, since dCas9 expression is driven by the IPTG-inducible promoter. (ii) The plasmid harbouring the sgRNA construct is relatively small (5.8 kb), thus allowing easy replacement of target sequences by inverse PCR. (iii) Multi-sgRNA plasmids, allowing simultaneous knockdown of several genes, can be constructed by combining existing sgRNA plasmids using BglBrick assembly (Anderson et al., 2010) (Fig. S2).
Using the CRISPRi system, we could construct combined deletion/depletion strains or double-depletion strains to study cells depleted of CozE proteins. Since all the different strains depleted of CozE proteins showed the same phenotypes, we could exclude that the conservative substitution in the gene thiI (detected by whole genome resequencing, see results) played any functional role. Low levels of CozEa and CozEb proteins have pleiotropic effects on the staphylococcal cells, including abnormal cell size homeostasis and nucleoid staining, frequent lysis and, most strikingly, the thickened cell wall and compromised timing and positioning of cell division (Fig. 4). Wild-type S. aureus cells divide in consecutive, perpendicular planes, i.e. the new septum is formed perpendicular to the previous and splitting of daughter cells (Monteiro et al., 2018) (popping) finishes before the next septum is formed (Pinho et al., 2013, Zhou et al., 2015, Monteiro et al., 2015). However, cells lacking CozEa and CozEb can initiate septum formation asynchronously in only one of the daughter cells before the previous division cycle finishes and non-perpendicular septa were also observed, resulting in elongated cells. This is reminiscent of elongating staphylococcal FtsZ mutant strains (Fig. 4) (Pereira et al., 2016) or staphylococci treated with antibiotics targeting the cell wall or cell division (Lund et al., 2018, Pinho et al., 2000, Sieradzki & Tomasz, 2006).
Despite having misplaced and thicker septa than wild-type, the cell wall composition does not appear to be altered in the CozEa/CozEb-depleted cells and the membrane proteins CozEa or CozEb are not directly interacting with any of the PBPs of S. aureus. An interaction between CozEa and MreC was detected, however, CozEa nor CozEb could interact with MreD. Although the CozEa-MreC interaction may be important for directing peptidoglycan synthesis, like in S. pneumoniae, it is worth noting MreC and MreD are non-essential in S. aureus (Tavares et al., 2015). The significance of the CozEa-MreC interaction thus remains unknown.
CozEa and CozEb might compromise cell division coordination and autolytic splitting by interfering directly with key cell division proteins. The detailed mechanism of action remains to be determined, but we show that CozEa and CozEb could interact with one of the early cell division proteins, namely EzrA, in two-hybrid interaction assays. Notably, the EzrA-GFP localization in S. aureus cells lacking CozEa and CozEb was severely perturbed. Interaction with EzrA could thus be a plausible way for CozEa and CozEb to mediate cell division control. EzrA is one of the first proteins binding to the Z-ring in the initiation of cell division. EzrA was identified as a negative regulator of FtsZ formation in B. subtilis (Levin et al., 1999), and is thought to be important for the switch between elongation and division growth in B. subtilis via protein-protein interactions with penicillin-binding proteins (Claessen et al., 2008). EzrA plays a similar role in S. pneumoniae (Rued et al., 2017). In S. aureus, EzrA is involved in a large number of protein-protein interactions. Bacterial two-hybrid interactions have been shown between EzrA and FtsZ, DivIB, DivIC, FtsA, FtsL, Pbp1-3, SepF, GpsB, RodA (Steele et al., 2011) and DivIVA (Bottomley et al., 2017). Although some of these interactions may be false positives, it clearly suggests that EzrA is a central protein for proper cell cycle progression and cell wall synthesis in S. aureus. It has indeed been shown that EzrA plays a key role in staphylococcal cell size homeostasis; different levels of EzrA in the cells influence the cell size (Steele et al., 2011, Jorge et al., 2011). We also observed the same when ezrA was targeted using the CRISPRi system (Fig. 5). Furthermore, lack of EzrA causes mislocalization of other key cell division proteins such as FtsZ, GpsB and PBPs (Jorge et al., 2011, Steele et al., 2011). Thus, disrupting the localization or functionality of EzrA, which may be the case in cells lacking CozEa and CozEb, will therefore likely have large pleiotropic effects on different cell cycle processes, and is consistent with the results of the current study. Note, however, that there are conflicting results in the literature regarding the essentiality of ezrA (Steele et al., 2011, Jorge et al., 2011). The results from our CRISPRi depletion suggest that ezrA is non-essential for growth under our experimental conditions. Thus, it is likely that CozEa/CozEb have other roles in S. aureus yet to be identified. Since the CozEa-EzrA and CozEb-EzrA interactions were found by testing a collection of proteins in a heterologous bacterial two-hybrid assay, there may be important CozEa/CozEb interaction partners that we have not yet identified. It also remains to be determined whether the abnormal nucleoid-staining pattern is directly affected by CozEa/CozEb, or if this is an indirect effect of the compromised cell division control.
