

Abstract
Streptococcus mutans, one of ∼600 bacterial species in the human oral cavity, is among the most acidogenic constituents of the plaque biofilm. Considered to be the primary causative agent of dental caries, S. mutans harbors a 25kDa SloR metalloregulatory protein which controls metal ion transport across the bacterial cell membrane to maintain essential metal ion homeostasis. The expression of SloR derives, in part, from transcriptional readthrough of the sloABC operon which encodes a Mn2+/Fe2+ ABC transport system. Herein, we describe the details of the sloABC promoter that drives this transcription, as well as a novel independent promoter in an intergenic region (IGR) that contributes to downstream sloR expression. RT-PCR studies support sloR transcription that is independent of sloABC expression, and the results of 5′ RACE revealed a sloR transcription start site in the IGR from which the −10 and −35 promoter regions were predicted. The results of gel mobility shift assays support direct SloR binding to the IGR, albeit with lower affinity than SloR binding to the sloABCR promoter. Function of the sloR promoter was validated in qRT-PCR experiments. Interestingly, sloR expression was not significantly impacted when grown in the presence of high manganese, whereas expression of the sloABC operon was repressed under these conditions. The results of in vitro transcription studies support SloR-mediated transcriptional-activation of sloR and -repression of sloABC. Taken together, these findings implicate SloR as a bifunctional regulator that represses sloABC promoter activity and encourages sloR transcription from an independent promoter.
Importance Tooth decay is a ubiquitous infectious disease that is especially pervasive in underserved communities worldwide. S. mutans-induced carious lesions cause functional, physical, and/or aesthetic impairment in the vast majority of adults, and in 60-90% of schoolchildren in industrialized countries. Billions of dollars are spent annually on caries treatment, and productivity losses due to absenteeism from the workplace are significant. Research aimed at alleviating S. mutans-induced tooth decay is important because it can address the socioeconomic disparity that is associated with dental cavities and improve overall general health which is inextricably linked to oral health. Research focused on the S. mutans SloR metalloregulatory protein can guide the development of novel therapeutics and so alleviate the burden of dental cavities.
Introduction
Dental caries are important indicators of oral health and overall general health in both children and adults. Despite significant public health efforts aimed at reducing caries incidence, approximately 60-90% of school-age children worldwide experience caries, with 91% of adult caries in the United States involving the permanent dentition (1, 2). Of particular concern are children of socioeconomically disadvantaged families who are twice as likely to experience rampant caries in comparison with their wealthier counterparts, and who often present with severe clinical outcomes later in life (3).
Among the early colonizers of the tooth surface is Streptococcus mutans, considered to be the primary causative agent of dental cavities in humans (4). Ongoing research aimed at alleviating or eliminating caries has given rise to valuable insights of S. mutans virulence properties, including genes that mediate its obligate biofilm lifestyle, its ability to tolerate acid and oxidative stress, and maintain metal ion homeostasis (5–12). The introduction of sucrose into the Western diet marked a turning point for S. mutans, which metabolizes carbohydrates for energy production and generates a lactic acid byproduct that demineralizes tooth enamel and drives the process of tooth decay (13, 14). Taken together, S. mutans’ arsenal of virulence attributes makes it an especially resolute dental pathogen, and in dysbiotic plaque a primary instigator of caries formation (15, 16).
Among the evolutionary responses that are paramount for S. mutans survival and pathogenesis in dental plaque is tight regulation of essential metal ion transport across the bacterial cell membrane. To this end, S. mutans is endowed with metal ion uptake machinery which enables the import of divalent cations such as Mn2+ and Fe2+ that are essential for cellular and subcellular functions, and ultimately for bacterial cell viability. Aberrant metal ion uptake, however, can result in over-accumulation of intracellular metal ions and bacterial cell death owing to Fenton chemistry and the elaboration of toxic oxygen radicals (17–20). To counteract these deleterious effects and achieve intracellular homeostasis, S. mutans has evolved transport mechanisms that function to maintain appropriate metal ion uptake, which is especially paramount during periods of feast and famine in the oral cavity when Mn2+ concentrations can vary greatly.
S. mutans metal ion uptake is mediated, in large part, by the sloABCR operon which encodes a SloABC Mn2+/Fe2+ transporter and, via transcriptional readthrough, a 25 kDa SloR metalloregulatory protein. As a transcription factor, SloR, modulates metal ion transport upon binding to DNA in response to manganese availability (7, 20, 21). For instance, between mealtimes exogenous metal ions are not readily available because they are sequestered to host proteins, such as lactoferrin in saliva. Hence, under fasting conditions, S. mutans upregulates the sloABC gene products which includes ATP-binding and -hydrolyzing proteins as well as a transmembrane SloC lipoprotein that scavenges metal ions for uptake (20, 21). In contrast, during a mealtime free metal ions are plentiful in the oral cavity, and in response S. mutans downregulates its metal ion importers so as to avoid over-accumulation of intracellular metal ions and their associated cytotoxic effects. We believe SloR mediates such metalloregulation by binding directly to Mn2+ which, in turn, promotes SloR dimerization and a subsequent conformational change at the N-terminus of the protein to facilitate DNA binding. Specifically, in a previous report we describe SloR-DNA binding upstream of the sloABC locus to a promoter-proximal SloR Recognition Element (SRE) that represses sloABC transcription, presumably via a mechanism that involves promoter exclusion to RNA polymerase (RNAP) (22, 23). Hence, the sloABC-encoded metal ion uptake machinery in S. mutans is subject to transcriptional repression by SloR under conditions of Mn2+ availability, and conversely to de-repression when Mn2+ becomes limiting. The transcriptional regulatory properties of the SloR protein are not limited to the sloABC locus. In fact, work in our laboratory suggests that the SloR protein, a member of the DtxR family of metalloregulators, may be involved in regulating as many as 200 genes in the S. mutans genome, either directly or indirectly (24). The genes that are subject to SloR control belong to a variety of different functional categories beyond metal ion homeostasis, and include gene products that mediate S. mutans oxidative stress and acid tolerance, biofilm formation, and genetic competence, all of which contribute to S. mutans virulence (7, 8, 24, 25). In addition, accumulating evidence in our laboratory supports SloR as more than just a repressor of S. mutans gene expression. While the results of expression profiling studies support SloR-mediated-repression of certain genes (called Class I genes), the binding of SloR to other gene loci can culminate in gene activation (called Class II genes) (24). The mechanism by which SloR encourages gene transcription is unknown and is currently under investigation in our laboratory.
Despite the central importance of SloR in promoting S. mutans survival and virulence gene expression, surprisingly little is known of the regulatory mechanism(s) that modulate SloR itself. To date, the regulation of SloR in S. mutans has been shown to be manganese-dependent and driven, in part, by the sloABC promoter via transcriptional read-through of a weak terminator that is located 3’ to the sloC gene (7, 20, 22). Hence, SloR levels that derive from sloABC promoter activity likely vary with Mn2+ availability between and during meal times. We propose however, that some constitutive baseline level of SloR is likely necessary to facilitate scavenging of essential Mn2+ and/or Fe2+ by the SloC metal ion importer regardless of exogenous metal ion availability, and that such fine-tuning could involve additional mechanisms of control.
In the present study, we set out to determine whether a 184 base pair intergenic region (IGR) that is located immediately downstream of the sloC coding region and upstream of the sloR gene, harbors a specific promoter that drives sloR expression independent of sloABC promoter activity. We propose that together with a unique and as-yet-unidentified SRE in this IGR, a bifunctional role for SloR as both a repressor and activator of S. mutans gene expression will be supported (26).
