

Summary
In animals, PIWI-interacting RNAs (piRNAs) protect the germline and regulate gene expression. Two pathways make piRNAs. Endonucleolytic cleavage of long transcripts generates phased precursor piRNAs (pre-piRNAs) that mature into primary piRNAs, whereas PIWI-catalyzed slicing initiates production of secondary piRNAs. We report that, in mice and flies, PIWI proteins direct the stepwise fragmentation of long piRNA-producing transcripts: the sites of endonucleolytic cleavage along the transcript reflect both the identity of the PIWI protein bound to the precursor transcript’s 5′ end and the availability of uridines at the prospective pre-piRNA 3′ end. In mice, slicing of precursor transcripts by pachytene-piRNA guided PIWI proteins produces new pachytene piRNAs. Thus, PIWI proteins both initiate piRNA biogenesis and position the subsequent endonucleolytic cleavage that yield phased pre-piRNAs. Our data define a central role of mouse PIWI proteins in generating their own guides and suggest a unified model for insect and mammalian piRNA production.
INTRODUCTION
PIWI-interacting RNAs (piRNAs) serve as guides for the PIWI clade of Argonaute proteins, an evolutionary conserved family of oligonucleotide-guided, RNA-binding proteins (Meister, 2013). To find their RNA targets, animal Argonaute proteins use small interfering RNAs (siRNAs; ~20–25 nt; Hammond et al., 2001; Tabara et al., 1999), microRNAs (miRNAs; 21–23 nt; Hutvagner and Zamore, 2002), or piRNAs (23–32 nt; Girard et al., 2006; Aravin et al., 2006; Vagin et al., 2006; Saito et al., 2006; Grivna et al., 2006). Processive cleavage of long double-stranded RNA (dsRNA) precursors by the dsRNA endonuclease Dicer produces siRNAs (Knight and Bass, 2001; Bernstein et al., 2001; Ketting et al., 2001). Transcripts containing intramolecularly paired hairpin structures produce miRNAs via cleavage first by the dsRNA endonuclease Drosha (Lee et al., 2003), and then by Dicer (Grishok et al., 2001). In contrast, piRNAs are made from single-stranded RNAs by single-strand-specific endonucleases (Brennecke et al.,2007; Gunawardane et al., 2007; Pane et al., 2007; Ipsaro et al., 2012; Nishimasu et al., 2012).
piRNAs complementary to transposons ensure genomic stability in the male or female germline (Siomi et al., 2011; Czech and Hannon, 2016). In many arthropods, piRNAs protect somatic tissues from both transposons and RNA viruses (Morazzani et al., 2012; Miesen et al., 2015; Vodovar et al., 2012; Lewis et al., 2017). Remarkably, in silk moth oocytes, a single piRNA helps determine sex (Kiuchi et al., 2014). In mouse, distinct classes of piRNAs (1) direct DNA methylation of transposon sequences in the embryonic testis (Aravin et al., 2008; Kuramochi-Miyagawa et al., 2008), (2) post-transcriptionally repress transposons later in spermatogenesis (Reuter et al., 2011; Inoue et al., 2017), and (3) ensure completion of meiosis and successful spermiogenesis (Gou et al., 2014; Goh et al., 2015; Zhang et al., 2015; Grivna et al., 2006; Girard et al., 2006; Li et al., 2013).
Studies of oogenesis in Drosophila melanogaster and spermatogenesis in mice suggest that germline piRNA biogenesis can be divided into two mechanisms. Primary piRNAs are generated from long, single-stranded RNAs that are transcribed from genomic loci called piRNA clusters (Brennecke et al., 2007; Li et al., 2013). The primary transcripts from these loci are thought to be fragmented by endonucleolytic cleavage, hypothesized to be catalyzed by Zucchini/PLD6, to yield tail-to-head strings of precursor piRNAs (pre-piRNAs; Ipsaro et al., 2012; Nishimasu et al., 2012; Mohn et al., 2015; Han et al., 2015; Ding et al., 2017). In fly oocytes and mouse neonatal spermatogonia, piRNA-directed, PIWI-protein-catalyzed slicing of piRNA-generating transcripts initiates production of such phased pre-piRNAs (Senti et al., 2015; Wang et al., 2015; Yang et al., 2016). Phased pre-piRNA production likely adds diversity to the piRNA population by enabling piRNAs to spread 5′-to-3′ from an initial cleavage site into sequences for which there are no preexisting, complementary piRNAs. What initiates phased pre-piRNA production in the postnatal mammalian testis is unknown.
Each pre-piRNA begins with a 5′ monophosphate, a prerequisite for loading RNA into nearly all Argonaute proteins (Nykanen et al., 2001; Ma et al., 2005; Parker et al., 2005; Wang et al., 2009; Frank et al., 2010; Boland et al., 2011; Kawaoka et al., 2011; Elkayam et al., 2012; Schirle and MacRae, 2012; Schirle et al., 2014; Cora et al., 2014; Wang et al., 2014; Matsumoto et al., 2016). Once bound to a PIWI protein, the 3′ ends of pre-piRNAs are trimmed by the single-stranded-RNA exonuclease, Trimmer/PARN-like domain-containing 1 (PNLDC1), to a length characteristic of the receiving PIWI protein (Kawaoka et al., 2011; Tang et al., 2016; Izumi et al., 2016; Ding et al., 2017; Zhang et al., 2017). (Flies lack a Trimmer/PNLDC1 homolog, and instead piRNA 3′ ends are resected by the miRNA-trimming enzyme Nibbler; Han et al., 2011; Liu et al., 2011; Feltzin et al., 2015; Hayashi et al., 2016.) Finally, the small RNA methyltransferase Hen1/HENMT1 adds a 2′-O-methyl moiety to the 3′ ends of the mature piRNAs (Kirino and Mourelatos, 2007b; Horwich et al., 2007; Saito et al., 2007; Kirino and Mourelatos, 2007a; Ohara et al., 2007; Lim et al., 2015). In mice, piRNAs loaded into MILI are characteristically 26–27 nt, while MIWI-bound piRNAs are 30 nt long. The differences in piRNA lengths have been proposed to reflect the length of RNA protected from PNLDC1 trimming by the specific PIWI protein bound to the pre-piRNA (Kawaoka et al., 2011; Izumi et al., 2016).
Primary piRNAs derive exclusively from genomic piRNA source loci (Brennecke et al., 2007; Li et al., 2013). In contrast, the secondary piRNA biogenesis pathway—the “ping-pong” cycle—uses piRNA target transcripts to generate piRNAs, thereby amplifying the abundance of preexisting piRNAs (Brennecke et al., 2007; Gunawardane et al., 2007). In the ping-pong cycle, piRNA-directed, PIWI-catalyzed slicing of a piRNA target creates an RNA fragment bearing a 5′ monophosphate (Wang et al., 2014). This RNA fragment acts as a pre-piRNA precursor (pre-pre-piRNA), loads into a PIWI protein, and generates a new piRNA with 10 nt complementarity to the piRNA that produced it. The new piRNA can now bind and cleave the original piRNA cluster transcript, generating a second copy of the initiating piRNA. In the fly germline, ping-pong amplification is required for the production of primary piRNAs (Mohn et al., 2015; Han et al., 2015; Senti et al., 2015; Wang et al., 2015). In mice, only piRNAs in the embryonic testis are believed to be produced by the secondary piRNA pathway (Aravin et al., 2007; Aravin et al., 2008; Kuramochi-Miyagawa et al., 2008); postnatal pachytene piRNAs are thought to be produced solely by the primary pathway (Reuter et al., 2011; Beyret et al., 2012).
Here, we report that PIWI proteins play a central role in the production of their own guide piRNAs. First, we show that in the biogenesis of pachytene piRNAs in the male germline of mice as well as in the production of transposon-silencing piRNAs in the female germline of flies, PIWI protein binding determines the site of the endonucleolytic cleavage that simultaneously establishes the 3′ end of the pre-piRNA and the 5′ end of the next primary piRNA. Because MILI and MIWI differentially position the endonuclease, they produce pre-piRNAs of different lengths before any trimming has occurred. In addition to being positioned by a PIWI protein, the pre-pre-piRNA-cleaving endonuclease is constrained to cleave 5′ to a uridine, explaining the 5′ U bias of mammalian primary piRNAs. Thus, heterogeneity in pre-piRNA length reflects both the identity of the PIWI protein bound to the pre-pre-piRNA and the availability of uridine residues near the 3′ end of the prospective pre-piRNA. Second, our data show that mouse MILI and MIWI proteins initiate primary pachytene piRNA biogenesis by slicing complementary piRNA precursor transcripts. These results suggest a unified model for insect and mammalian piRNA production in which PIWI slicing and binding separately establish the two ends of a pre-piRNA.
RESULTS
MILI- and MIWI-bound Pre-piRNAs Differ in Length
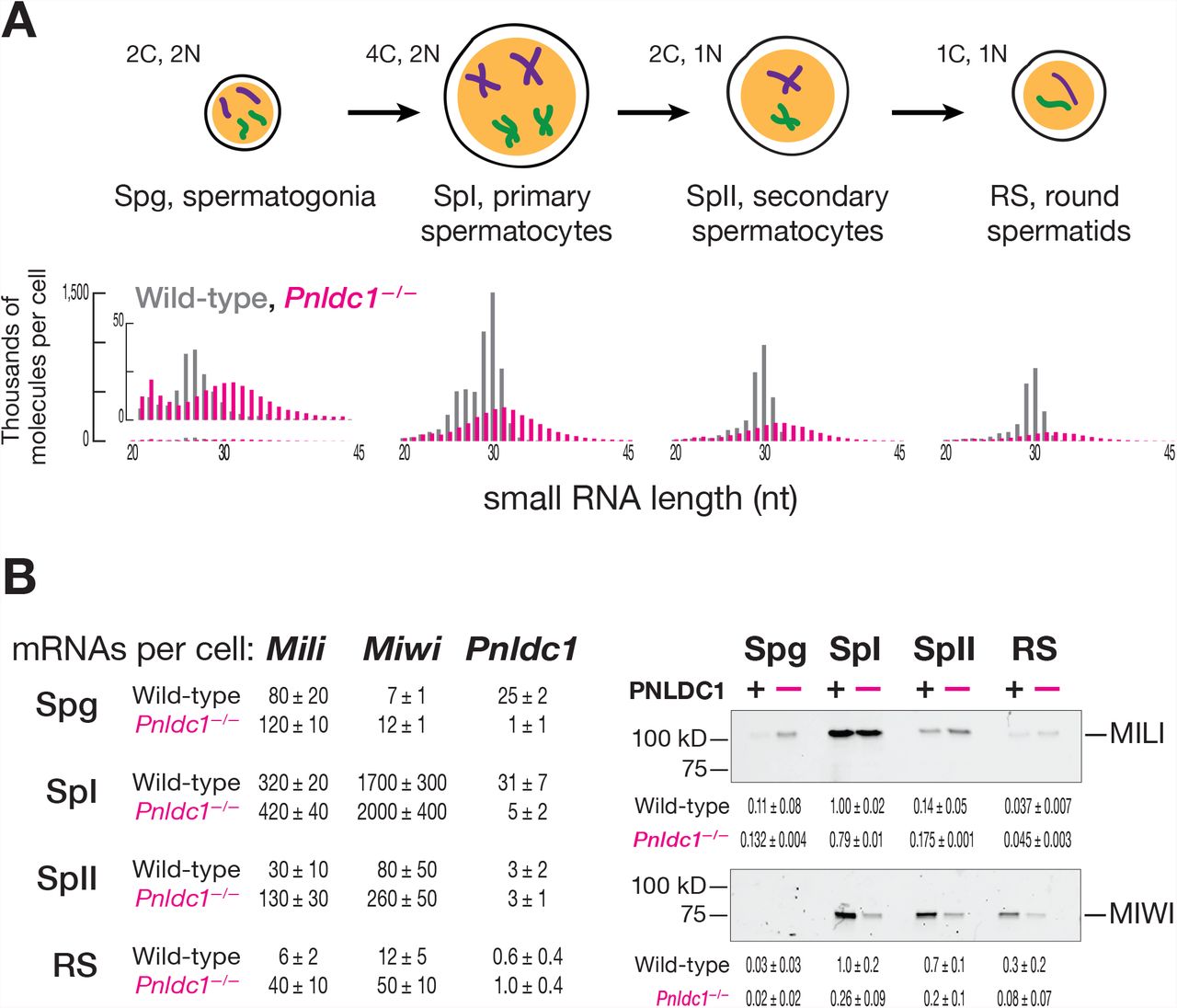
The current model for primary piRNA biogenesis in mouse male germ cells (Figure 1) predicts that in the absence of the 3′-to-5′ trimming enzyme PNLDC1, piRNAs should be replaced by longer, untrimmed pre-piRNAs. To test this, we generated Pnldc1−/− mutant mice and sequenced small RNA from FACS-purified postnatal spermatogonia, primary spermatocytes, secondary spermatocytes and round spermatids. Consistent with previous studies using neonatal and postnatal whole testis (Ding et al., 2017; Zhang et al., 2017), the purified cell types all contained 25–40 nt small RNAs rather than their normal complement of mature 25–31 nt piRNAs (Figure 2A).