Our results also show that the influence of CozE on cell division observed in S. aureus was conserved in ovococcal S. pneumoniae. Just like in S. aureus, CozESpn could interact with EzrASpn in bacterial two-hybrid assays and depletion of CozESpn in S. pneumoniae caused aberrant cell division placement as observed by mislocalization of EzrASpn-GFP. In line with this, EzrASpn interacts with FtsZSpn, GpsBSpn and DivIVASpn and is important for coordination of septal and peripheral cell wall synthesis in ovococcal S. pneumoniae cells (Rued et al., 2017, Fleurie et al., 2014b). Depletion of ezrASpn expression in S. pneumoniae (Fig. 5) also resulted in cells with variable sizes and nucleoid staining pattern as well as multiple or misplaced septa (Liu et al., 2017). Notably, both cozEa and cozEb could complement the ΔcozESpn in S. pneumoniae, although the functionality of the staphylococcal proteins was reduced compared to the native CozESpn.
CozESpn was identified as an essential regulator of cell elongation in S. pneumoniae, working through interactions with the MreCDSpn and PBP1aSpn (Fenton et al., 2016). S. aureus also appears to elongate slightly during the cell cycle, but little is known about this and a machinery for peripheral peptidoglycan synthesis is lacking (Monteiro et al., 2015). The results presented here suggest that CozE proteins in bacteria have additional functionalities to what was found for CozESpn and that they may act at an earlier stage of cell division to mediate proper spatial and temporal control. During the bacterial cell cycle, DNA replication, chromosome segregation, cell division and cell wall synthesis need to be coordinated spatially and temporally. The uncontrolled cell division occurring in S. aureus cells lacking CozEa and CozEb, may thus result in the pleiotropic effects we observed, such as aberrant chromosome replication/segregation, cell lysis and variable cell sizes. Future studies are required to unravel in more detail the molecular mode of action by which CozE proteins work. The CozE-mediated control of cell division seems to be a conserved feature between spherical S. aureus and ovococcal S. pneumoniae. Since genes encoding these proteins are widespread and found in bacteria with different cellular morphologies (Fenton et al., 2016), it will be interesting to unravel how CozE proteins function in bacterial cells of various shapes.
Experimental procedures
Bacterial strains, growth conditions and transformation
Bacterial strains used in this study are listed in S2 Table. S. aureus was routinely grown at 37°C in brain-heart-infusion BHI broth with shaking or on BHI agar plates at 37°C. When appropriate, 5 μg/ml erythromycin, 10 μg/ml chloramphenicol or 100 μg/ml spectinomycin was added for selection. For induction of gene expression, different concentrations of IPTG was added. S. pneumoniae was grown in C medium (Lacks & Hotchkiss, 1960) at 37°C without shaking or on Todd-Hewitt (TH) agar plates at 37°C. When appropriate, 400 μg/ml kanamycin, 200 μg/ml streptomycin or 100 μg/ml spectinomycin was added to the growth medium for selection. Escherichia coli was grown at 37°C in LB medium with shaking or on LA plates at 37°C with 100 μg/ml ampicillin or 50 μg/ml kanamycin added for selection.