Results
SloR homodimers bind to a 72bp SRE
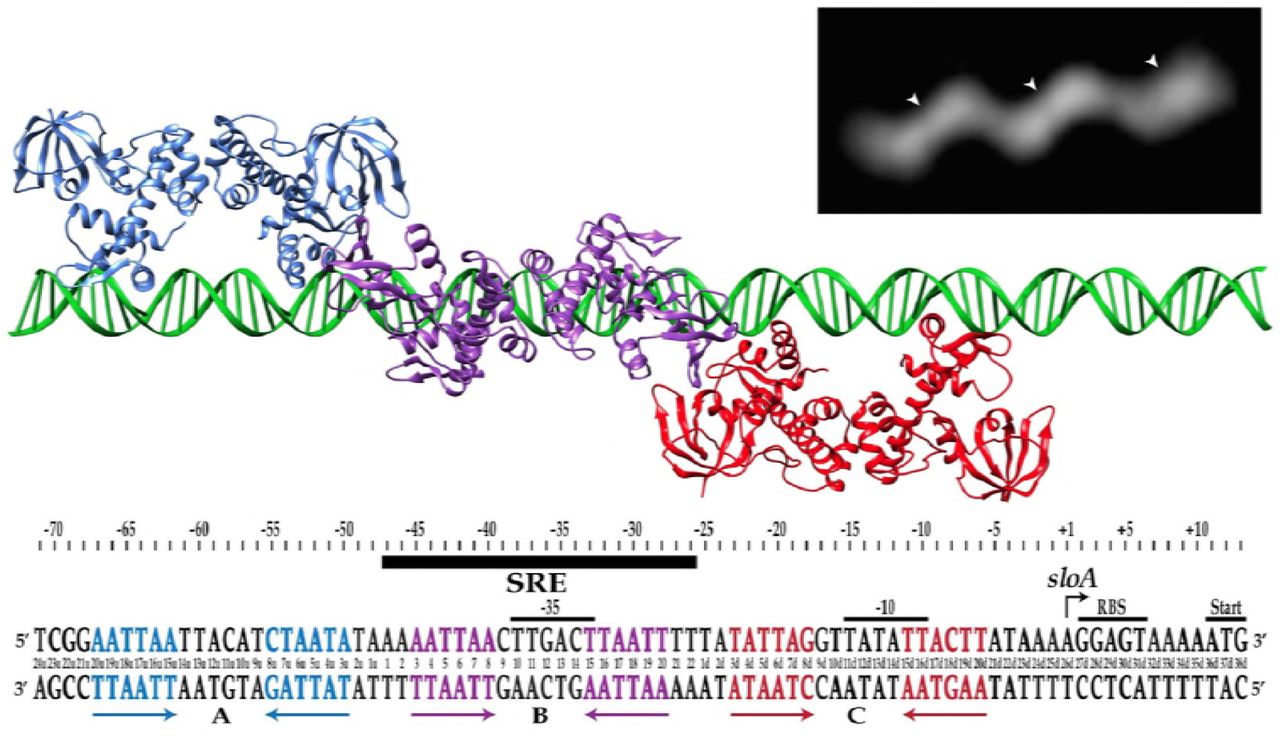
In a previous report, we describe the thermodynamic binding properties of the S. mutans SloR metalloregulator to its cognate SRE within the sloABC promoter region. Herein, the results of fluorescence anisotropy studies conducted with 1mM manganese and various SRE-containing DNA fragments revealed tight binding of the SloR protein to a 72bp DNA fragment (Kd = 32nM). Combined with the results of EMSA and DNA footprinting experiments which support the binding of at least two SloR dimers to this 72bp sequence, we hypothesized that SloR may bind as a set of homodimers to each of three inverted hexameric repeats on this 72bp target DNA, each with the consensus sequence AATTAA or some modification thereof (Figure 1). To test this hypothesis, we used the SloR protein, 1mM Mn2+, and the predicted 72bp SloR recognition element in negative staining and electron microscopy experiments. The resulting dataset comprised of 55 total images was classified into two-dimensional class averages using a PARTICLE software package (www.image-analysis.net/EM/). The dominant pattern that was revealed by the class averages share a common shape with three distinct ellipsoidal regions presumed to be SloR dimers (Figure 1 inset, arrowheads), tilted off of the DNA axis by ∼32°. The SloR binding pattern occupies a total length of 239 Angstroms on the DNA with each SloR dimer measuring ∼90 Angstroms across, and the distance from the center of one dimer to the center of the next measures 75 Angstroms (equal to 22bp). Taken together, the binding pattern and low-resolution image of the SloR-SRE interaction are consistent with the binding of three SloR dimers to the 72bp target sequence.

Shown are each of three inverted hexameric repeats (in blue, purple, and red) that span 72bp in the sloABC promoter region, and to which SloR presumably binds as three homodimers. Affinity binding studies support preferential binding of SloR homodimers to the hexameric repeat in region B (designated by the black bar), followed by cooperative binding of SloR dimers to regions A and C. Inset: Negative staining of the SloR-SRE interaction in vitro supports homodimeric binding of SloR to a 72bp SloR recognition element (SRE). Shown are three SloR homodimers bound to a DNA filament containing the 72bp SloR binding element. The arrowheads denote the center of mass for each SloR homodimer.
SloR binding to the 72bp SRE is cooperative
Equilibrium binding of SloR to fluoresceinated duplex DNA containing sequences from the sloABC promoter region (Table S1) was measured using fluorescence anisotropy with SloR binding to fluorescently-labeled duplex DNA containing either one, two, or three 22bp sequences identified previously on the 72bp SRE (labeled A, B and C in Figure 1). Region B, which includes a pair of inverted repeats, was the only site that demonstrated SloR-specific binding under conditions as high as 250mM NaCl (data not shown). Sequences A and C, which deviate from the consensus 22bp sequence at two and three nucleotide positions respectively, do not measurably bind SloR under these same assay conditions. Saturation binding to these sequences was observed however, when the salt concentration was lowered to 50mM, albeit along with some non-specific interactions. With that said, when SloR was titrated into solutions containing 46bp duplexes harboring two contiguous pairs of inverted repeats, either sloA_AB or sloA_BC, specific cooperative binding was observed under high salt conditions, with Hill coefficients of 1.8 and 1.7, respectively. This result indicates that SloR is capable of binding to the A and C sites with high affinity if the B site is already occupied, consistent with SloR interactions at the B-site that recruit the additional dimers to the flanking A and C sites. In addition, we measured 50% occupancy of the two sites on sloA_AB and sloA_BC at 26nM and 10nM SloR, respectively (data not shown). This result corroborates the observed binding of SloR dimers to all three sites on the 72bp duplex described above in the negative staining experiments.
Differential expression of the S. mutans sloABC and sloR genes suggests the presence of a sloR-specific promoter
To determine whether expression of the sloABC and sloR genes is coordinated under conditions of low versus high manganese, we performed qRT-PCR experiments, the results of which reveal different transcription profiles (Table 2) despite the derivation of these genes from a single polycistronic mRNA. Specifically, expression of the sloABC operon was repressed 3-fold under conditions of high manganese availability (Student’s t-test, p<0.05), whereas transcription of the sloR gene was not significantly altered under these same conditions (Student’s t-test, p>0.05). That expression of the sloABC and sloR genes is different under conditions of high Mn2+ suggests an additional control mechanism for sloR transcription that may involve an independent promoter. We predict this promoter is located within the 184bp intergenic region (IGR) that separates the sloC gene of the sloABC operon from the downstream sloR gene on the S. mutans chromosome.