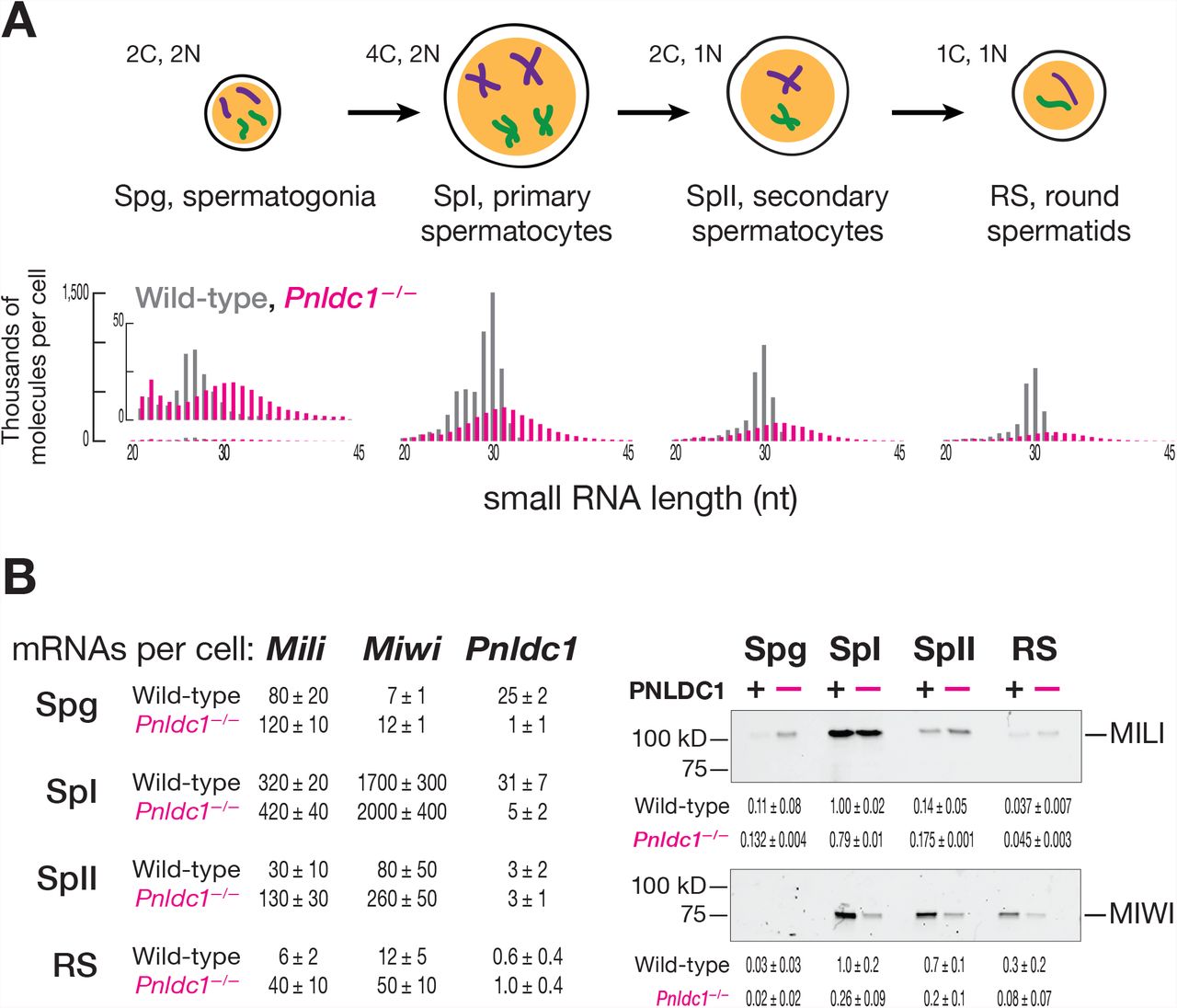
(A) Length profiles of total small RNAs in male germ cells of wild-type and Pnldc1 −/− mice. Data are presented in thousands of molecules per cell from a single biological sample for fully genome-matching reads (reads with non-templated 3′ nucleotides were excluded). C, cell chromosome content; N, ploidy. (B) At left, absolute steady-state level of Mili, Miwi, and Pnldc1 mRNAs in male germ cells of wild-type and Pnldc1 −/− mice: Spg, spermatogonia; SpI, primary spermatocytes; SpII, secondary spermatocytes; RS, round spermatids. Data are presented in thousands of transcripts per cell as the mean of three biological replicates with standard deviation. At right, relative abundance and quantified relative abundance of MILI (n = 2) and MIWI (n = 3) proteins assessed by Western blotting. Each lane contains lysate from ~11,000 cells. Uncropped gel images and the second biological replicate can be found in Figures S1A and S1B.
Mature piRNA length heterogeneity has been presumed to arise from different extents of PNLDC1-mediated trimming of MILI-versus MIWI-bound pre-piRNAs (Kawaoka et al., 2011; Izumi et al., 2016). In the absence of trimming, both pre-pachytene (Mohn et al., 2015; Han et al., 2015) and pachytene (Ding et al., 2017) phased pre-piRNAs are produced: the 5′ end of each succeeding pre-piRNA begins immediately after the 3′ end of the preceding pre-piRNA. Despite this tail-to-head phasing of pre-piRNAs, MILI- and MIWI-bound pre-piRNAs have heterogeneous, 25– 40 nt lengths (Mohn et al., 2015; Han et al., 2015; Zhang et al., 2017; Ding et al., 2017; Saxe et al., 2013). Such heterogeneity was assumed to reflect both imprecision in the generation of pre-piRNAs and the requirement for the pre-piRNA-producing endonuclease to cleave 5′ to a uridine (Mohn et al., 2015; Han et al., 2015). In this view, the narrow length distributions characteristic of individual PIWI proteins in wild-type mice results from pre-piRNA trimming by PNLDC1, homogenizing the initial, broad pre-piRNA length distribution (Figure 2A). A key prediction of this model is that without PNLDC1, untrimmed pre-piRNAs bound to MILI or MIWI will have similar length distributions (Figure 1).
To test this idea, we examined pre-piRNAs from Pnldc1−/− primary spermatocytes, a cell type expressing both MILI and MIWI (Figures 2B, and S1; Table S1). Surprisingly, pre-piRNAs bound to MILI (31 nt modal length) and MIWI (34 nt modal length) had different length distributions (Figure 3A). How could a common pre-piRNA-producing machinery generate distinct lengths of pre-piRNAs for MILI and MIWI? One idea is that pre-piRNAs bound to MILI or MIWI might undergo two successive trimming steps, with an as-yet-unidentified nuclease first shortening the pre-piRNA to ~31 or ~34 nt, followed by PNLDC1 further trimming the pre-piRNA to the characteristic 26–27 nt length of MILI-bound piRNAs and the 30 nt length of MIWI-bound piRNAs.

(A) Length profiles of MILI- and MIWI-bound piRNAs in wild-type (left) and pre-piRNAs in Pnldc1−/− primary spermatocytes (right). Abundance is normalized to all genome mapping reads and is reported in parts per million (ppm). Data are from a single biological sample for fully genome-matching reads (reads with non-templated 3′ nucleotides were excluded). (B) Probability of distances from the 3′ ends of MILI- or MIWI-bound piRNAs to the 5′ ends of MILI- or MIWI-bound piRNAs on the same genomic strand in wild-type and the same for pre-piRNAs in Pnldc1−/− primary spermatocytes. Data are for piRNAs and pre-piRNAs derived from pachytene piRNA loci from a single biological sample; see Figure S2A for an analysis of all genome mapping piRNAs and pre-piRNAs. (C) Probability of distances from the 5′ ends of MILI-bound piRNAs to the 5′ ends of MIWI-bound piRNAs on the same genomic strand (left), and from the 3′ ends of MILI-bound piRNAs to the 3′ ends of MIWI-bound piRNAs on the same genomic strand (right) in wild-type (top) and the same for pre-piRNAs in Pnldc1−/− primary spermatocytes (bottom). Numbers indicate the total frequency of 5′ or 3′ ends of MILI-bound small RNAs residing before, after, or coinciding with the 5′ or 3′ ends of the MIWI-bound small RNAs. Data are from a single biological sample for piRNAs and pre-piRNAs from the pachytene piRNA loci; see Figure S2D for an analysis of all genome mapping piRNAs and pre-piRNAs. (D) Distance between the most frequent 3′ end of the MILI-bound small RNA group and the most frequent 3′ end of the corresponding paired MIWI-bound small RNA group. Data are for piRNAs and pre-piRNAs deriving from pachytene piRNA loci for a single biological sample; see Figure S2E for an analysis of all genome mapping piRNAs and pre-piRNAs.
Such a mechanism would blur the phasing of MILI- or MIWI-bound pre-piRNAs in Pnldc1−/− primary spermatocytes, because some trimming of pre-piRNAs would still occur. Our data argue against this idea. The probability of 3′-to-5′ distances between all pre-piRNAs bound to MILI or MIWI in Pnldc1−/− mutants peaked sharply at 0, evidence that pre-piRNAs are produced tail-to-head, one after another (Figure S2A). We obtained similar results analyzing only pre-piRNAs from pachytene piRNA loci—i.e., excluding pre-piRNAs bound to MILI prior to the onset of meiosis (Figure 3B). Finally, for both MILI- and MIWI-bound pre-piRNAs, the genomic nucleotide immediately 3′ to the pre-piRNAs was most often uridine (Figure S2B), a hallmark of primary piRNA biogenesis (Mohn et al., 2015; Han et al., 2015). We conclude that PNLDC1 alone trims pre-piRNAs in the mouse postnatal testis.
Our data also show that a MIWI-bound pre-piRNA often lies tail-to-head with a MILI-bound pre-piRNA and vice versa. Analysis of the probabilities of 3′-to-5′ distances between MIWI- and MILI-bound pre-piRNAs and between MILI- and MIWI-bound pre-piRNAs also gave a sharp peak at 0 (Figure 3B, pachytene piRNA loci; Figure S2A, all genome mappers). Thus, the same pre-pre-piRNA molecule can generate both MILI- and MIWI-bound phased pre-piRNAs (Figure 1).