Transformation of E. coli was performed with a standard heat shock protocol. S. aureus was transformed with electroporation using plasmid DNA isolated from E. coli DC10B (Monk et al., 2012) or IM08B (Monk et al., 2015). Preparation of electrocompetent cells and electroporation were performed essentially as described before (Lofblom et al., 2007). Constructs were introduced into S. pneumoniae using natural transformation as described before (Stamsås et al., 2017).
Construction of plasmids for the CRISPRi system
Construction of plasmid pLOW-dCas9. The dcas9 gene was amplified from plasmid pJWV102-dcas9 (Liu et al., 2017) using primers mk41 and mk42. The fragment and the vector pLOW-ftsZ-m(sf)gfp were both digested with SalI and NotI and ligated to produce the pLOW-dCas9 construct where dcas9 is placed downstream of an IPTG-inducible promoter. The ligation was transformed into E. coli IM08B with ampicillin selection and correct construct was verified by PCR and sequencing. All plasmids in this study are listed in S3 Table, while all primers are listed in S4 Table.
Constructions of plasmids expressing single guide RNA. The single guide RNA (sgRNA) construct, containing a transcriptionally isolated sgRNA (see Fig. 2) driven by a constitutive promoter, was cut out from vector pPEPX-sgRNA(luc) (Liu et al., 2017) using PstI and BamHI. The fragment was ligated into the corresponding sites of vector pCG248 (Helle et al., 2011) (thus removing the xyl/tet regulatory system from this vector) to produce pCG248-sgRNA(luc). The construct was verified by sequencing.
New sgRNAs were then made by replacing the 20 nt base-pairing region with an inverse PCR approach; using the pCG248-sgRNA(luc) as template, new sgRNA-plasmids were amplified using one reverse phosphorylated primer mk200 annealing immediately upstream of the sgRNA, combined with a gene-specific forward primer containing the base-pairing region as overhangs. The product was treated with DpnI to remove the template plasmid and ligated using T4 DNA ligase prior to transformation into E. coli IM08B with ampicillin selection. Constructs were verified by sequencing. The resulting plasmids were named pCG248-sgRNA(x), where x denotes the name of the gene to be targeted. Selection of the gene-specific base pairing region to be used was done using established criteria (Liu et al., 2017, Peters et al., 2016).
For construction of the double-sgRNAs targeting both cozEa and cozEb, the fragment containing the sgRNA(cozEb) was cut out from the plasmid pCG248-sgRNA(cozEb) using restriction sites PstI and BamHI. The resulting fragment was ligated into the PstI and BglII sites of plasmid pCG248-sgRNA(cozEa). The resulting plasmid, pCG248-sgRNA(cozEa-cozEb), expresses two sgRNAs targeting both cozEa and cozEb. See also Fig. S2.
CRISPR interference. In order to obtain S. aureus strains for CRISPRi, the plasmid pLOW-dCas9 was first introduced with erythromycin selection. Then, in a second step, the sgRNA-containing plasmid pCG248(x), was introduced with combined chloramphenicol and erythromycin selection (in order to retain both plasmids). Cells were then grown in the presence of IPTG to induce expression of dcas9.
S. aureus plasmid and strain construction
Construction of strain with constitutive GFP expression. A fragment containing a spectinomycin resistance gene and a gene encoding a monomeric superfolder GFP, m(sf)gfp, was first assembled by overlap extension PCR. The spectinomycin resistance cassette was amplified from pCN55 (Charpentier et al., 2004) using primers mk203 and mk204. The m(sf)gfp gene was amplified from plasmid pMK17 (Kjos et al., 2016) using primer im84 and im2. The primers mk204 and im84 contain overlapping sequence, and the spc-m(sf)gfp fragment could then be assembled in a second amplification step with outer primers mk203 and im2. The resulting fragment contains EcoRI sites on both ends introduced by overhangs in the primers. The fragment was digested with EcoRI and ligated into the corresponding site of plasmid pMAD-int2-luc. The ligation was transformed in E. coli IM08B with ampicillin selection. The resulting construct, pMAD-int2-luc-spc-gfp, was verified by PCR and sequencing. The temperature sensitive pMAD-derivative vector (Arnaud et al., 2004) was transformed in S. aureus RN4220 at 30°C with erythromycin and X-gal selection. Integration of the plasmid into the chromosome and excision to construct the integration of P3-luc-spc-gfp in the int-locus (Fagerlund et al., 2014) was performed as described (Arnaud et al., 2004) with spectinomycin selection.