The results of 5’ RACE reveal the location of a transcription start site in the intergenic region between the S. mutans sloC and sloR genes
To investigate whether a promoter might exist on the IGR that separates the sloABC and sloR genes, we performed 5′ RACE experiments to identify a putative +1 transcription start site. The results revealed that transcription of the 654bp sloR-specific transcript begins at an adenosine residue located 19bp upstream of the ATG translation start codon. Mapping the cDNA sequence back to the S. mutans UA159 reference genome allowed us to predict and annotate the −10 and −35 sites of the predicted sloR promoter (Figure 2). The putative −10 site aligns precisely with the conserved prokaryotic consensus sequence (TATAAT) whereas there is variation in the sequence between the predicted −35 site and its consensus sequence in other prokaryotes. A putative extended −10 element which is characterized by a TGN sequence and the presence of two poly T tracts in the spacer region may compensate for degeneracy in the −35 promoter.

The nucleotide sequence of this region was aligned with the S. mutans UA159 genome from the NCBI GenBank Database (RefSeq accession number NC_004350.2). The +1 transcription start site (designated by the arrow) marks the transcription start site of the sloR gene as defined by 5’RACE, and defines a 19bp 5’ untranslated region (UTR). Also shown are the predicted −35 and −10 promoter regions, the predicted ribosome binding site (RBS), and the start codon (SC) of the 654bp sloR gene. A putative extended −10 element is denoted by the dashed line.
The S. mutans sloR gene is transcribed even in the absence of a functional sloABC promoter
To investigate the impact of promoter/SRE variants on transcription of the S. mutans sloABCR operon, we introduced transition mutations into the 72bp SRE at positions 11 and 11d, both of which share overlap with the −35 and −10 promoter sites upstream of sloABC, respectively. Notably, T-to-C mutations at these sites in the resulting GMS611 and GMS611d strain variants culminated in significantly compromised sloABC transcription (Student’s t-test, p<0.0001) in qRT-PCR experiments, with Cq values approaching those of the no template and reverse transcriptase-minus controls (data not shown). This is consistent with disruption of the sloABC promoter in the GMS611 and GMS611d strain variants. While sloABC transcription in these mutant variants was greatly reduced, transcription of sloR was diminished to a much lesser extent (Student’s t-test, p<0.05) that we propose is the result of continued transcription from an independent, sloR-specific promoter (Table 2).
The S. mutans sloR gene is transcribed from the sloABC promoter as well as from an independent promoter on the 184bp IGR
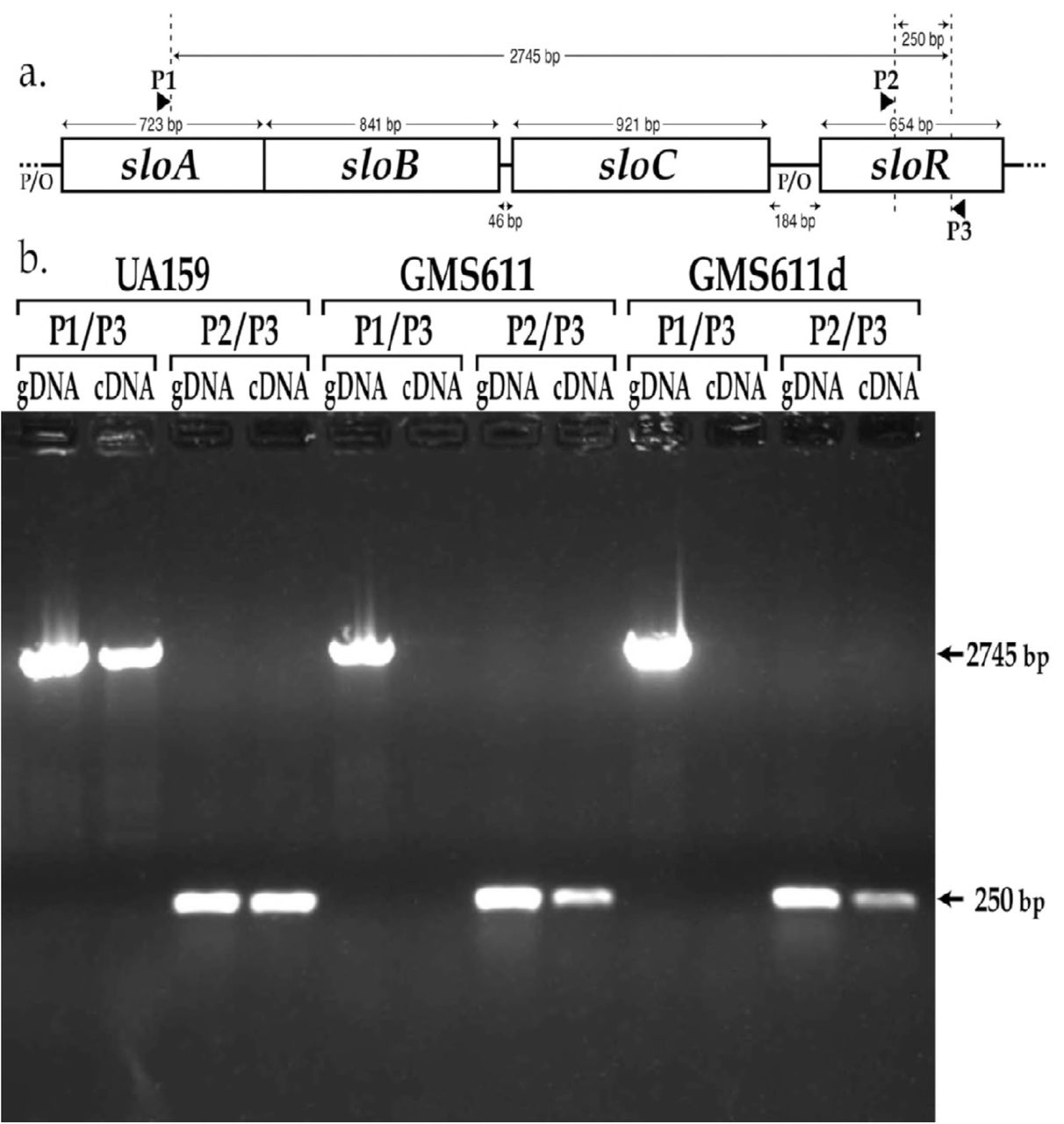
To determine whether sloR transcription is indeed driven by an independent sloR-specific promoter, we performed reverse transcriptase PCR (RT-PCR) experiments with cDNAs that we generated from the S. mutans GMS611 and GMS611d sloABC promoter variants and their UA159 wild-type progenitor. As noted above, expression of the sloABC operon in the GMS611 and GMS611d is virtually nil, indicating successful disruption of the sloABC promoter in these strains. In S. mutans, expression of the sloABC and sloR genes derives from a 3.4Kb polycistronic mRNA owing to transcription off of the UA159 chromosome that is driven by the upstream sloABC promoter and transcriptional readthrough of a weak terminator at the 3’ end of the sloC gene (21, 32, 33). Herein, we expect to generate a 2.7Kb polycistronic mRNA by RT-PCR given the positioning of the P1 and P3 primers that span the sloABC operon and the downstream IGR (Figure 3a). In fact, the results of RT-PCR indicate the presence of a 2.7kb cDNA product in S. mutans UA159, and the absence of this product in GMS611 and GMS611d (Figure 3b). In addition to this polycistronic mRNA however, we noted the presence of a 250bp cDNA product in all three S. mutans strains with the P2/P3 primer pair. The presence of this amplicon in GMS611 and GMS611d indicates the production of a sloR-specific transcript even in the absence of a functional sloABC promoter and supports sloR transcription from an independent promoter. Importantly, PCR products deriving from genomic DNA when used as the amplification template confirm the specificity of the respective primer pairs (Figure 3b).