If pre-piRNAs are trimmed only by PNLDC1, then we expect a pre-piRNA to be followed by a pre-pre-piRNA not yet converted to a pre-piRNA. Such pre-pre-piRNAs would have 5′ monophosphorylated ends and be longer than pre-piRNAs. To identify pre-pre-piRNAs, we sequenced 5′ monophosphorylated, single-stranded RNA fragments ≥150 nt from wild-type primary spermatocytes. As predicted for authentic pre-pre-piRNAs, the most likely distance from the 3′ ends of MILI- or MIWI-bound pre-piRNAs to the 5′ ends of the long, 5′ monophosphorylated RNAs was 0 (Figure S2C). We conclude that (1) pre-piRNAs are generated tail-to-head by a single endonucleolytic cleavage event that simultaneously establishes the 3′ end of each pre-piRNA and the 5′ end of the downstream pre-pre-piRNA, and (2) PNLDC1 is the only enzyme trimming mouse pachytene pre-piRNAs.
MILI and MIWI Participate in Pre-piRNA Production
Another explanation for the difference in the modal length of MILI- and MIWI-bound pre-piRNAs is that PIWI proteins themselves position the endonucleolytic cleavage that converts pre-pre-piRNAs into pre-piRNAs. To test this, we asked whether the genomic positions of the 5′ or 3′ ends of the pre-piRNAs bound to MILI differ from those of the pre-piRNAs bound to MIWI. For both piRNAs and pre-piRNAs, we calculated the probabilities of the 5′ ends of MILI-bound RNAs residing before, after, or coinciding with the 5′ ends of MIWI-bound RNAs. Similarly, we calculated the probabilities for MILI- or MIWI-bound piRNA and pre-piRNA 3′ ends. In wild-type primary spermatocytes, MILI- and MIWI-bound mature piRNAs were more likely to share 5′ ends than 3′ ends (0.43 for 5′ ends vs. 0.12 for 3′ ends; Figure 3C, pachytene piRNA loci; Figure S2D, all genome mappers). Similarly, in Pnldc1−/−, MILI- and MIWI-bound pre-piRNAs were more likely to share 5′ ends than 3′ ends (0.36 for 5′ ends vs. 0.13 for 3′ ends; Figures 3C, pachytene piRNA loci; Figure S2D, all genome mappers).
Consistent with the idea that the length of a mature piRNA is defined by the footprint of its PIWI partner (Kawaoka et al., 2011; Izumi et al., 2016), the 3′ ends of MILI-bound piRNAs were more likely to be upstream of the 3′ ends of MIWI-bound piRNAs in wild-type primary spermatocytes (0.55 upstream vs. 0.33 downstream; Figure 3C, pachytene piRNA loci; Figure S2D, all genome mappers). Surprisingly, in Pnldc1−/− mutants, the probability of the 3′ ends of MILI-bound pre-piRNAs lying upstream of the 3′ ends MIWI-bound piRNAs was also higher (0.52 upstream vs. 0.35 downstream; Figure 3C, pachytene piRNA loci; Figure S2D, all genome mappers). These data suggest that although MILI-bound pre-piRNAs are likely to share 5′ ends with MIWI-bound pre-piRNAs, those pre-piRNAs bound to MILI are generally shorter than MIWI-bound pre-piRNAs.
These analyses compared all combinations of 3′ ends of pre-piRNAs, including those that do not share a common 5′ end. To examine the 3′ ends of pre-piRNAs produced from a common pre-pre-piRNA, we grouped together all unambiguously mapping pre-piRNAs with the same 5′, 25-nt prefix and used their read abundance to identify the most frequent 3′ end for each group (Figure S3A). We then paired the groups of MILI- and MIWI- bound wild-type piRNAs or Pnldc1−/− pre-piRNAs with the same 5′, 25-nt prefix (Figure S3A; in two independent experiments, 87,748–88,155 paired groups account for 78% of all MILI-bound piRNAs, and 76–77% of all MIWI-bound piRNAs; 62,214–80,568 paired groups account for 59–69% of all MILI-bound pre-piRNAs, and 58–67% of all MIWI-bound pre-piRNAs). Analysis of the paired groups of MILI- and MIWI- bound pre-piRNAs showed that the most frequent 3′ end of the MILI- bound pre-piRNA group was either the same (46% of paired groups; Figure 3D, pachytene piRNA loci; Figure S2E, all genome mappers) or upstream of the most frequent 3′ end of the corresponding MIWI-bound pre-piRNA group (41% of paired groups; Figure 3D, pachytene piRNA loci; Figure S2E, all genome mappers). That is, when MILI binds the 5′ end of a pre-pre-piRNA, it is rare for the resulting pre-piRNA to be longer than the pre-piRNA generated when MIWI binds the same pre-pre-piRNA (13% of paired groups; Figure 3D, pachytene piRNA loci; Figure S2E, all genome mappers).
Like mature piRNAs in wild-type spermatocytes, pre-piRNAs in Pnldc1−/− mice overwhelmingly start with uridine (5′ U bias; Figure S3B; Aravin et al., 2006; Girard et al., 2006). We took advantage of this 5′ U bias to further test the hypothesis that MILI and MIWI position the endonucleolytic cleavage that generates the 3′ ends of pre-piRNAs. Inspection of individual pre-piRNAs from pachytene piRNA loci suggested that when MILI- and MIWI-bound pre-piRNAs share the same 5′ end but differ in their 3′ ends, both 3′ ends are followed by a uridine in the genomic sequence (Figure 4, at right). For MILI- and MIWI-bound pre-piRNAs with identical 5′ and 3′ ends, the U following their shared 3′ end is the only uridine present in that genomic neighborhood.
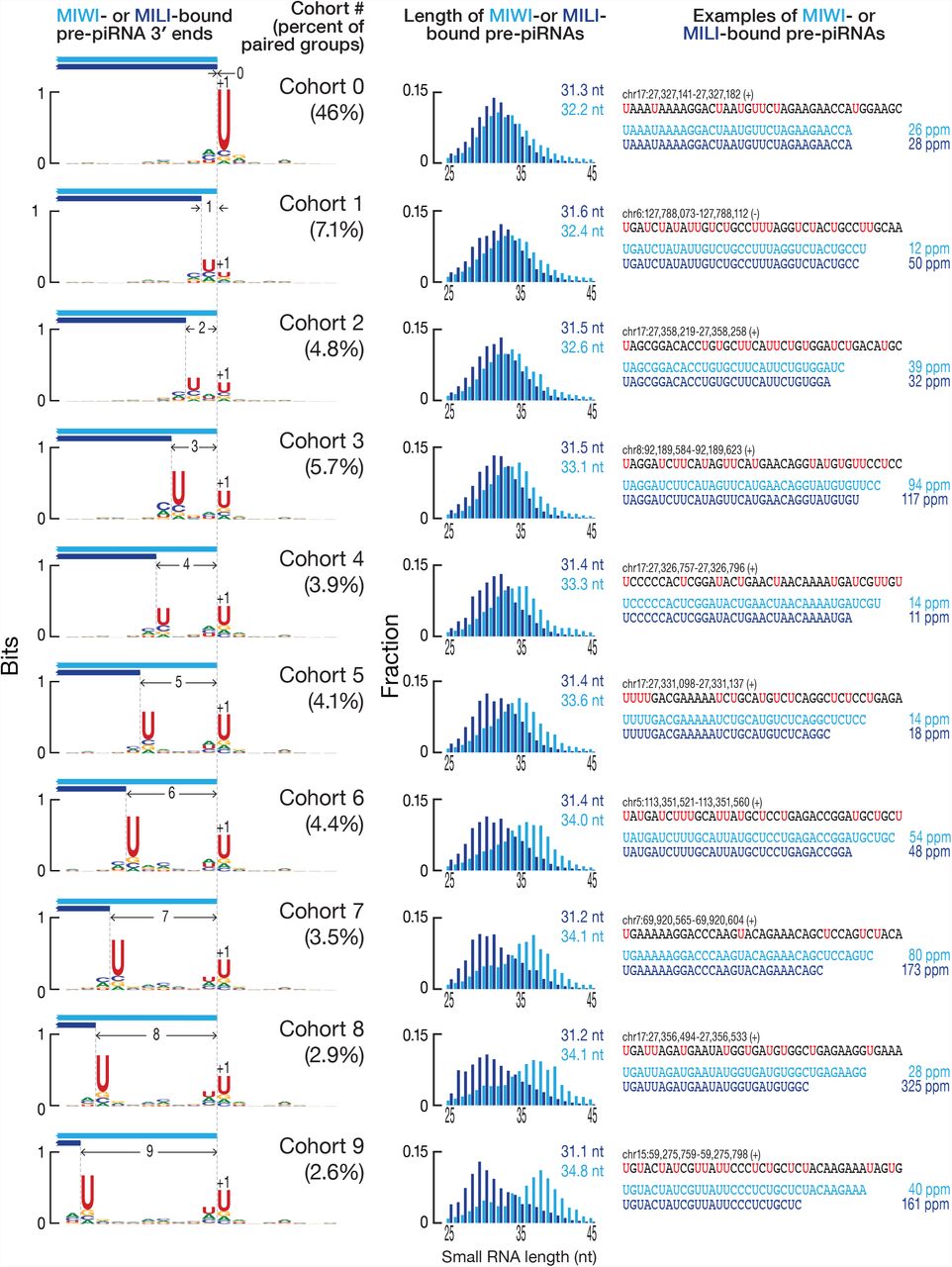
At right, a comparison of the most frequent 3′ ends of individual MILI- and MIWI-bound pre-piRNAs sharing the same 5′ end in Pnldc1−/− primary spermatocytes. Data are in parts per million (ppm) for pre-piRNAs derived from pachytene piRNA loci from a single biological sample. At left, the nucleotide bias of the genomic neighborhood around the most frequent 3′ ends of paired MILI- and MIWI-bound pre-piRNA groups in Pnldc1−/− primary spermatocytes. In the center, length profiles of the corresponding paired MILI- and MIWI-bound pre-piRNA groups in Pnldc1−/− primary spermatocytes. Data are from a single biological sample; see Figure S3C for an analysis of all genome mapping pre-piRNAs.
To quantify these observations, we sorted the paired groups of pre-piRNAs bound to MILI and MIWI into cohorts according to the number of nucleotides separating the most frequent MILI- and MIWI-bound pre-piRNA 3′ ends (Figure S3A). Thus, cohort 0 contained paired groups of pre-piRNAs whose most frequent 3′ end was identical for MILI- and MIWI-bound pre-piRNA. Similarly, in cohort 1 the most frequent MILI-bound pre-piRNA 3′ end lay 1 nt upstream of the 3′ end of the pre-piRNA bound to MIWI (Figure S3A). For each cohort, we measured the uridine frequency at each position in the genomic neighborhood of the 3′ ends. Strikingly, whenever two separate peaks of high uridine frequency resided in a genomic neighborhood, the most frequent 3′ end of MILI-bound pre-piRNA groups lay immediately before the upstream uridine, while the most frequent 3′ end of MIWI-bound pre-piRNA groups lay before the downstream uridine (Figure 4, at left, pachytene piRNA loci; Figure S3C, all genome mappers). When a single peak of uridine was surrounded by a uridine desert, the 3′ ends of the MILI- and MIWI-bound pre-piRNAs coincided at the nucleotide immediately before the single uridine peak (Figure 4, at left, pachytene piRNA loci; Figure S3C, all genome mappers).