Construction of ΔcozEa∷spc and ΔcozEb∷spc. Vectors for deletion of cozEa and cozEb were made in pMAD. The constructions cozEa∷spc and cozEb∷spc were first assembled by overlap extension PCR as follows: The spectinomycin resistance cassette (spc) was amplified from plasmid pCN55 using primers mk188 and mk189. The cozEa upstream region was amplified with primers mk182 and mk184 and the downstream fragment with primers mk185 and mk187. The three fragments were assembled using overlap extension PCR and amplified using the outer primers mk183 and mk186. The outer primers contain restriction sites for NcoI and BamHI, and the cozEa_up – spc – cozEa_down fragment was ligated into the NcoI and BamHI sites of pMAD. The ligation was transformed into E. coli IM08B and correct transformants containing the pMAD-cozEa∷spc plasmid were verified by PCR and sequencing.
pMAD-cozEb∷spc plasmid was constructed in a similar way. The spc fragment was amplified in the same manner as above. The cozEb up- and downstream regions were amplified using primers mk190 and mk192, and mk193 and mk195, respectively. The resulting fragments were fused by overlap extension PCR using primers mk191 and mk194, and the resulting fragment (cozEb_up – spc – cozEb_down), was ligated into the NcoI and BamHI sites of pMAD.
Finally, the pMAD-cozEa∷cam plasmid was constructed by amplifying the upstream region with primers mk183 and mk259 and the downstream region with primers mk260 and mk186. A chloramphenicol resistance cassette was amplified from plasmid pRAB11 (Helle et al., 2011), using primers mk257 and mk258. The fragments were fused by overlap extension PCR and ligated into the NcoI and BamHI sites of pMAD.
Construction of the deletion strains was done as previously described for the temperature sensitive pMAD system (Arnaud et al., 2004). Briefly, the plasmids were transformed into S. aureus SH1000 with erythromycin selection with incubation at permissive temperature of 30°C. X-gal was also added to the transformation plates and blue colonies were re-streaked once at 30°C. One colony was then picked and grown in medium without selection at 30°C for 2 hours before the tube was transferred to non-permissive temperature for plasmid replication (43°C) for 6 hours. The culture was then plated on TSA with spectinomycin and X-gal at 43°C. White colonies, where double crossover had taken place to replace the gene of interest with the spectinomycin cassette were re-streaked on two separate plates to verify that they were spectinomycin resistant and erythromycin sensitive. Correct constructs were further verified by PCR and sequencing. The ΔcozEa∷spc deletion strain was named SAMK24 and the ΔcozEb∷spc deletion strain SAMK21.
Construction of pLOW-cozEa-m(sf)gfp and pLOW-cozEb-m(sf)gfp. m(sf)gfp, was first inserted into the plasmid pLOW-FtsZ-GFP (Liew et al., 2011) (replacing the gfp gene). The m(sf)gfp gene with linker was amplified from plasmid pMK17 (Kjos et al., 2016) using primers im1 and im2 and ligated into the BamHI and EcoRI sites of plasmid pLOW-FtsZ-GFP. The resulting construct, pLOW-ftsZ-m(sf)gfp, was verified by PCR and sequencing. To construct pLOW-cozEa-m(sf)gfp and pLOW-cozEb-m(sf)gfp, ftsZ was replaced with cozEa or cozEb in this vector. cozEa was amplified using primers im10 and im11, while cozEb was amplified using primers im12 and im13, both using genomic DNA from SH1000 as template. The fragments were digested with SalI and BamHI and ligated into the respective sites of vector pLOW-ftsZ-m(sf)gfp. The constructs were verified by PCR and sequencing.