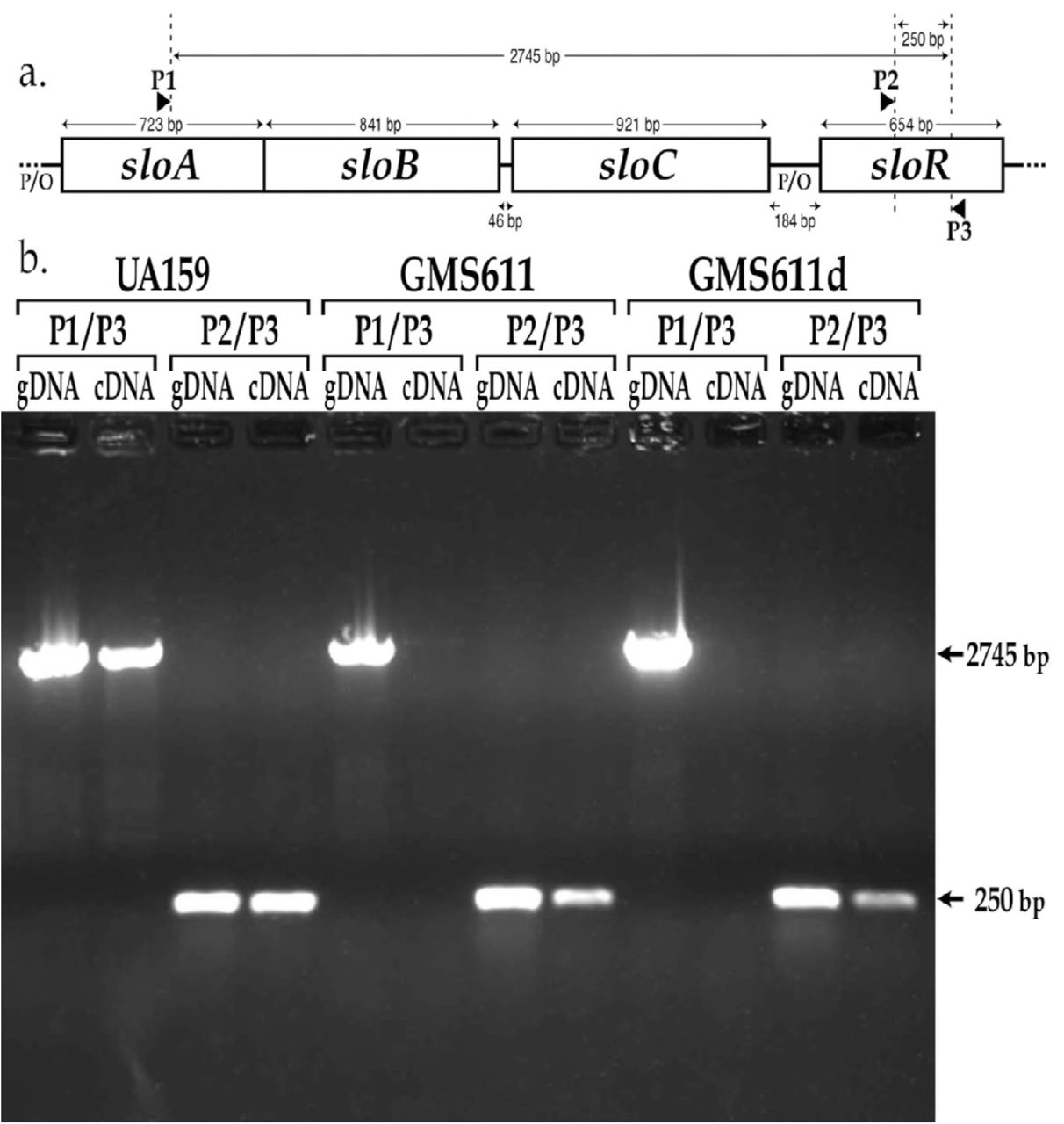
a.) Map of the sloABCR operon and location of primer annealing sites. Primer P1 (sloA.RT_PCR.F) anneals within the sloA coding sequence, and primers P2 and P3 (sloR.RT_PCR.F and sloR.RT_PCR.R, respectively) anneal within the sloR coding sequence. b.) Products of reverse transcriptase PCR resolved on a 0.8% agarose gel. Amplification of cDNA with the P1/P3 primer pair generated a 2745bp amplicon in UA159 but not in GMS611 or GMS611d, consistent with disruption of the sloABC promoter in the mutant strains. In contrast, cDNA amplification with the P2/P3 primer pair gave rise to a 250bp amplicon even in the sloABC promoter mutants GMS611 and GMS611d, indicating the presence of a sloR-specific promoter in the 184bp intergenic region that separates the sloABC operon from the downstream sloR gene (gDNA = genomic DNA; cDNA = copy DNA).
SloR binds directly to the intergenic region between the S. mutans sloC and sloR genes
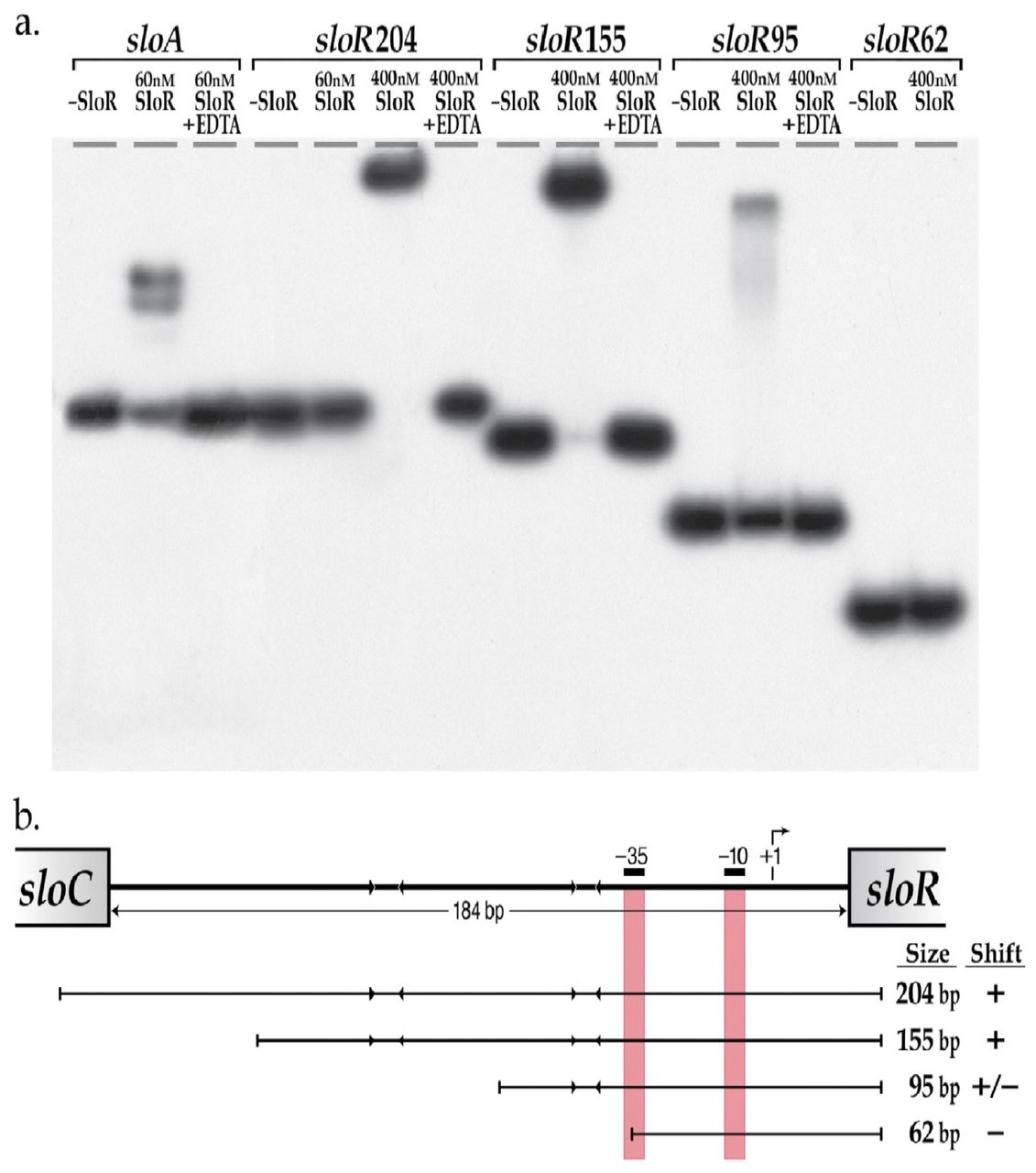
To determine whether the impact of SloR binding on sloR transcription is direct, we performed EMSA experiments, the results of which support direct SloR binding to the intergenic region between the S. mutans sloC and sloR genes. Specifically, we observed protein-IGR binding when SloR was provided at concentrations as low as 400nM, but not at concentrations of 200nM or less (Figure 4a). This contrasts with the SloR binding we observed at the sloABC promoter region which occurred with as little as 60nM SloR protein. These findings support SloR binding to the IGR with lower affinity than that of SloR binding to the sloABC promoter region. In addition, SloR-IGR binding was abrogated upon the addition of 1.5mM EDTA, consistent with the metal ion-dependence of the interaction.