To test whether the length of pre-piRNAs from different cohorts is homogenized after trimming to their mature length, we compared the paired groups of MILI- and MIWI-bound pre-piRNAs from Pnldc1−/− mice with the corresponding paired groups of MILI- and MIWI-bound mature piRNAs from wild type. Regardless of the cohort to which a paired pre-piRNA group belonged, the lengths of the corresponding mature trimmed piRNAs were determined solely by the identity of their bound PIWI protein (Figure S3D).
For the pachytene piRNA loci, 46% of paired groups of MILI- and MIWI-bound pre-piRNAs belonged to cohort 0 (Figures 3D and 4), i.e., although a range of 3′ ends was present for these paired groups, the most frequent 3′ end for a MILI-bound pre- piRNA group was the same as the most frequent 3′ end for the corresponding MIWI-bound pre-piRNA group. In theory, this observation could reflect an underlying non-random distribution of uridines in primary piRNA transcripts. Pachytene piRNA precursor transcripts are 27.4% U, which is expected for a near random uridine distribution. However, these transcripts could hypothetically contain many uridine-free regions, forcing the pre-pre-piRNA cleaving endonuclease to cut 5′ to the only uridine available in the genomic region. To test this idea, we used random sampling to estimate the probability of uridine surrounded by non-uridine stretches (VVVVUVVVV) and the probabilities of the two closest uridines being separated by a given length of non-uridine sequence (UU, UVU, UVVU, UVVVU, etc.; Figure 5A). The data obtained from random sampling fit well to the geometric distribution, P(x) = p(100−p)x−1, where p = 27.4 (Pearson’s = 0.996; data not shown), demonstrating that uridines are spread randomly across the pachytene piRNA transcripts. Uridines were also found to be randomly distributed, when analysis was confined to the genomic neighborhood around the paired groups of MILI- and MIWI-bound pre-piRNAs (data not shown). Therefore, the distribution of uridine in pachytene piRNA precursor transcripts cannot explain why for 46% of paired groups of MILI- and MIWI-bound pre-piRNAs, their most frequent 3′ ends coincide (cohort 0 in Figure 5A). We conclude that the underlying mechanism of pre-piRNA production, not the sequence of pachytene piRNA precursor transcripts, determines the distribution of pre-piRNA 3′ ends.

(A) Probability of a single uridine surrounded by non-uridine stretches (VVVVUVVVV), and probabilities of lengths of non-uridine nucleotide stretches between two nearest uridines (UU, UVU, UVVU, etc.) in pachytene piRNA loci estimated by random sampling of the data from pachytene piRNA cluster transcripts. Mean and standard deviation are presented for the randomly sampled data based on 1,000 iterations. Data for pre-piRNA cohorts are for a single biological sample from Figure 3D. (B) Model for pre-piRNA production in which MILI and MIWI proteins direct endonucleolytic cleavage by binding 5′ ends of pre-pre-piRNAs: protein footprints limit the availability of uridines for the pre-pre-piRNA cleaving endonuclease (vertical arrows). Because MILI footprint is smaller than that of MIWI, the endonuclease will have access to more upstream uridines if pre-pre-piRNA is bound by MILI compared to MIWI. MILI footprint will still place a 5′ limit on the upstream shift in the uridine availability windows for MILI and MIWI.
MILI and MIWI Position the Endonuclease by Binding Pre-pre-piRNA 5′ End
If PIWI proteins position the endonuclease during primary piRNA transcript fragmentation, they may do so by binding the 5′ end of pre-pre-piRNA. In this case, much of the sequence of the prospective pre-piRNA would be occluded by the PIWI protein and inaccessible for cleavage. Only uridines located 3′ to the PIWI protein’s footprint could be recognized by the endonuclease. Based on pre-piRNA length data, the footprint of MILI is expected to be ~3 nt smaller than that of MIWI (Figure 3A), giving the endonuclease access to more upstream uridines when it is positioned by MILI rather than MIWI. In this view, the endonuclease is constrained by the PIWI protein to cleave at the nearest uridine not masked by the protein’s footprint. Thus, when only a single exposed uridine is present locally, the resulting MILI-bound and MIWI-bound pre-piRNAs share a common 3′ end (cohort 0 in Figure 4).
This model makes two predictions. Imagine a MIWI-bound pre-piRNA corresponding to the shortest length permitted by MIWI’s footprint (Figure 5B). By definition, when this pre-piRNA is a member of cohort 0, the corresponding MILI-bound pre-piRNA has the same 3′ end—i.e., the two pre-piRNAs are identical. The model predicts that no uridines will be found in the last three nucleotides of the pre-piRNA, but that there may be uridines ≥4 nt upstream of the pre-piRNA 3′ end. These uridines are inaccessible to the endonuclease, because they are concealed within the footprint of MILI. In other words, because of MILI’s footprint, cohort 0 is expected to incorporate all instances in which uridines are ≥4 nt upstream of the uridine used by the endonuclease positioned by MIWI (Figure 5B).
Because all paired pre-piRNA groups are equally represented in our analysis (i.e., abundance is not considered), we can test this prediction using the pachytene piRNA cluster uridine distribution data obtained by random sampling (Figure 5A) and inferring the MIWI footprint from the modal (34 nt; Figure 3A) or mean (34.5 ± 0.3 nt) length of MIWI-bound pre-piRNAs. We calculated the cumulative probabilities of non-uridine nucleotide stretches separating the two nearest uridines with length ≥ N nt. For example, for UU, N = 1, while for UVU, N = 2. Consistent with the model’s prediction, the size of cohort 0 for 34–35 nt long MIWI-bound pre-piRNAs is best predicted by the cumulative probability when N = 4. That is, when the distance from the pre-piRNA 3′ end to the nearest upstream uridine is ≥4 nt, cohorts 4 and greater mainly collapse into cohort 0 (Figure 5B and Figure S3E, at right).
The model makes a second prediction, that the MILI-bound pre-piRNAs present in cohorts 4 and greater (Figure 4) will be paired with extremely long, MIWI-bound pre-piRNAs. Indeed, for MIWI-bound pre-piRNAs ≥37 nt long, their MILI-bound counterparts are often both shorter by ≥4 nt (Figure S3E, at left). Consistently, the mean length of MIWI-bound pre-piRNAs in cohorts 4–9 (33.3–34.8 nt; Figure 4) is longer than that in cohorts 0–3 (32.2–33.1 nt; Figure 4). Thus, the data support the idea that the footprints of MILI and MIWI restrict which uridines can be used to generate pre-piRNA 3′ ends.
We conclude that MILI and MIWI differentially position the pre-pre-piRNA endonuclease when it establishes pre-piRNA 3′ ends. Both MILI and MIWI direct the endonuclease to cleave 5′ to the nearest uridine, but MILI positions the endonuclease upstream of the site dictated by MIWI. When PIWI proteins bind the 5′ ends of pre-pre-piRNAs to initiate this process, the distinct sizes of the MILI and MIWI footprints differentially restrict the uridines available to the endonuclease.
Pre-piRNAs are Unlikely to be Sorted between MILI and MIWI
An alternative and simpler explanation for the difference in the modal length of MILI- and MIWI-bound pre-piRNAs is that before PNLDC1 trimming, pre-piRNAs are sorted by length between the two PIWI proteins. This alternative model predicts that in Pnldc1−/− mutants, both short and long pre-piRNAs should be produced throughout spermatogenesis, regardless of presence of MILI or MIWI. Our data do not support a sorting model: the modal length of all pre-piRNAs increases from 31 nt in spermatogonia, where only MILI is present (mean length of ≥25-nt pre-piRNAs = 31.81 ± 0.09 nt), to 32 nt in round spermatids, where MIWI is more abundant than MILI (mean length of ≥25-nt pre-piRNAs = 32.15 ± 0.04 nt; Figures 2A and 2B). We cannot formally exclude more complex mechanisms, such as models in which long pre-piRNAs bound to MILI are selectively degraded. Nonetheless, the simplest interpretation consistent with our data is that MILI and MIWI directly participate in the production of pre-piRNA 3′ ends rather than that multiple lengths of pre-piRNAs are first generated and then sorted between MIWI and MILI.
TDRKH and PNLDC1 Act in the Same Step of Primary piRNA Biogenesis
TDRKH (Papi in flies) is a Tudor domain-containing protein required for pre-piRNA trimming (Saxe et al., 2013; Mohn et al., 2015; Han et al., 2015). In Tdrkh−/− mutants, spermatogenesis arrests before the pachytene stage, so these mice produce only MILI-bound, pre-pachytene pre-piRNAs. Consistent with the current model of phased pre-piRNA biogenesis (Figure 1), pre-piRNAs in FACS-sorted spermatogonia from both Tdrkh−/− and Pnldc1−/− are often found tail-to-head (Figure S4A) and frequently begin with U and end immediately before a U in the genomic sequence (Figures S4B and S4C; Mohn et al., 2015; Han et al., 2015; Ding et al., 2017). As expected, pre-piRNAs in Tdrkh−/− spermatogonia are longer than mature piRNAs, and are indistinguishable in length from pre-piRNAs in Pnldc1−/− spermatogonia (mean length of ≥25-nt pre-pachytene pre-piRNAs = 32.0 ± 0.3 nt in Tdrkh−/− and 31.8 ± 0.1 nt in Pnldc1−/− spermatogonia; Figure S4D).
To test further whether TDRKH and PNLDC1 are required for the same step in pre-piRNA production, we examined the length of pre-piRNAs produced from the same pre-pre-piRNA in Tdrkh−/− and Pnldc1−/− spermatogonia. We identified the unambiguously mapping pre-piRNAs in Tdrkh−/− and Pnldc1−/− spermatogonia and for each genotype grouped together the pre-piRNAs sharing a common 5′, 25-nt prefix. When we paired the pre-piRNA groups from Tdrkh−/− and Pnldc1−/−, the most frequent 3′ end in Tdrkh−/− was most often the same as the most frequent 3′ end in Pnldc1−/− (47% of paired groups; Figure S4E). In contrast, when we paired pre-piRNA groups from Pnldc1−/− or Tdrkh−/− spermatogonia with mature piRNA groups from wild-type animals, the most frequent 3′ ends of mature piRNAs were typically found upstream of the most frequent 3′ ends of untrimmed pre-piRNAs (77% of paired groups for Pnldc1−/− vs. wild-type; 73% of paired groups for Tdrkh−/− vs. wild-type). These data suggest that both TDRKH and PNLDC1 act in the same step of the primary piRNA biogenesis pathway. At least in a genetic sense, TDRKH and PNLDC1 are required to trim the 3′ ends of pre-piRNAs to produce mature piRNAs, as reported for silkmoth piRNA biogenesis (Izumi et al., 2016).
D. melanogaster Piwi and Aub Also Participate in Pre-piRNA Production
D. melanogaster makes three PIWI proteins—Piwi, Aubergine (Aub), and Argonaute3 (Ago3). Ago3 and Aub collaborate to generate secondary piRNAs via the ping-pong cycle, whereas Piwi and, to a lesser extent, Aub bind primary piRNAs (Mohn et al., 2015; Han et al., 2015; Senti et al., 2015; Wang et al., 2015). The modal lengths of Piwi- and Aub-bound piRNAs differ by just one nucleotide (26 and 25 nt, respectively). To test if PIWI-protein binding similarly positions the endonuclease that establishes the 3′ ends of pre-piRNAs in D. melanogaster, we analyzed data from fly ovaries.