Strain construction for S. pneumoniae
Construction of PcomX-cozESpn, PcomX-cozEa, PcomX-cozEb and deletion of cozESpn. The ectopic PcomX-cozESpn construct integrated in the cpsO-cpnN locus of S. pneumoniae has been described previously (Straume et al., 2017).
For construction of PcomX-cozEa and PcomX-cozEb, primers gs693/gs694 were used to amplify the cozEa gene and primers GS691/GS692 were used to amplify the cozEb gene, both using genomic DNA from S. aureus SH1000 as template. Using strain S. pneumoniae SPH131 as template, the PcomX and 800 bp upstream region in the cpsO-cpsN locus were amplified with primers khb31/khb33 and the cpsO-cpsN downstream fragment was amplified with primers khb34/khb36. The three fragments contain overlapping sequences introduced in the primers, and they were assembled by overlap extension PCR to create PcomX-cozEa and PcomX-cozEb. The constructs were transformed into strain SPH131 (containing a Janus cassette in the cpsO-cpsN locus) and transformants were selected on plates with streptomycin. The resulting strains were named GS1169 and GS1170.
The native pneumococcal cozESpn (spr0777) gene was replaced with a Janus cassette in strains GS1169, GS1170 and KHB432 as described before (Straume et al., 2017). Since spr0777 is essential, different concentrations of the transcription inducer ComS (0, 0.2 and 2 μM) were added during all transformation steps to induce expression of the various cozE genes from the PcomX promoter. Transformants were selected on plates containing kanamycin. The number of colonies were counted and the transformants were screened for the presence of the pneumococcal cozESpn gene with primers gs337 and gs338 for each ComS concentration.
Construction of ezrASpn-yfp. An ezrA-yfp_spc fragment was assembled by overlap extension PCR. The ezrA_up fragment was amplified from S. pneumoniae R6 using primers mk288 and mk289, while the ezrA_down fragment was amplified using primers mk292 and mk293. The yfp_spc fragment was amplified from strain MK123 using primers mk290 and mk291. Due to overhangs in the primers, the three fragments could be assembled using outer primers mk301 and mk302, to produce the ezrA-yfp_spc fragment, which integrates in the pneumococcal chromosome to replace the native ezrA gene with an ezrA-yfp fusion gene. The fragment was transformed into S. pneumoniae and transformants were selected on plates with spectinomycin. Correct transformants were verified by PCR.
Total RNA isolation, cDNA synthesis and qPCR
Overnight cultures were diluted to OD600 = 0.05 in 20 ml BHI containing 10 μg/ml chloramphenicol and 5 μg/ml erythromycin. There were two cultures of each strain, one of them was induced with 150 μM IPTG. Cells were harvested from 10 ml culture at OD600 = 0.4 by centrifugation at 4000 × g at 4°C for one minute, and the pellets were immediately frozen in liquid nitrogen. The cells were lysed by mechanical disruption in Lysing Matrix B, 2 mL tubes (MP Biomedicals) by FastPrep®-24 (MP Biomedicals). The disruption was done at maximum speed for 3 × 20 seconds, with cooling on ice between the runs. Total RNA was extracted using RNeasy Mini kit following the manufacturers’ description (Qiagen). Eluted RNA was treated with DNase I for removal of residual DNA, following the description of the manufacturer (Invitrogen). Thereafter, DNase was removed by Phenol-chloroform extraction. cDNA was synthesized using Superscript™ III Reverse Transcriptase (Invitrogen). Twenty-five ng cDNA was used as template for qPCR performed with PowerUp™ SYBR™ Green Master Mix (Applied Biosystems) in a StepOne Plus machine (Applied Biosystems). The setup included triplicates for each of the target genes for every sample. Primers im126 and im127 were used to target the reference gene, pta (Valihrach & Demnerova, 2012). Primers im130 and im131 were used to target cozEa, and primers im132 and im133 to target cozEb. The differential expression of cozEa and cozEb between non-induced and induced conditions was calculated according to the Pfaffl-method (Pfaffl, 2001).