a.) Shown are the results of EMSA which support direct SloR binding to 204bp and 155bp fragments of the sloC-sloR intergenic region at protein concentrations as low as 400nM. SloR binding to a 95bp IGR derivative was relatively compromised however, and completely absent when a 62bp deletion derivative was used as the binding template. A 205bp amplicon that includes the sloABC promoter was used as a positive control for SloR binding. EDTA was added to select reaction mixtures in an attempt to abrogate metal ion-dependent binding. 12% nondenaturing polyacrylamide gels were run at 300 volts for 1.5 hours. Film exposure in the presence of an intensifying screen proceeded for 48 hours at −80°C before development. b.) SloR binding to serial deletion fragments of the S. mutans IGR. The arrowheads facing inward represent AATTAA hexameric repeats to which SloR putatively binds. The vertical red bars denote the positioning of the −10 and −35 promoter sequences of the s/oR-specific promoter. Whether or not SloR binds to the IGR fragment is shown with a (+) or (−) designation.
We also used EMSA to determine the region on the IGR to which SloR binds. To this end, we generated a series of IGR fragments with serial deletions at their 5’ or 3’ ends (Figure 4a). Interestingly, robust band shifts were generated with DNA fragments harboring promoter-distal hexameric repeat sequences that are located at least 62bp upstream of the +1 transcription start site, but not with DNA fragments less than 62bp from the +1 start site that lack these repeat sequences (Figure 4b).
Expression of the S. mutans sloR gene is subject to positive autoregulation
Direct binding of SloR to promoter-proximal sequences at the sloABC locus and to promoter-distal sequences at the sloR locus, coupled with their negative and positive effects on global gene expression respectively, led us to hypothesize a role for SloR as a bifunctional regulator in S. mutans. To validate such a dual role for SloR, we performed in vitro transcription (IVT) experiments, the results of which demonstrate unequivocally that sloABC transcription is repressed by SloR while transcription of the sloR gene is facilitated by SloR (Figure 5). These findings, quantified with ImageJ software and in combination with the results of binding studies, indicate that SloR can either repress or encourage gene expression via direct binding to DNA. To our knowledge, this is the first experimental evidence to demonstrate bifunctional regulation of gene expression by the SloR metalloregulator in S. mutans.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Transcription of the sloABC and sloR genes was quantified using ImageJ software. SloR (75nM) was added to select reaction mixtures containing RNAse inhibitor and either sloA or sloR template DNA. Pixel counting was performed with ImageJ software and does not include unincorporated α32P-UTP. Higher pixel counts indicate greater band intensity. Shown are the results of a single representative experiment (from a total of 3 independent experiments) which support repression of sloA and activation of sloR transcription by the SloR metalloregulator.
Discussion
Until recently, work in our laboratory was focused primarily on understanding the mechanism(s) of SloR binding at the sloABC locus, which is subject to transcriptional repression by SloR. Herein we present evidence that supports an updated model for SloR binding at this locus, consistent with the direct binding of SloR homodimers to binding sites that span a 72bp region of DNA which includes the −10 and −35 sloABC promoter. In previous work (22), we described a pattern for SloR binding at the sloABC promoter site that involved two SloR dimers binding to two sets of inverted repeats, each 6bp in length and separated by 8bp. Subsequent analysis of the 72bp region upstream of the sloABC operon suggested that, in fact, SloR binds to three distinct but abutting 22bp sites in that region, referred to herein as A, B, and C. Each binding site is comprised of two inverted hexameric repeats with an AATTAA consensus separated by 6bp, thereby defining a putative 6-6-6 motif for SloR binding (Figure 1). This sequence pattern (xxAATTAAxxxxxxTTAATTxx, where “x” is a non-conserved nucleotide) is similar to that of the SloR homolog in S. gordonii, called ScaR, which binds to two adjacent 22bp sites upstream of the scaABC operon (34) and for which the binding pattern was confirmed by negative staining and electron microscopy (unpublished observations). We therefore expanded what we previously thought to be a 42bp SRE in the sloABC promoter region to include at least 30 additional base pairs.
While SloR binds to the central 22bp SRE (region B) with strong affinity when provided as a template in isolation, we measured cooperative interactions between SloR homodimers when bound to adjacent SRE sequences (regions A and C). These cooperative interactions are strong enough to support high affinity binding between SloR homodimers, presumably because of interactions involving the initial binding of SloR homodimers to the B site. Such cooperativity has likewise been observed for the S. gordonii ScaR protein (32), suggesting that this property may be a common feature among interactions involving other streptococcal manganese-dependent regulators and their promoter/operator sequences.
In the present study, we extend our SloR binding observations to include the details of protein binding to the IGR that immediately precedes the S. mutans sloR gene. Based on accumulating evidence presented herein, we propose that the location of the SloR-DNA binding element relative to the promoter sequences that modulate downstream sloABC and sloR gene transcription contributes to SloR’s ability to differentially down-regulate sloABC promoter activity and up-regulate sloR promoter activity. Interestingly, an in silico analysis of the 184bp IGR that precedes the S. mutans sloR gene failed to reveal a recognizable SRE like the one we describe above for the sloABC locus, consistent with differential regulation by SloR at these two loci.
The transcription start site for the S. mutans sloR gene occurs within the 184bp IGR that separates sloR from the sloABC operon immediately upstream, as determined in 5’RACE experiments. From the +1 transcription start site, −10 and −35 promoter regions were predicted and a 19bp 5’ untranslated region (UTR) was defined. Notably, the hexameric −10 region shares 100% sequence identity with the canonical prokaryotic consensus sequence for a −10 promoter (TATAAT) (35, 36). An in silico analysis of the sloR promoter revealed a putative extended −10 element in the IGR which is absent from the sloABC promoter region. Reports in the literature describe such a TGN motif immediately upstream of the −10 sequence as an element that could facilitate downstream transcription by stabilizing the open complex during initiation, and by shortening the distance between the −10 and −35 sites (34, 37). The contact that RNA polymerase makes with the nonamer that defines the extended −10 site could compensate for the suboptimal contact that the polymerase makes with the degenerate −35 sequence (37). The TGN motif that we noted on the sloR-containing IGR is similarly located 14-16 nucleotides upstream of the transcription start site. Previous studies have also noted that short poly(T) tracts are characteristic to the spacer region of E. coli TG promoters (38). We similarly noted the presence of two poly(T) tracts centered at positions −18 and −29 in the spacer region of sloR’s TG promoter.