Fly piRNAs are less extensively trimmed than those in mice, so the sequences of mature piRNAs more easily reveal the mechanics of pre-piRNA biogenesis. Analysis of the 3′-to-5′ distance between mature piRNAs showed that Piwi- and Aub-bound piRNAs are often found tail-to-head in any combination of the two proteins (Figure S5A). In addition, the probability of common 5′ ends between Piwi- and Aub-bound piRNAs was higher than the probability of common 3′ ends (0.34 for 5′ vs. 0.17 for 3′ ends; Figure S5B). To analyze the 3′ ends of piRNAs produced from a common pre-pre-piRNA, we separately grouped Piwi- and Aub-bound piRNAs sharing a common 5′, 23-nt prefix (Figure S3A). After pairing the Piwi and Aub groups, we found that the most frequent 3′ ends of Aub-bound piRNA groups often coincided (66% of paired groups; Figure S5B) or lay upstream (26% of paired groups; Figure S5B) of the most frequent 3′ ends of the corresponding Piwi-bound piRNA groups. For just 8% of paired groups, the most frequent 3′ ends of Aub-bound piRNAs were downstream of the most frequent 3′ ends of the corresponding Piwi-bound piRNAs.
Next, we then divided the paired Piwi- and Aub-bound piRNA groups into cohorts according to the distance between the most abundant 3′ ends for Piwi- and Aub-bound piRNAs. When the most frequent 3′ ends of Piwi- and Aub-bound piRNA groups were identical, they lay immediately before a single uridine in the genomic neighborhood (Figure 6A). Conversely, when there were two peaks of uridine, the most frequent 3′ ends of Aub-bound piRNA groups lay immediately before the upstream peak, while the most frequent 3′ ends of Piwi-bound piRNA groups were immediately before the downstream peak (Figure 6A). Confining the analyses to piRNAs arising from germline-specific transposons (Wang et al., 2015) produced essentially the same results (data not shown). Analysis of published data sets (Hayashi et al., 2016) of piRNAs bound to Aub or Piwi in wild-type fly ovaries further supported these conclusions (Figures S5D, S5E, S5F, S5G).
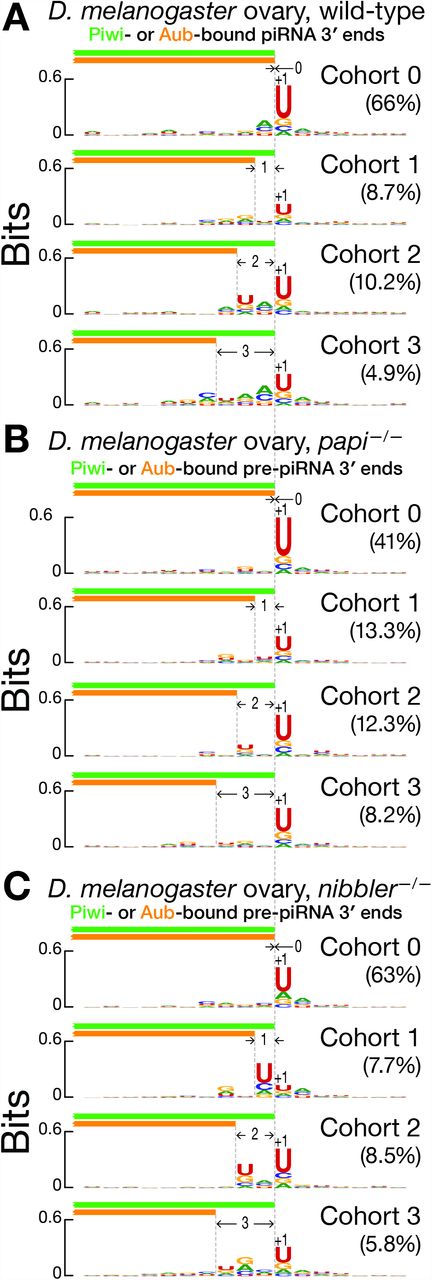
(A, B, C) Nucleotide bias of the genomic neighborhood around the most frequent 3′ ends of paired Piwi- and Aub-bound piRNA groups in wild-type (A), and pre-piRNA groups in papi −/− (B) and nibbler −/− (C) D. melanogaster ovary. Data are for all unambiguously mapping RNAs from a single biological sample.
Like mice, flies require Papi for pre-piRNA trimming: without Papi, fly pre-piRNAs have a median length 0.35 nt longer than wild-type piRNAs (Han et al., 2015; Hayashi et al., 2016). However, flies lack a PNLDC1 homolog (Hayashi et al., 2016). Instead, the miRNA-trimming enzyme Nibbler (Han et al., 2011; Liu et al., 2011) trims fly pre-piRNAs (Feltzin et al., 2015; Wang et al., 2016; Hayashi et al., 2016). In papi−/− and nibbler−/− mutant ovaries (Hayashi et al., 2016), Piwi- and Aub-bound pre-piRNAs are phased (Figure S5H). As in mice, phasing occurs between Aub- and Piwi-bound, Aub- and Aub- bound, Piwi- and Piwi-bound, and Piwi- and Aub-bound piRNAs, supporting the idea that a single pre-pre-piRNA molecule can generate both Aub- and Piwi-bound pre-piRNAs. Piwi- and Aub-bound pre-piRNAs are more likely to share 5′ ends than 3′ ends (papi−/−: 0.21 for 5′ ends vs. 0.09 for 3′ ends; nibbler−/−: 0.28 for 5′ ends vs. 0.12 for 3′ ends; Figure S5I). Moreover, the 3′ ends of Aub-bound pre-piRNAs are more likely to be found upstream than downstream of the 3′ ends of Piwi-bound pre-piRNAs (papi−/−: 0.51 upstream vs. 0.40 downstream; nibbler−/−: 0.51 upstream vs. 0.37 downstream; Figure S5I). Grouping and pairing unambiguously mapping Piwi- and Aub-bound pre-piRNAs with common 5′, 23-nt prefix revealed that the most frequent 3′ end of Aub-bound pre-piRNA groups either coincides with the most frequent 3′ end of the corresponding Piwi-bound pre-piRNA group (papi−/−: 41% of paired groups; nibbler−/−: 63% of paired groups; Figure S5J) or lies upstream of the most frequent 3′ end of the Piwi-bound pre-piRNA group (papi−/−: 43% of paired groups; nibbler−/−: 28% of paired groups; Figure S5J). For only a small minority of paired groups are Aub-bound pre-piRNAs longer than pre-Piwi-bound piRNAs (papi−/−: 16% of paired groups; nibbler−/−: 9% of paired groups; Figure S5J). Similar to mice, when a genomic neighborhood contained a single uridine frequency peak, the most frequent 3′ ends of Piwi- and Aub-bound pre-piRNA groups were identical and mapped immediately before the peak (Figures 6B, 6C): i.e., the most frequent Piwi- and Aub-bound pre-piRNAs were identical. In contrast, when two uridine frequency peaks were present, the most frequent 3′ ends of Aub-bound pre-piRNA groups preceded the upstream peak, while the most frequent 3′ ends of the corresponding Piwi-bound pre-piRNA groups lay before the downstream peak.
Collectively, these data suggest that, as for MILI and MIWI in mice, Aub and Piwi participate in pre-piRNA biogenesis by positioning the endonuclease that simultaneously generates the pre-piRNA 3′ end and the 5′ end of the succeeding pre-pre-piRNA. Thus, in mice and flies, PIWI proteins directly participate in establishing the pattern of phased pre-piRNA biogenesis.
MILI and MIWI Slicing of Primary piRNA Transcripts Initiates Pre-Pre-piRNA Biogenesis
Primary spermatocytes, secondary spermatocytes and round spermatids from Pnldc1−/− testes contain approximately half as many total small RNAs as wild-type, whereas miRNA abundance is unchanged (Figure S6A). Although piRNA cluster transcript abundance, measured by RNA-seq and RT-qPCR of RNA from whole testes, has been reported to be unchanged in Pnldc1−/− mice (Zhang et al., 2017), our RNA-seq data from FACS-sorted germ cells show that without PNLDC1, the steady-state levels of many piRNA cluster transcripts increase. Absolute transcript abundance increased both in secondary spermatocytes (FDR ≤ 0.1; median increase 3.0-fold) and round spermatids (FDR ≤ 0.1; median increase 4.5-fold) for the ten loci that produce the most pachytene piRNAs and which account for 52% of all piRNAs in meiotic and post-meiotic wild-type cells. At the same time, the absolute abundance of piRNAs from these loci decreased both in secondary spermatocytes (FDR ≤ 0.1; median decrease 2.4-fold) and round spermatids (FDR ≤ 0.1; median decrease 3.6-fold).
A possible explanation for the increased steady-state levels of pachytene piRNA cluster transcripts and the decreased amount of pre-piRNAs in Pnldc1−/− mutants is that piRNAs themselves may initiate processing of piRNA cluster transcripts into phased pre-piRNAs. Indeed, in the Drosophila female germline, primary piRNA biogenesis is typically initiated by a slicing event directed by an Ago3-bound secondary piRNA generated via the ping-pong amplification pathway (Mohn et al., 2015; Han et al., 2015; Senti et al., 2015; Wang et al., 2015). Thus, secondary piRNAs counterintuitively initiate phased primary piRNA production by cleaving a piRNA cluster transcript to generate a pre-pre-piRNA. Although PIWI-protein initiated, phased piRNA production plays a role in the production of MILI- and MIWI2-bound piRNAs by MILI-MILI ping-pong amplification in the neonatal mouse testis (Yang et al., 2016), such a mechanism is not generally thought to play a role in piRNA biogenesis in the postnatal male germline of mice. We reexamined this presumption.
To identify the pre-pre-piRNAs from which phased pre-piRNAs are generated in mouse primary spermatocytes, we selected from our data set of ≥150-nt, 5′ monophosphorylated RNA sequences from wild-type cells, those RNAs derived from the pachytene piRNA loci. Many of these shared 5′ ends with mature piRNAs, consistent with their corresponding to pre-pre-piRNAs (Figure S6B; 79% shared 5′ ends with MILI-bound mature piRNAs, 77% shared 5′ ends with MIWI-bound mature piRNAs). We separated the putative pre-pre-piRNAs—the long RNAs sharing their 5′ end with a mature piRNA—from those for which no piRNA with the same 5′ end could be found (control, Figure 7A). As expected for precursors of pre-piRNAs, most putative pre-pre-piRNAs began with uridine (65% of pre-pre-piRNAs sharing 5′ ends with MILI-bound piRNAs, 67% of those sharing 5′ ends with MIWI-bound piRNAs; Figure 7B). Unexpectedly, putative pre-pre-piRNAs also showed a significant enrichment for adenine at their tenth nucleotide (39% of pre-pre-piRNAs sharing 5′ ends with MIWI-bound piRNAs and 38% of those sharing 5′ ends with MILI-bound piRNAs; Figure 7B). Such a 10A bias is the hallmark of PIWI-protein catalyzed target slicing and reflects the intrinsic preference of some PIWI proteins for adenine at the t1 position of their target RNAs (Wang et al., 2014; Matsumoto et al., 2016).