Bacterial two-hybrid analysis
Construction of plasmids. Genes of interest were fused in frame to either 5` end or 3` end of either the T18 or the T25 domain of adenylate cyclase from Bordetella pertussis using the four vectors (pKT25, pKNT25, pUT18, pUT18C) provided by the manufacturer (Euromedex). Primers used for amplification of the genes are listed in S4 Table. The amplified fragments were digested (restriction sites indicated in the S4 Table) and ligated into the corresponding restriction sites in the vectors. Ligations were transformed into E. coli XL1-Blue cells, and selected on 1 % glucose LA plates containing either 50 μg/ml kanamycin or 100 μg/ml ampicillin. Correct plasmids were verified by PCR and sequencing.
Bacterial two-hybrid assay. Bacterial two-hybrid assays (Karimova et al., 2005) were performed as described by the manufacturer (Euromedex). Briefly, two plasmids, one containing a fusion to the T18 domain and the other a fusion to the T25 domain, were co-transformed into E. coli BTH101. The transformants were selected on LA plates containing 50 μg/ml kanamycin and 100 μg/ml ampicillin for selection. Five random colonies were picked per assay and grown in liquid LB containing kanamycin and ampicillin to OD600 0.3, before 2.5 μl of the cell culture was spotted on LA plates supplemented with 50 μg/ml kanamycin, 100 μg/ml ampicillin, 40 μg/ml of X-gal and 0.5 mM IPTG. The plates were incubated protected from light at 30°C for 20 h to 48 h. Positive interactions are indicated by appearance of blue colonies, while white colonies indicate no interaction. All interaction assays were repeated with at least five independent replicates.
Isolation of peptidoglycan and HPLC-analysis
Strains GS1167 and SAMK15 were inoculated in 60 ml BHI containing 10 μg/ml chloramphenicol and 5 μg/ml erythromycin. At OD600 0.2 these cells were transferred to 1.5 liters of BHI containing 10 μg/ml chloramphenicol, 5 μg/ml erythromycin and 150 μM IPTG. When reaching OD600 = 0.3, cells were harvested at 8000 x g for 10 minutes. Peptidoglycan was isolated according to the protocol described by Vollmer (Vollmer, 2007). The isolated peptidoglycan was lyophilized and resuspended in water to a final concentration of 50 mg/ml.
HPLC analysis of muropeptides was performed as described by Vollmer (Vollmer, 2007) and Carvalho et al (Carvalho et al., 2015) with minor changes. Briefly, to remove cell wall teichoic acids, ten milligrams of purified peptidoglycan were treated with 1.5 ml 48 % HF at 4°C for 48 hours with gentle mixing. The HF-treated peptidoglycan was collected by centrifugation at 20 000 x g for 30 minutes and washed two times with 1.5 ml of dH2O, once with 1.5 ml of 50 mM Tris-HCl (pH 7.4) and finally twice with 1.5 ml of dH2O. One mg of HF-treated peptidoglycan was digested with 5000 U mutanolysin at 37°C for 18-20 hours in a final volume of 100 μl containing 12.5 mM NaH2PO4 (pH 5.5). The sample was boiled for 20 minutes before insoluble material was removed by centrifugation at 20 000 x g for 30 minutes. The supernatant was added with 0.5 M Na-borate pH 9.0 (1:1 volume) and treated with 1-2 mg of Na-borohydride for 30 minutes at room temperature to reduce the sugars. The reaction was stopped by adjusting the pH to 2.0 using 20 % phosphoric acid. Muropeptides were separated on a C18 column (Vydac 218TP C18 5 mm, Grace Davison Discovery Sciences) at 52°C using a linear 155-minutes gradient of methanol from 5-30 % in 0.1 M NaH2PO4 (pH 2.0) at a flow rate of 0.5 ml/min. Eluted muropeptides were detected at 206 nm.