In contrast to the −10 promoter region, the predicted −35 site (TATCCA) shares only 50% sequence identity with the typical prokaryotic promoter sequence (TTGACA) (35). This is not surprising given frequent reports of sequence variation in and around the −35 promoter region within and across prokaryotic species (39). Since promoter strength is, in part, determined by conservation of the −10 and −35 promoter sequences (36) one might expect RNAP to have only moderate binding affinity for the relatively divergent sloR promoter (TATAAT and TATCCA) as compared to RNAP binding at the more highly conserved sloABC promoter (TATATT and TTGACT) (22), and accordingly, weaker transcription from the former as compared to the latter. The results of DNA binding and expression profiling experiments reported herein support these predictions and suggest a role for divergent sloABC and sloR promoter sequences in fine-tuning metal ion transport and minimizing the toxic effects of metal ion hyper-accumulation. Additional layers of gene control involving SloR likely evolved at these loci given the importance of maintaining metal ion homeostasis under conditions as transient as those in the oral cavity.
The absence of a recognizable SloR binding motif in the IGR that precedes sloR is consistent with differential gene regulation and SloR binding at this locus versus that at the sloABC locus. That is, three adjacent palindromes on the 72bp SRE appear to be absent from the IGR, although a pair of consensus palindromes with the sequence AATTAA appear to be uniquely located 44-50bp and 94 −100bp distal to the sloR promoter, respectively. Interestingly, reports in the literature describe AT-rich sequences, including palindromes like those in the sloABC and sloR promoters, that can engender intrinsic curvatures in the DNA (24, 38). To assess inherent DNA curvature in the sloABC and sloR promoter regions, we applied a BEND algorithm (40) to the 72bp sloABC SRE and the 184bp IGR, the results of which revealed high fidelity alignment of AT-rich palindromes with predicted sites for SloR binding (data not shown). Hence, DNA curvature that localizes to the paired palindromic repeats at the sloABC and sloR loci supports a SloR-DNA interaction that is not strictly defined by nucleotide sequence specificity, but by DNA conformation as well. EMSA studies are currently underway to determine what impact, if any, these AT-rich palindromes may have on SloR-DNA binding, and whether the DNA curvature they can instigate contributes to SloR’s function as a repressor versus an enhancer of gene transcription.
Differential expression of the sloABC and sloR genes was especially pronounced in in vitro transcription (IVT) assays where we used the 5’ end of the sloA or sloR coding regions and up to 200bp of upstream DNA sequence as the DNA template. Pixel counting of the resulting mRNA transcripts on autoradiograms, performed with Image J, revealed considerably more robust sloR transcription in the presence of exogenous SloR than in its absence (Figure 5). This contrasts with the in vitro transcription we observed for the upstream sloABC operon which, as expected, was repressed by the presence of SloR. Taken together with the EMSA results that support direct SloR binding at these loci, these data demonstrably support a role for SloR as a bifunctional regulator of S. mutans gene transcription. While the mechanism for sloABC repression likely derives from SloR binding to an SRE that shares overlap with the sloABC promoter, thereby excluding RNAP from promoter access, sloR transcription is the likely result of de-repression with SloR binding to promoter-distal sites. In future work, we will consider Mn2+ status as a potential contributor to differential SloR binding and gene transcription outcomes. In fact, binding of the MntR metalloregulator to different sequences upstream of the mntABCD locus in Bacillus subtilis is known to be Mn2+ concentration-dependent (41).
In summary, the results of the present study support SloR-mediated transcriptional events at the sloR locus that are different from those at the sloABC locus and lend further support to a role for SloR as a bifunctional regulator of gene transcription. It’s tempting to suggest that the sloR-specific promoter on the 184bp IGR evolved to ensure at least some level of constitutive SloR production. Accordingly, when free metal ions are introduced into the oral cavity during a mealtime, S. mutans is poised and ready to modulate the controlled uptake of the exogenous Mn2+ and Fe2+ it needs for survival. Hence, while the sudden introduction of metal ions into the mouth could prove damaging to some constituents of the oral microbiota, S. mutans can exploit these conditions with a metal ion-dependent SloR regulator that can repress the cytotoxic effects of excessive metal ion import, while maintaining baseline levels of SloR from an ancillary promoter. Such fine-tuned gene regulation can impact cell function beyond the scope of metal ion homeostasis, and influence processes like adherence, acid production and the oxidative stress response that more directly contribute to S. mutans-induced disease (7, 8, 24, 25). In conclusion, we propose that S. mutans coordinates the regulated expression of its metal ion transport machinery with that of its virulence attributes. An improved understanding of sloR autoregulation is significant because it can elucidate the mechanisms that fine-tune the regulated expression of metal ion homeostasis and virulence in an important oral pathogen. Moreover, from these investigations we may better elucidate the details of SloR-mediated gene regulation that can benefit the design of an anti-SloR therapeutic aimed at alleviating and/or preventing caries.
Materials & Methods
Bacterial strains, plasmids, and primers
The bacterial strains used in this study are listed in Table 1. Working stocks of each bacterial strain were prepared from overnight cultures and stored in 20% or 50% sterile glycerol at −20°C or −80°C, respectively.
The primers used in this study are shown in Table 1. All primers were designed using the Primer Blast tool from the NCBI website. A RefSeq record was used as the input with forward and reverse primer locations specified by the user. Lack of secondary structure was confirmed using the Oligo Evaluator tool (Sigma) and primers were checked for specificity against the S. mutans UA159 genome with the NCBI Basic Local Alignment Search Tool (BLAST).
Negative-staining and EM analysis
A SloR-Mn2+-72bp SRE complex was prepared in vitro assuming a 3:1 stoichiometry of dimers to DNA. The reaction mixtures were then systematically diluted to 25, 50 and 100nM for negative-stain EM grid preparation using 1% uranyl acetate. After screening for the optimal staining quality and particle concentration, single-particle data were collected on the 50 nM specimen grid using an FEI T12 electron microscope at 120KeV and 68,000x nominal magnification, producing 108 micrographs at 3.15 Å/pixel in the image with varying defocus between 0.9 and 1.8 μm. Then, 8,500 particles of the SloR-DNA complex were selected for 2D classification using the PARTICLE (www.sbgrid.org/software/titles/particle) program.
Construction of S. mutans promoter variants
To generate specific mutations in the promoters that drive sloABC transcription, we adopted a markerless mutagenesis strategy similar to that described by Xie et al. (27). This involved constructing promoter variants with an IFDC2 cassette inserted within the sloABC promoter region for subsequent CSP-transformation into S. mutans UA159 to generate the erythromycin-resistant and p-4-chlorophenylalanine-sensitive GMS602 strain. The double-crossover event was confirmed by polymerase chain reaction (PCR) and Sanger sequencing. Derivatives of GMS602 were generated by overlap extension PCR (OE-PCR) with the reverse primers harboring a single point mutation in the predicted 72bp SRE that precedes the sloABC genes (Table 1). S. mutans GMS611 was generated with degenerate primers that introduced a point mutation into the SRE at nucleotide position 11 that we predict shares overlap with the sloABC-specific −35 promoter region (22). Likewise, GMS611d was generated with a different degenerate primer set that introduced a point mutation at position 11d into the SRE, which we predict shares overlap with the sloABC-specific −10 promoter region. Thymine to cytosine transition mutations were generated in both S. mutans strains and validated by Sanger sequencing (22).
Chromosomal DNA Isolation
S. mutans grown overnight at 37°C and 5% CO2 in 14ml Todd-Hewitt Yeast Extract (THYE) broth were pelleted by centrifugation at 7000rpm for 5 minutes in a Sorvall RCB centrifuge after which the cells were resuspended in 1ml Tris-EDTA buffer (10mM Tris, 1mM EDTA) and chemically disrupted according to established protocols (8, 22). Genomic DNA was purified in subsequent rounds of phenol-chloroform extraction, ethanol precipitated, and resuspended overnight in nuclease-free water (Ambion) at 4°C with gentle agitation (28). Nucleic acid yield and purity were assessed with a NanoDrop Lite spectrophotometer (Thermo Fisher Scientific) and the samples were stored at −20°C.