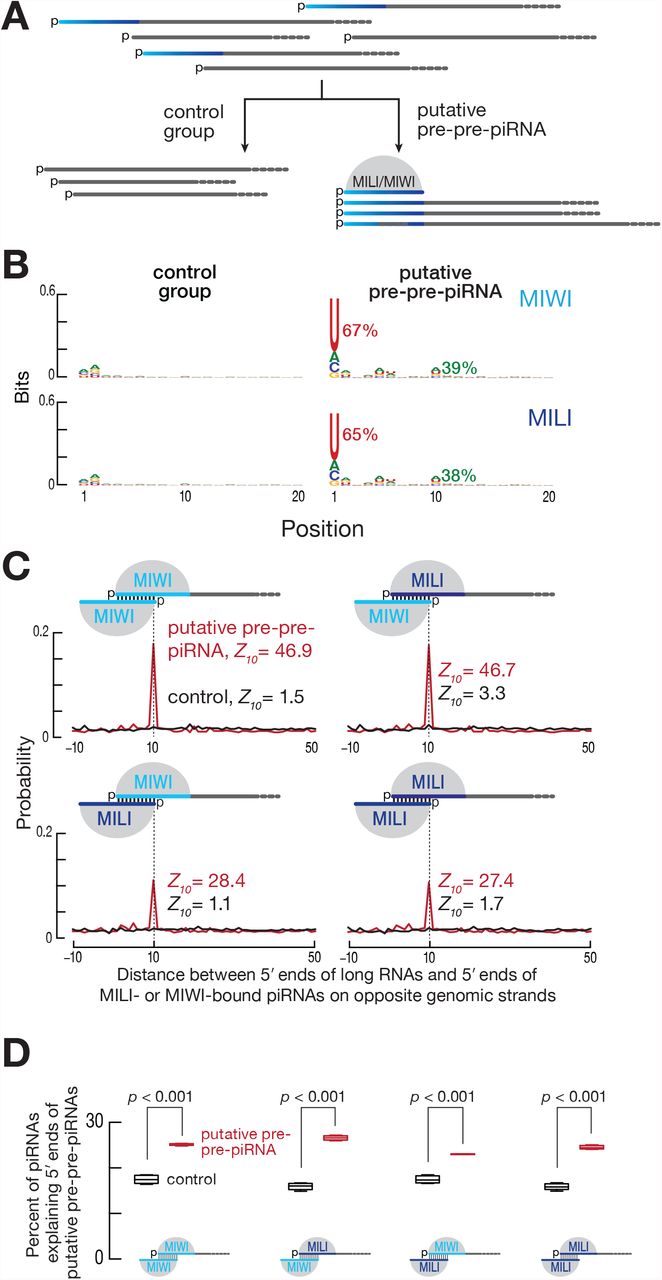
(A) Strategy for the selection of putative pre-pre-piRNAs from the data set of ≥150-nt, 5′-monophosphorylated RNAs. Only RNAs sharing 5′ ends with MILI- or MIWI-bound mature piRNAs were classified as putative pre-pre-piRNAs. The remaining long RNAs serve as the control group. (B) Nucleotide bias of the 5′ ends of putative pre-pre-piRNAs compared to the control group. Fraction of the initial uridine and the tenth adenine is shown in percent. Data are for long, 5′ monophosphorylated RNAs derived from pachytene piRNA loci and sharing their 5′ ends with either MILI- or MIWI-bound piRNAs for a single biological sample. (C) Probability of distances between the 5′ ends of long RNAs (putative pre-pre-piRNAs or control) and the 5′ ends of MILI- or MIWI-bound piRNAs on opposite genomic strands. Only the first 10 nucleotides of guide piRNAs were required to be complementary to the target long RNAs. Data are for long RNAs and MILI- or MIWI-bound piRNAs derived from pachytene piRNA loci for a single biological sample. (D) Percent of piRNAs explaining the 5′ ends of either putative pre-pre-piRNAs or the control RNAs. Only the first 10 nucleotides of guide piRNAs were required to be complementary to the target long RNAs. Data are for long RNAs and MILI- or MIWI-bound piRNAs derived from pachytene piRNA loci. All possible pairwise combinations of data sets from two biological replicates were used to calculate medians. Whiskers correspond to minimum and maximum values. Wilcoxon rank-sum test was used to assess statistical significance.
To test the idea that MILI- or MIWI-bound piRNAs direct slicing of piRNA cluster transcripts to generate pre-pre-piRNAs, we searched for MILI- and MIWI-bound piRNAs that could have guided production of the 5′ ends of the putative pre-pre-piRNAs. Unlike siRNAs and miRNAs, the extent of base-pairing required between a piRNA and its target RNA to support PIWI-protein catalyzed target slicing is incompletely understood (Reuter et al., 2011; Gou et al., 2014; Goh et al., 2015; Zhang et al., 2015). Minimally, the first 10 nucleotides of the piRNA are expected to pair with the target RNA. We calculated the probability of piRNAs with ten-nucleotide complementarity to the first 10 nt of putative pre-pre-piRNAs or to the control RNAs, and observed a statistically significant, 10-nt, 5′ overlap between MILI- and MIWI-bound piRNAs and putative pre-pre-piRNAs, but detected no such overlap with the control RNAs (Figures 7C and 7D). This result is unlikely to reflect canonical cis ping-pong, in which the piRNA and target pair overlap in the genome sequence: >>99% of piRNAs contributing to the 10-nt, 5′ overlap do not map to the same genomic location as their targets. That is, the majority of piRNAs act in trans to cleave targets transcribed from a piRNA-producing locus different from their own.
To account for variance in sample sizes and the different contributions of species with high and low read abundance, we repeated the analyses using random resampling of the data. This bootstrapping analysis confirmed the presence of the 10-nt ping-pong signature for the putative pre-pre-piRNAs but not for the control RNAs (Figures S6C and S6D). Because structural studies of Argonautes suggest that the first nucleotide of guide RNA is not expected to pair with the target (Ma et al., 2005; Parker et al., 2005; Wang et al., 2009; Frank et al., 2010; Boland et al., 2011; Elkayam et al., 2012; Cora et al., 2014; Matsumoto et al., 2016), we also performed the analysis requiring complementarity to the target at only piRNA nucleotides g2–g10. The results were indistinguishable from requiring g1–g10 (data not shown).
Transposons are a possible source of piRNAs capable of targeting piRNA precursor transcripts, because the sequence of a transposon can potentially bear short but sufficient stretches of complementarity with genomic copies of the same or a distant family of the transposon either in the same or a different piRNA locus. For the pachytene piRNA loci, we found that twice as many transposon-derived piRNAs contributed to the 10-nt, 5′ overlap with putative pre-pre-piRNAs than expected by chance: 35.8% observed vs. 17.4% expected, based on the fraction of transposon-mapping piRNAs for MILI-bound piRNAs, and 47.3% observed vs. 20.9% expected for MIWI-bound piRNAs. This finding is particularly striking given that the transcribed regions of the pachytene piRNA loci (32.9%) contain fewer transposon sequences than the genome as a whole (43.9%).
Together, these data support the idea that in mice, as in the fly germline, piRNA-directed slicing initiates the production of phased primary piRNAs. Just as A-MYB, the transcription factor that turns on the pachytene piRNA loci, drives its own transcription via a positive feedback loop (Li et al., 2013), so too pachytene piRNAs promote their own production. In flies, piRNAs provided by the mother during oogenesis likely initiate piRNA production in her offspring (Brennecke et al., 2008; Le Thomas et al., 2014), but inherited piRNAs are unlikely to play a role in mice. Might pachytene piRNA biogenesis be initiated from pre-pachytene piRNAs produced in spermatogonia? To test the idea that a subset of pre-pachytene piRNAs might guide MILI to slice pachytene piRNA transcripts, we sequenced MILI-bound piRNAs from FACS-sorted wild-type spermatogonia. Surprisingly, there was significant 5′, 10-nt overlap of MILI-bound, pre-pachytene piRNAs from spermatogonia with primary spermatocyte putative pre-pre-piRNAs but not with the control RNAs (Figures S6E and S6F). Bootstrapping analysis supported this finding (Figures S6G and S6H). As observed for pachytene piRNAs, the fraction of transposon-derived pre-pachytene piRNAs contributing to the 5′, 10-nt overlap was higher than expected by chance (67% observed vs. 37.5% expected based on the fraction of transposon-mapping MILI-bound pre-pachytene piRNAs). These results suggest that MILI-bound pre-pachytene piRNAs initiate pachytene piRNA biogenesis by converting pachytene piRNA precursor transcripts into pre-pre-piRNAs.
Collectively, these data show that PIWI slicing plays a central role in initiating phased primary piRNA biogenesis in animals evolutionarily as distant as flies and mice. They also suggest that transposon-derived piRNAs can function in the production of piRNAs not participating in transposon silencing.
Origin of 5′ U Bias of Primary piRNAs
The 5′ U bias of primary piRNAs is thought to arise from the specificity of the pre-pre-piRNA cleaving endonuclease producing phased pre-piRNAs. However, the 5′ U bias could also reflect the preference of PIWI proteins to bind guide RNAs beginning with uridine (Kawaoka et al., 2011; Matsumoto et al., 2016). If PIWI proteins contribute to 5′ U bias of primary piRNAs, then the selection for initial uridines should occur when MILI or MIWI binds the 5′ end of a pre-pre-piRNA. The frequency of 5′ U for pre-piRNAs bound to MILI or MIWI and sharing their 5′ ends with putative pre-pre-piRNAs is 91%, higher than the frequency of 5′ U for putative pre-pre-piRNAs (65% for those sharing 5′ ends with MILI-bound pre-piRNAs, 67% for those sharing 5′ ends with MIWI-bound pre-piRNAs). These data suggest that not all pre-pre-piRNAs are converted to pre-piRNAs with equal efficiency.
To determine whether first nucleotide identity influences the potential of pre-pre-piRNAs to produce pre-piRNAs, we ask if highly abundant pre-piRNAs were more likely to derive from pre-pre-piRNAs beginning with uridine. First, we sorted pre-piRNA species by their abundance into 10 equally sized bins. We then calculated the percent 5′ U for the putative pre-pre-piRNAs sharing their 5′ ends with the pre-piRNAs in each bin. For each bin, we also determined the ratio of pre-piRNA abundance to the abundance of the corresponding pre-pre-piRNAs. Consistent with the idea that the guide-binding preference of PIWI proteins contributes to the 5′ uridine bias of piRNAs, putative pre-pre-piRNAs were more likely to produce pre-piRNAs when the pre-pre-piRNAs started with uridine (Figure S6I). Notably, the frequency of 5′ U for mature piRNAs bound to MILI or MIWI and sharing their 5′ ends with putative pre-pre-piRNAs was the same as for untrimmed pre-piRNAs (91%). That no change in 5′ U bias is introduced at the trimming step agrees well with the current piRNA maturation model in which trimming of pre-piRNAs occurs after their loading into PIWI proteins.
In Mice, piRNA Methylation is Uncoupled from Trimming
The final step in piRNA maturation is 3′ terminal 2′-O-methylation. Biochemical experiments in silkworm BmN4 cell lysate suggest that piRNA methylation is coupled to trimming; in the absence of trimming, piRNAs are unmethylated (Kawaoka et al., 2011; Izumi et al., 2016). However, in D. melanogaster and C. elegans, piRNA methylation and trimming are uncoupled: the 3′ ends of untrimmed pre-piRNAs are nonetheless 2′-O-methylated (Tang et al., 2016; Hayashi et al., 2016). We asked whether the untrimmed pre-piRNAs in Pnldc1−/− mice were 2′-O-methylated. To assess the relative fraction of 2′-O-methylated pre-piRNAs, we sequenced NaIO4-oxidized small RNAs from wild-type and Pnldc1−/− primary spermatocytes. (NaIO4 oxidation prevents incorporation of 2′,3′ hydroxy small RNAs into a sequencing library; a 2′-O-methyl group blocks oxidation.) To quantify 2′-O-methylation of piRNAs, we added a mix of 2′,3′ hydroxy and 2′-O-methyl, 3′ hydroxy synthetic RNAs to the total mouse RNA before preparing sequencing libraries. The extent of pre-piRNA 2′-O-methylation in Pnldc1−/− mutants was similar to that observed for piRNAs in wild-type: 66–70% vs. 73–89% (Figure S7A). This was also true for piRNAs bound to a PIWI protein: the MILI- and MIWI-bound piRNAs in wild-type and the pre-piRNAs in Pnldc1−/− were 2′-O-methylated to a similar extent: 48–86% vs 45–88% (Figure S7A). Finally, the fraction of 2′-O-methylation of pre-piRNAs in Tdrkh−/− spermatogonia was similar to that of mature piRNAs from wild-type spermatogonia (Figure S7A). Thus, 2′-O-methylation of both pre-pachytene and pachytene piRNAs is uncoupled from pre-piRNA trimming in mice.