Phase contrast and fluorescence microscopy
Microscopy was performed on a Zeiss AxioObserver with ZEN Blue software. Images were captured with an ORCA-Flash4.0 V2 Digital CMOS camera (Hamamatsu Photonics) through a 100x PC objective. For fluorescence microscopy, HPX 120 Illuminator (Zeiss) was used as a light source. Image analysis was performed using MicrobeJ (Ducret et al., 2016) and plotting was done in RStudio.
Transmission electron microscopy
Strains SH1000, SAMK21 and SAMK24 were grown to OD600 = 0.4 prior to sample preparation. GS1167 and SAMK15 were pre-grown to OD600 = 0.1, after which the cultures were diluted 64-fold in medium with or without 150 μg/ml IPTG and grown until OD600 = 0.3. Cells were fixed by adding a solution of 4 % parafomaldehyd (w/v) and 5 % glutardialdehyd (w/v) in 1 x PBS pH 7.4 to the cell culture in a 1:1 ratio. The fixation mix was incubated 1 hour in room temperature and kept overnight at 4 °C. The next day the cells were washed three times in PBS and three times in cacodylate buffer (CaCo) before being post-fixed for one hour in 1 % OsO4 in 0.1 M CaCo. Cells were washed three times in CaCo buffer, infiltrated in 3 % agarose and washed again three times in CaCo buffer. The samples were then dehydrated in a gradient series of 70 %, 90 %, 96 %, and 100 % ethanol (15 min for each ethanol concentration). Infiltration in LR White resin was then performed in multiple steps; LR White resin:EtOH in a ratio 1:3 was first incubated overnight, then a ratio of 1:1 for 7 hours, a ratio of 3:1 overnight and finally 100 % LR White resin overnight. Then the samples were embedded in 100 % LR White resin at 60°C for 72 hours. Thin sections were made and stained with uranyl acetate and potassium permanganate. The samples were analyzed in a FEI MORGAGNI 268 electron microscope.
Growth assays
Growth assays were performed in a Synergy H1 Hybrid Reader (BioTek) microtiter plate reader at 37°C. Five ml of cell culture were grown to exponential phase, OD600 = 0.4 before being harvested, resuspended in fresh BHI medium and diluted to OD600 = 0.05. Appropriate antibiotics were always present. Each well in the microtiter plate was added 280 μl diluted cell culture. IPTG (150 μM) was added to the wells when appropriate. Measurements of OD600 were taken every 10th minute throughout growth.
Genome resequencing and analysis
Genomic DNA was isolated from S. aureus SH1000, SAMK21 and SAMK24 using the NucleoBond AXG 100 kit (Macherey-Nagel). For S. aureus SH1000, library for sequencing was created using the Nextera XT DNA library preparation kit (Illumina), and the sequencing was performed using an in-house Illumina MiSeq. For SAM21 and SAMK24, PCR-free library preparation and sequencing (HiSeq4000 PE151) was performed by BGI Hong Kong. Sequences assembly to the S. aureus NCTC8325 reference genome and SNP detection were done using Geneious version 10.1 (Kearse et al., 2012).
Author contributions
I.M, J.W.V, L.S.H. and M.K. conceived the study. G.A.S., I.M. D.S., Z.S. and M.K. performed experiments. G.A.S, I.M. and M.K. wrote the manuscript. D.S., J.W.V., L.S.H. edited the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
We would like to thank Lene C. Hermansen at the Imaging Center, NMBU, for help with transmission electron microscopy, Davide Porcellato, NMBU, for help with genome sequencing and Simon J. Foster and Katarzyna Wacnik, University of Sheffield, for providing the strain SH4639 (SH1000, ezrA-gfp). The work was funded by the Research Council of Norway (www.forskningsradet.no, grant number 250976 awarded to M.K).
References