RNA Isolation
RNA was isolated from S. mutans strains according to established protocols (8). Cells were grown to mid-logarithmic phase (OD600nm = 0.4-0.6) before pelleting by centrifugation as described above and resuspending in RNAProtect (Qiagen). Total intact RNA was purified following cell lysis and DNase I treatment with a Qiagen RNeasy kit after which nucleic acid yield and purity were assessed with a NanoDrop Lite spectrophotometer (Thermo Fisher Scientific). RNA samples were analyzed for integrity by agarose gel electrophoresis and stored at −80°C.
RT-PCR
Reverse-transcriptase PCR was performed with cDNAs that were reverse-transcribed from RNA using a First Strand cDNA Synthesis kit according to the recommendations of the supplier (Thermo Fisher Scientific) or else with chromosomal DNA isolated from S. mutans UA159, GMS611, or GMS611d according to a modification of Idone et al. (29). Q5 Hi-Fidelity Polymerase was used for PCR in accordance with the recommendations of the supplier (New England Biolabs). Each 50 μL PCR reaction consisted of 10 μL 5X Q5 Reaction buffer, 1 μL 10mM dNTPs, 2.5 μL sloA.RT_PCR.F or sloR.RT_PCR.F, 2.5 μL sloA.RT_PCR.R or sloR.RT_PCR.R [Table 1]), 200 ng of genomic DNA or 1 μL of cDNA product, nuclease-free water up to 49.5 μL, and 0.5 μL of Q5 Hi-Fi Polymerase added to the reaction just prior to amplification. PCR conditions were as follows: initial denaturation at 98°C for 30 s, followed by 25 cycles of denaturation at 98°C for 10 s, annealing at 67°C for 30 s, and extension at 72°C, concluding with a final extension at 72°C for 80 s. An aliquot of each sample was visualized on 0.8% agarose gels as described above.
5’ RACE
To reveal the sloR transcription start site and predict the −10 and −35 regions of the sloR promoter we performed 5′ Rapid Amplification of cDNA Ends (5’ RACE) with an Invitrogen 5′ RACE Kit in accordance with the manufacturer’s protocol unless otherwise specified. RNA isolated from S. mutans GMS611 and GMS611d was reverse-transcribed into cDNA using a sloR.[R].GSP1.A reverse primer and 200 U of Superscript II Reverse Transcriptase. After S.N.A.P. column purification, the cDNA was poly(-dC) tailed using terminal deoxynucleotidyl transferase as described in the manufacturer’s protocol. The cDNA products were PCR amplified using the kit-supplied Abridged Anchor Primer (AAP), a sloR.[R].GSP2.B reverse primer, and Platinum Hi-Fi Taq polymerase, in a BioRad PCR machine programmed at 94°C for 2 minutes, followed by 35 cycles of 94°C for 15 s, 58°C for 30 s, and 68°C for 1 minute, and a final hold at 4°C. An aliquot of the resulting PCR product was analyzed by agarose gel electrophoresis and the remainder was purified using a MinElute PCR Purification kit in accordance with the recommendations of the supplier (Qiagen). The purified amplicons were quantified using a NanoDrop Lite spectrophotometer (Thermo Fisher Scientific) and sequenced (Eurofins) using the reverse primers sloR.GSP2.A or sloR.nested.GSP. The 5′ sequence of the mRNA transcript was aligned with the S. mutans UA159 reference genome (RefSeq accession number NC_004350.2) from NCBI to identify the nucleotide that immediately follows the poly(-dC)-tail as the transcription start site (+1 site), predict the −10 and −35 promoter sequences in the 184bp intergenic region, and reveal the 5′ untranslated region of the sloR transcript (30).
EMSA
Electrophoretic mobility shift assays (EMSAs) were performed according to established protocols to determine whether SloR binding to the intergenic region upstream of the sloR gene is direct, and to narrow down the region of SloR binding at this locus (8, 22, 23). Primer design for the DNA binding template spanned the 184bp intergenic region (IGR) between sloC and sloR and included serial deletions thereof. PCR amplification was performed with Q5 polymerase according to the manufacturer’s protocol (New England Biolabs) using the following thermal cycling conditions: initial denaturation at 98°C for 30 s, 35 cycles at 98°C for 10 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s, with a final extension at 72°C for 2 minutes. The resulting amplicons were PCR purified as described earlier, confirmed by agarose gel electrophoresis, and quantified by NanoDrop spectrophotometry.
The resulting amplicons were end-labeled with [γ-32P]-dATP (Perkin-Elmer) in the presence of T4 polynucleotide kinase (New England BioLabs) after which they were centrifuged through a TE Select-D G-25 spin column (Roche Applied Science) to remove the unincorporated 32P-dATP. Binding reactions were prepared as described previously (23) in a 16-μl total volume containing 1 μl of end-labeled amplicon, purified native SloR protein at concentrations ranging from 60 nM to 400nM, and 3.2 μl of 5× binding buffer (42 mM NaH2PO4, 58 mM Na2HPO4, 250 mM NaCl, 25 mM MgCl2, 50 μg/ml bovine serum albumin, 1 mg sonicated salmon sperm DNA, 50% [vol/vol] glycerol, and 37.5 μM MnCl2). EDTA was added to a separate reaction mixture at a final concentration of 1.5mM to validate SloR binding that is metal ion-dependent. An end-labeled sloA promoter-containing amplicon was used as a positive control for SloR binding (7, 22, 23). Samples were loaded onto 12% nondenaturing polyacrylamide gels (3 ml 20× bis-Tris borate [pH 7.4], 74 μl 100 nM MnCl2, 1.5 ml 100% glycerol, 24 ml 30% acrylamide [37.5:1 acrylamide-bis], 31 ml Millipore H2O, 300 μl 15% ammonium persulfate [APS], 90 μl TEMED [N,N,N′,N′-tetramethylethylenediamine]) and resolved at 300 volts for 1.5 hours. Gels were exposed to Kodak BioMax film for 24-72 hours at –80°C in the presence of an intensifying screen prior to autoradiography.
Fluorescence anisotropy
Equilibrium binding of SloR to regions within the sloABC promoter/operator region was probed by titrating SloR onto fluoresceinated DNA and monitoring binding by fluorescence anisotropy. Duplex DNA fragments containing relevant sequences were prepared from oligonucleotides obtained from Integrated DNA Technologies (Coralville, IA). A 5’ fluoresceinated oligonucleotide was annealed with a 10% excess of its unlabeled complementary strand in 25mM HEPES, pH7.9, 50mM NaCl by heating to 90°C and cooling slowly to room temperature (Table S1). Titrations were performed in either high or low salt buffers; the high salt buffer contained 25mM HEPES, pH 8.0, 250mM NaCl, 10% glycerol and 1mM MnCl2 whereas the low salt buffer contained 25mM HEPES, pH7.9, 50mM NaCl, and 1mM MnCl2. SloR was titrated into 1ml of 1nM fluoresceinated duplex DNA in buffer and anisotropy measurements were made at 25°C using a Beacon 2000 fluorescence polarization instrument. Data were fit to one of several equations related to simple 1:1 binding stoichiometry, where Kd was greater than 10nM (equation 1), less than 10nM (equation 2) or to the Hill equation (equation 3).



In these equations, r is anisotropy, Δr is the total change in anisotropy at saturation, Kd is the dissociation constant, Kh is the concentration of SloR dimers giving 50% maximal binding under cooperative conditions, n is the Hill coefficient, and rmin is the anisotropy obtained before addition of SloR. P is the concentration of SloR dimers and D is the concentration of duplex DNA. In addition, where non-specific binding prevented signal saturation, a term to model the slow linear increase in anisotropy was added to equations 1 or 2, with the form KnsP where Kns is the non-specific binding constant. All curve fitting was performed in R software.