DISCUSSION
Together, the analyses presented here suggest a revised model for pachytene piRNA biogenesis in the mouse testis (Figure 1). As previously established, production of pachytene piRNAs begins with the A-MYB-dependent transcription of the pachytene piRNA loci, generating 5′ capped, spliced, 3′ polyadenylated transcripts (Li et al., 2013). Our data suggest that MILI- and MIWI-catalyzed, piRNA-guided slicing of these long transcripts produces pre-pre-piRNAs, initiating their stepwise conversion into mature piRNAs. The involvement of PIWI-catalyzed piRNA-directed slicing in the production of mouse pachytene piRNAs unites the models for fly and mammalian germline piRNA biogenesis (Mohn et al., 2015; Han et al., 2015; Senti et al., 2015; Wang et al., 2015). In both animals, slicing of primary piRNA transcripts by a PIWI protein, guided by transposon-deriving piRNAs, creates a pre-pre-piRNA whose monophosphorylated 5′ end serves as an entry point for further phased pre-piRNA production. In flies, this new 5′ end makes a secondary piRNA, typically bound to Aub, that can produce a new copy of the initial, Ago3-bound piRNA. We do not yet know for mice whether the new 5′ end generates a secondary piRNA that can initiate further piRNA production.
The essential role of 5′ phosphate recognition for loading guide RNAs into PIWI and other Argonaute proteins (Nykanen et al., 2001; Ma et al., 2005; Parker et al., 2005; Wang et al., 2009; Frank et al., 2010; Boland et al., 2011; Kawaoka et al., 2011; Elkayam et al., 2012; Schirle and MacRae, 2012; Schirle et al., 2014; Cora et al., 2014; Wang et al., 2014; Matsumoto et al., 2016), makes it unlikely that MILI or MIWI can bind a pre-pre-piRNA downstream of its 5′ end. In other words, generation of the 5′ end of a pre-pre-piRNA likely precedes the loading of the RNA into a PIWI protein. Binding of MILI or MIWI to the newly generated pre-pre-piRNA 5′ end allows the PIWI protein to position an endonuclease—perhaps PLD6 (Zucchini in flies)—near the 3′ end of the future pre-piRNA. The endonuclease then cleaves 5′ to the first accessible uridine. For MILI-bound pre-pre-piRNAs, the endonuclease typically cuts 3 nucleotides 5′ to where it cuts for MIWI-bound pre-pre-piRNAs. For both PIWI proteins, endonuclease positioning is flexible, responding to local variation in the availability of uridine residues. The combination of the frequency of uridines in the primary piRNA transcripts and the size difference in the MILI and MIWI footprints explains pre-piRNAs with the same 3′ end for the two PIWI proteins: when only a single uridine is present in the genomic neighborhood not concealed by PIWI protein footprint, the endonuclease most frequently cuts MILI- and MIWI-bound pre-pre-piRNAs at the same site, generating identical pre-piRNAs. Our data also suggests that the 5′ U bias of phased pre-piRNAs is probably created by the combined action of the endonuclease cleaving 5′ to uridines and the preference of PIWI proteins for pre-pre-piRNAs beginning with uridine.
The resulting pre-piRNAs, still bound to MILI or MIWI, can then be trimmed to their characteristic length by PNLDC1 and 2′-O-methylated by HENMT1, generating a mature, functional piRNA ready to guide the PIWI protein. We find that in the mouse testis, piRNA trimming and 2′-O-methylation are uncoupled: untrimmed pre-piRNAs in Pnldc1−/− mutants are nonetheless largely 2′-O-methylated. We also confirm that in mice TDRKH acts in the same step of piRNA maturation as PNLDC1, i.e., trimming of the pre-piRNA 3′ end. However, we note that the abundance of pre-piRNAs in Tdrkh−/− is half that in Pnldc1−/− spermatogonia (Figure S6A). TDRKH contains a Tudor domain, a module associated with dimethyl arginine binding. PIWI proteins often contain dimethyl arginines, and RNA-independent interaction of TDRKH with MIWI and, to a lesser extent, with MILI has been demonstrated previously (Chen et al., 2009; Vagin et al., 2009; Saxe et al., 2013). We speculate that its Tudor domain allows TDRKH to tether MILI- and possibly MIWI-bound pre-pre-piRNAs to a locale with a high concentration of the pre-piRNA-producing endonuclease, such as the outer face of mitochondria, thereby promoting phased pre-piRNA biogenesis.
Although the revised model explains the data presented here, the details of individual steps remain to be established. Assigning specific functions to all the proteins known to act in processing long single-stranded piRNA precursor transcripts into mature piRNAs remains a formidable challenge. Our model no doubt underestimates the complexity of piRNA biogenesis, whose individual steps occur in at least two cellular compartments: perinuclear nuage and mitochondria. Moreover, we do not understand what genetic or epigenetic determinants destine pachytene piRNA cluster transcripts, which are generated by conventional RNA Pol II transcription from euchromatic loci, to become piRNAs. Most importantly, there is still no consensus on what function, if any, pachytene piRNAs serve (Reuter et al., 2011; Vourekas et al., 2012; Watanabe et al., 2014; Gou et al., 2014; Zhang et al., 2015; Goh et al., 2015).
The model proposed here establishes the central role of PIWI-clade Argonautes in piRNA biogenesis: PIWI proteins both initiate primary piRNA transcript processing and direct generation of their own guide piRNAs. Perhaps, PIWI-clade Argonautes emerged first in evolution, generating their own guides without assistance, and only later came to be aided by proteins such as the phased pre-piRNA-producing endonuclease (Zuc/PLD6), helicases (Vas/DDX4, Armi/MOV10L1), factors driving primary piRNA transcript generation (Rhi, Moon, A-MYB), and the myriad Tudor-domain containing proteins. Finally, despite differences among animals in the sources and functions of piRNAs, our results underscore the common descent of the piRNA-producing machinery in metazoan evolution.
ACCESSION NUMBERS
Sequencing data are available from the National Center for Biotechnology Information Small Read Archive using accession number PRJNA421205.
AUTHOR CONTRIBUTIONS
I.G., C.C., and P.D.Z. conceived and designed the experiments. I.G., C.C., and K.C. performed the experiments. I.G. analyzed the sequencing data. I.G., C.C., and P.D.Z. wrote the manuscript.
STAR Methods
Mice
C57BL/6J mice (Jackson Labs, stock number 664) were maintained and used according to the guidelines of the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School. Tdrkh−/− mice were obtained from Jackson Labs and Mili −/− mice (Kuramochi-Miyagawa et al., 2004) were a gift from Dr. Shinichiro Chuma (Kyoto University, Japan).
To create Pnldc1−/− mutants, three sgRNAs targeting sequences in exon 1, exon 2, and intron 2 of Pnldc1 (5′-GUC CCA GGG CGC GCC GGA UGC GG)-3′, 5′-UGU CUC GGC CCC AAC AGA UCA GG)-3′, and 5′-UAA CUA AGG AGA CAC CGG UGA GG-3′, underlining denotes the PAM) were transcribed using T7 RNA Polymerase and purified by electrophoresis through a 10% polyacrylamide gel. Superovulated female C57BL/6J mice (7–8 weeks old) were mated to C57BL/6J stud males, and fertilized embryos were collected from oviducts. A mix of Cas9 mRNA (50 ng/μl, TriLink Biotechnologies, L-7206) and the 3 sgRNAs (each 20 ng/μl) were injected into the cytoplasm or pronucleus of fertilized eggs in M2 medium (Sigma, M7167). The injected zygotes were cultured in KSOM with amino acids at 37°C under 5% CO2 until the blastocyst stage (3.5 days). Thereafter, 15–25 blastocysts were transferred into uterus of pseudopregnant ICR females at 2.5 dpc.
For genotyping, genomic DNA extracted from tail tissues was analyzed by PCR using primers 5′-TTC CCA GCA TGA GAA GAT CA-3′ and 5′-CCA CTC AGA TGG CAA GTC AA-3′. PCR products were Sanger-sequenced using the sequencing primer 5′-TGA CAC GTG CAC GAG CTT TA-3′. The male sterility phenotype reported previously (Ding et al., 2017; Zhang et al., 2017) was confirmed by the presence of epididymal sperm, testis weight, and histological assessment of the testes (Figures S7B, S7C, and S7D).
For histological assessment, testes were collected and fixed using Bouin′s solution at 4°C overnight, then washed three times with 70% (v/v) ethanol and stored in 70% ethanol until tissue sectioning. Fixed tissues were paraffin-embedded, cross-sectioned at a thickness of 5 μm, and stained with hematoxylin and eosin by the UMass Morphology Core.
Isolation of Mouse Germ Cells by FACS
Testes were isolated, decapsulated, and rotated in 1× Gey′s Balanced Salt Solution (GBSS, Sigma, G9779) containing 0.4 mg/ml collagenase type 4 (Worthington LS004188) at 150 rpm for 15 min at 33°C. Seminiferous tubules were then washed twice with 1× GBSS and rotated in 1× GBSS with 0.5 mg/ml Trypsin and 1 μg/ml DNase I at 150 rpm for 15 min at 33°C. After incubation, the tubules were homogenized by pipetting with a Pasteur pipette for 3 min on ice, and fetal bovine serum (FBS; 7.5% f.c., v/v) was added to inactivate trypsin. The cell suspension was then strained through a pre-wetted 70 μm cell strainer and pelleted at 300✕ g for 10 min. The supernatant was removed, cells were resuspended in 1× GBSS containing 5% (v/v) FBS, 1 μg/ml DNase I, and 5 μg/ml Hoechst 33342 (Thermo Fisher, 62249) and rotated at 150 rpm for 45 min at 33°C. Propidium iodide (0.2 μg/ml, f.c.; Thermo Fisher, P3566) was added, and cells strained through a single pre-wetted 40 μm cell strainer. Cell sorting was performed at the UMass Medical School FACS Core as described previously (Bastos et al., 2005; Cole et al., 2014). Cell viability was assessed using live phase contrast microscopy. Purity check of sorted fractions was performed by immunostaining aliquots of cells.
For immunostaining, cells were incubated for 20 min in 25 mM sucrose and then fixed on a slide with 1% (w/v) paraformaldehyde solution containing 0.15% (v/v) Triton X-100 for 2 h at room temperature in a humidifying chamber. Slides were then sequentially washed for 10 min in (1) 1× PBS containing 0.4% (v/v) Photo-Flo 200 (Kodak, 1464510), (2) 1× PBS containing 0.1% (v/v) Triton X-100, (3) 1× PBS containing 0.3% (w/v) BSA, 1% (v/v) donkey serum (Sigma, D9663), and 0.05% (v/v) Triton X-100. After washing, slides were incubated with primary antibodies in 1× PBS containing 3% (w/v) BSA, 10% (v/v) donkey serum, and 0.5% (v/v) Triton X-100 in a humidifying chamber overnight at room temperature. Primary antibodies used in this study were rabbit polyclonal anti-SYCP3 (Abcam, ab15093, 1:1000 dilution), and mouse monoclonal anti-gH2AX (Millipore, 05-636, 1:1000 dilution). Slides were washed again as described above and incubated with secondary donkey anti-mouse IgG (H+L) Alexa Fluor 594 (Thermo Fisher, A-21203, 1:2000 dilution) or donkey anti-rabbit IgG (H+L) Alexa Fluor 488 (Thermo Fisher, A-21206, 1:2000 dilution) antibodies for 1 h in a humidifying chamber at room temperature. After incubation, slides were washed three times for 10 min in 1× PBS containing 0.4% (v/v) Photo-Flo 200 and once for 10 min in 0.4% (v/v) Photo-Flo 200. Finally, slides were dried, mounted with ProLong Gold Antifade Mountant with DAPI (Thermo Fisher, P36931), and covered with a cover slip.
Western Blotting
Sorted germ cells were homogenized in lysis buffer (20 mM Tris-HCl pH 7.5, 2.5 mM MgCl2, 200 mM NaCl, 0.05% (v/v) NP-40, 0.1 mM EDTA, 1 mM 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride, 0.3 μM Aprotinin, 40 μM Bestatin, 10 μM E-64, 10 μM Leupeptin) and centrifuged at 20,000✕ g for 20 min at 4°C. The supernatant was moved to a new tube, an equal volume of loading dye (120 mM Tris-Cl, pH 6.8, 4% (w/v) SDS, 20% (v/v) glycerol, 2.5% (v/v) 2-Mercaptoethanol, 0.2% (w/v) bromophenol blue) was added, the sample incubated at 90°C for 5 min and resolved through a 4– 20% gradient polyacrylamide/SDS gel electrophoresis (Bio-Rad Laboratories, 5671085). After electrophoresis, proteins were transferred to PVDF membrane (Millipore, IPVH00010), the membrane blocked in Blocking Buffer (Rockland Immunochemicals, MB-070) at room temperature for 2 h and then incubated overnight at 4°C in Blocking Buffer containing primary antibody (anti-PIWIL2/MILI, Abcam, ab36764, 1:1000 dilution; anti-PIWIL1/MIWI, Abcam, ab12337, 1:1000 dilution). Next, the membrane was washed three times with Blocking Buffer at room temperature for 30 min and incubated for 2 h at room temperature with donkey anti-rabbit IRDye 680RD secondary antibody (diluted 1:20,000; LI-COR, 926-68073) in Blocking Buffer. Then the membrane was washed three times with Blocking Buffer at room temperature for 30 min and the signal was detected using Odyssey Infrared Imaging System. Data was obtained for two independent biological replicates.
Small RNA Immunoprecipitation
Mouse total testis or sorted germ cells were homogenized with lysis buffer (20 mM Tris-HCl, pH 7.5, 2.5 mM MgCl2, 200 mM NaCl, 0.05% (v/v) NP-40, 0.1 mM EDTA, 1 mM 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride, 0.3 μM Aprotinin, 40 μM Bestatin, 10 μM E-64, 10 μM Leupeptin) and then centrifuged at 20,000x g for 20 min at 4°C, retaining the supernatant. Anti-MIWI (2C12, monoclonal Anti-PIWIL1, Wako, 017-23451, ~5 μg; Nishibu, 2012) or anti-MILI (Abcam, ab36764, ~5 μg) antibodies were incubated with rotation with 30 μl of Protein G Dynabeads (Thermo Fisher, 10003D) in 1× PBS containing 0.02 % (v/v) Tween 20 (PBST) at 4°C for 1 h. The bead-antibody complex was washed with PBST. Freshly prepared testis or cell lysate were added to the bead-antibody complex and incubated with rotation at 4°C overnight. The next day, the beads were washed once with lysis buffer and three times with 0.1 M Trisodium Citrate. After washing, RNA was purified with Trizol reagent (Thermo Fisher, 15596026) and used for small RNA library preparation. Each experiment was conducted for two independent biological replicates. The specificity of the commercial anti-MILI antibody was confirmed by immunoprecipitation from lysate of Mili−/− whole testis (data not shown).
Small RNA-seq Library Preparation and Analysis
Total RNA from sorted mouse germ cells was extracted using mirVana miRNA isolation kit (Thermo Fisher, AM1560). Small RNA libraries were constructed as described (Han et al., 2015) with several modifications. Briefly, before library preparation, a set of 18 synthetic RNA oligonucleotides was added to each RNA sample to enable absolute quantification of small RNAs (Gainetdinov, Colpan, and Zamore, manuscript in preparation). To reduce ligation bias, a 3′ adaptor with three random nucleotides at its 5′ end was used (5′-rApp NNN TGG AAT TCT CGG GTG CCA AGG /ddC/-3′). After 3′ adaptor ligation, RNA was purified by 15% urea polyacrylamide gel electrophoresis (PAGE), selecting for 15–55 nt small RNAs (i.e., 29–79 nt with 3′ adaptor). The rRNA 2S blocking step before 5′ adaptor ligation was omitted. Small RNA-seq libraries for two independent biological replicates were sequenced using a NextSeq 500 (Illumina) to obtain 75 nt single-end reads.
The sequence of the 3′ adapter, including the three random nucleotides, was removed from raw reads, which were further filtered by requiring their Phred quality score to be ≥20 for all nucleotides. Sequences of synthetic spike-in oligonucleotides were identified allowing no mismatches and used for absolute quantification of small RNAs in each library. Reads not fully matching the genome were analyzed using the Tailor pipeline (Chou et al., 2015) to account for non-templated tailing of small RNAs.
Distances between small RNAs were calculated using all possible alignments of either in all genome-matching reads (mouse and fly data sets) or matching only to annotated pachytene piRNA loci (mouse data sets only; Li et al., 2013), taking into account the number of times a small RNA sequence occurs in the library divided by the number of locations where this small RNA maps in the genome (i.e., multi-mapping reads were apportioned). Z0 scores for phasing were calculated as described (Han et al., 2015) using distances from –10 to –1 and from 1 to 50 as background. All analyses for spermatogonial total piRNAs and pre-piRNAs were conducted for ≥25-nt long reads. Sequence motif charts for genomic neighborhoods around 5′ and 3′ ends of small RNAs were generated with motifStack (Ou et al., 2018) using alignments in pre-pachytene or pachytene piRNA loci only (Li et al., 2013) and apportioning reads.
Grouping of piRNAs or pre-piRNAs (≥1 ppm) with the same 5′, 23-nt or 25-nt prefix was done for all unambiguously mapping piRNAs (mouse and fly data sets) or mapping to the pachytene piRNA loci only (mouse data sets only; Li et al., 2013). Most frequent 3′ end in each group was identified based on the number of times 3′ ends are found in the library. Pairing MILI- and MIWI-bound small RNA groups was done based on their 5′, 25-nt prefix. Pairing Aub- and Piwi-bound small RNA groups was done based on their 5′, 23-nt prefix. Comparisons in the most frequent 3′ end between MIWI and MILI in mice and Piwi and Aub in flies were performed for each paired group without taking into account the number of times a small RNA sequence is found in the library (species-based approach). Species-based approach was used not to conflate the analysis with production and degradation rates of individual piRNAs or pre-piRNAs. Uridine frequencies and sequence motif charts for genomic neighborhood around the most frequent 3′ ends were also generated using the species-based approach.
RNA-seq Library Preparation and Analysis
Total RNA from sorted germ cells was extracted using mirVana miRNA isolation kit (Thermo Fisher, AM1560) and used for library preparation as described previously (Zhang et al., 2012) with several modifications (Wu, Fu, and Zamore, manuscript in preparation). Briefly, before library preparation, 1 μl of 1:100 dilution of ERCC spike-in mix 1 (Thermo Fisher, 4456740, LOT00418382) was added to 0.5–1 μg total RNA to enable absolute quantification of mRNA. For ribosomal RNA depletion, RNA was hybridized in 10 μl with a pool of 186 rRNA antisense oligos (0.05μM/each; Morlan et al., 2012; Adiconis et al., 2013) in the presence of 10 mM Tris-Cl (pH 7.4) and 20 mM NaCl, heating the mixture to 95°C, then cooling it at –0.1°C/sec to 22°C, and incubating at 22°C for 5 min. Ten units of RNase H (Lucigen, H39500) were added and the mixture incubated at 45°C for 30 min in 20 μl containing 50 mM Tris-Cl (pH 7.4), 100 mM NaCl, and 20 mM MgCl2. RNA was further treated with 4 units DNase (Thermo Fisher, AM2238) in 50 μl at 37°C for 20 min. After DNase treatment, RNA was purified using RNA Clean & Concentrator-5 (Zymo Research, R1016). RNA-seq libraries for three independent biological replicates were sequenced using a NextSeq 500 (Illumina) to obtain 75 + 75 nt, paired-end reads.
RNA-seq analysis was performed with piPipes (Han et al., 2014). Briefly, RNAs were first aligned to ribosomal RNA sequences using Bowtie2 (v2.2.0; Langmead and Salzberg, 2012). Unaligned reads were then mapped using STAR to mouse genome mm10 (v2.3.1; Dobin et al., 2013). Sequencing depth and gene quantification was calculated with Cufflinks (v2.1.1; Trapnell et al., 2010). Differential expression analysis for primary piRNA transcripts (Li et al., 2013) was performed using DESeq2 (v1.18.1; Love et al., 2014). In parallel, raw reads were aligned to an index of ERCC spike-in transcripts (Thermo Fisher, 4456740, LOT00418382) using Bowtie (v1.0.0; Langmead et al., 2009) to enable assessment of the absolute quantity of transcripts in each library.
Cloning and Sequencing of 5′ Monophosphorylated Long RNAs
Total RNA from sorted mouse germ cells was extracted using mirVana miRNA isolation kit (Thermo Fisher, AM1560) and used to prepare a library of 5′ monophosphorylated long RNAs as described (Wang et al., 2014). Libraries for two independent biological replicates were sequenced using a NextSeq 500 (Illumina) to obtain 75 + 75 nt, paired-end reads. Bioinformatics analysis was performed with piPipes (Han et al., 2014). Briefly, RNAs were first aligned to ribosomal RNA (rRNA) sequences using Bowtie2 (v2.2.0; Langmead and Salzberg, 2012). Unaligned reads were then mapped using STAR to mouse genome mm10 (v2.3.1; Dobin et al., 2013) and alignments with soft clipping of ends were removed with SAMtools (v1.0.0; Li et al., 2009; Li, 2011). The number of overlaps between small RNAs and the 5′ ends of long RNAs on opposite strands was calculated with read apportioning, analyzing only sequences mapping to the pachytene piRNA loci (Li et al., 2013). Ping-pong Z10 score was calculated as described (Zhang et al., 2011) using distances from –10 to 9 and from 11 to 50 as background. Bootstrapping was performed with 1,000 iterations for randomly resampled sets of 5′ monophosphorylated long RNAs. The size of the resampled sets (100,000 species) was based on the median size of sets of 5′ monophosphorylated long RNAs.
SUPPLEMENTAL ITEM TITLES
Table S1. Relative and absolute steady-state levels of transcripts. Data are for all annotated genes in spermatogonia, primary spermatocytes, secondary spermatocytes and round spermatids of wild-type and Pnldc1−/− mice. Relative data are presented in fpkm (Fragments Per Kilobase of transcript per Million mapped reads), absolute data are presented in molecules per cell.
ACKNOWLEDGEMENTS
We thank S. Pechhold, T. Giehl, B. Gosselin, Y. Gu, and T. Krumpoch at UMass FACS Core for help sorting mouse germ cells; J. Gosselin and J. Gallant at UMass Transgenic Animal Modeling Core for help generating Pnldc1−/− mice; A. Boucher, C. Tipping, and G. Farley for technical assistance; Z. Weng, Y. Fu, and members of the Zamore laboratory for discussions and critical comments on the manuscript. This work was supported in part by National Institutes of Health grants GM65236 and P01HD078253 to P.D.Z.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}