Preparation of S. mutans RpoD
An E. coli DH5α strain harboring plasmid pIB611 (kind gift of Indranil Biswas, University of Kansas) was streaked for isolation on L-agar plates supplemented with ampicillin (100 μg/mL) and incubated overnight at 37°C. Resident on pIB611 is the S. mutans rpoD gene cloned directly downstream of an inducible operon on vector pET-23d(+) and upstream of a C-terminal His-tag (31). Importantly, the 6x His-tag was shown in in vitro transcription experiments to have no physiological impact on RpoD functionality (31). Plasmid pIB611 was purified with a Qiagen miniprep kit according to the recommendations of the supplier and mapped by restriction digestion (New England Biolabs).
Next, pIB611 was used to transform E. coli BL21 DE3 cells in accordance with the manufacturer’s protocol (Invitrogen). Successful transformants were selected after overnight growth on L-agar plates supplemented with 100ug/mL ampicillin and used to inoculate a 25 mL starter culture for yet another overnight incubation. This culture was then used to inoculate 500 mL of pre-warmed L-ampicillin (100ug/ml) broth in a 2.8 L Fernbach flask which was incubated at 37°C with continuous shaking at 200rpm. When the cells reached mid-log phase (OD600=0.4), isopropyl β-D-1-thiogalactopyranoside (IPTG) was added at a final concentration of 0.5mM to induce expression of the T7 RNA polymerase in the BL21 DE3 cells. Protein induction proceeded with continuous shaking for an additional 3.5 hours after which the cells were centrifuged as previously described and stored as dry pellets at −80°C.
Cell pellets were resuspended in His-Binding Buffer (0.5M NaCl, 20mM Tris-HCl, 5mM imidazole) with Halt EDTA-free protease inhibitor at 1x concentration (Thermo Fisher Scientific). The cell suspension was sonicated (Ultrasonic Power Corporation, model number: 2000U) at 60% power for six 30-second cycles using a 0.5 second pulse, with samples maintained on ice between runs. Cells were pelleted by centrifugation at 10,000 x g for 30 minutes at 4°C, after which aliquots of the supernatant and 2X Laemmli buffer (4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromophenol blue and 0.125 M Tris HCl, pH 6.8) were mixed in equal proportions and resolved on 10% Bis-Tris polyacrylamide gels in 3-(N-morpholino) propanesulfonic acid (MOPS) buffer. Proteins were visualized with Sypro Ruby Gel Stain (Thermo Fisher Scientific) according to the manufacturer’s instructions. Polyacrylamide gels were fixed for 45 minutes in fixative solution (50% methanol, 7% acetic acid) with gentle shaking on an orbital shaker. The fixative was subsequently decanted and replaced with 80 mL of Sypro Ruby Gel Stain. Gels were covered and left to stain on an orbital shaker overnight at room temperature. The gel stain was decanted and wash solution (10% methanol, 7% acetic acid) added before UV visualization.
The remaining cell lysate was further purified by Ni2+-Nitrilotriacetic acid (Ni-NTA) column chromatography (Thermo Fisher Scientific) at 4°C according to the manufacturer’s instructions. The columns were placed on a rotating platform for 30 minutes at 4°C to encourage RpoD binding to the Ni-NTA resin before centrifugation to remove unbound protein from the column. After elution, protein yield was determined using both NanoDrop Lite spectrophotometry and a bicinchoninic acid (BCA) protein determination assay (Thermo Fisher Scientific). RpoD purity was assessed on SDS-PAGE gels following Sypro Ruby staining. Select fractions containing RpoD were dialyzed using G2 Slide-A-Lyzer Cassettes with a 10 kDa molecular weight cut off (Thermo Fisher Scientific) in dialysis buffer (25% glycerol in 1x PBS). RpoD was assayed for concentration as described above and stored at −20°C.
In vitro transcription
In vitro transcription was performed according to an adaptation of Kajfasz et al (9). First, genomic DNA spanning approximately 100-200bp of the sloA or sloR coding region and about 150-200bp of upstream sequence was amplified by PCR with Q5 polymerase (New England Biolabs) and either PM.IVT.sloA.F and PM.IVT.sloA.R or PM.IVT.sloR.F and PM.IVT.sloR.R (Table S1) according to the following thermal cycling conditions: initial denaturation at 98°C for 30 s, 35 cycles at 98°C for 10 s, annealing (60°C for sloA, 67°C for sloR) for 30 s, and extension at 72°C for 30 s, with a final extension at 72°C for 2 minutes. The resulting amplicons were PCR purified as described earlier and confirmed by agarose gel electrophoresis. DNA concentration was determined spectrophotometrically on a NanoDrop Lite spectrophotometer. Next, in 1.5 mL microfuge tubes, 10 nM DNA template (sloA or sloR), 1U E. coli RNA Polymerase core enzyme (New England Biolabs), 20U of SUPERase RNase Inhibitor (Thermo Fisher Scientific), 25 nM S. mutans RpoD extract (based on BCA assay), +/− 75nM purified SloR were mixed in Reaction Buffer (10 Mm Tris-HCl [pH 8.0], 50 mM NaCl, 5 mM MgCl2, 50 μg/mL bovine serum albumin) to yield a final volume of 17.8 μL and incubated at 37°C for 10 minutes. To generate an mRNA transcript, 2.2 μL of Nucleotide Mixture (200 μM ATP, 200 μM GTP, 200 μM CTP, 10 μM UTP) and 5 μCi α32P-UTP (Perkin Elmer) were then added and the reaction was incubated at 37°C for 10 minutes. 10 μL of Stop Solution (1M ammonium acetate, 0.1 mg/mL yeast tRNA (Ambion), 0.03M EDTA) was added to terminate transcription, after which 90 μL of ice-cold 99% EtOH was added for ethanol precipitation overnight at 4°C. The following day samples were pelleted at 16,200 x g for 30 minutes, followed by three rounds of washing with 70% EtOH, with additional centrifugation between washes. A final wash with 99% EtOH was performed after which the samples were lyophilized in a vacuum centrifuge (Eppendorf) for 10 minutes. The radiolabeled cell pellet was resuspended in 5 μL of formamide dye (0.3% xylene cyanol, 0.3% bromophenol blue, 12mM EDTA, dissolved in formamide) and the samples were heated to 70°C in a water bath for 2 minutes before placing on ice for gel loading. Samples were resolved on a Novex 10% TBE-7M Urea Gel in 1X TBE Buffer (10.8 g Tris, 5.5g boric acid, 0.01M EDTA [pH 8.0]) and mRNA transcripts were visualized via autoradiography using BIOMAX XAR Film (Thermo Fisher Scientific) exposed for up to 4 hours at −80°C in the presence of an intensifying screen. Film was developed according to standard protocols and ImageJ software was used to quantify the band intensities between samples.
Semi-quantitative real-time PCR (qRT-PCR)
Total intact RNA was isolated from mid-logarithmic phase S. mutans cultures of the wild-type UA159 strain grown in a semi-defined medium (SDM) (22) supplemented with either 5uM (low) or 125uM (high) MnSO4. The resulting RNAs were analyzed for integrity on 0.8% agarose gels before reverse-transcribing 100ng of each RNA sample into cDNA as described above. The cDNAs were used as templates for qRT-PCR which was performed in accordance with established protocols (22) in a CXR thermal cycler (BioRad). Specifically, sloABC and sloR transcription was assessed in three independent qRT-PCR experiments, each performed in triplicate and normalized against the expression of gyrA (8,22).
To assess the impact of transition mutations in the SRE that precedes the sloABC operon on downstream sloR transcription, S. mutans GMS611 and GMS611d were grown as described above except without supplemental Mn2+. Total intact RNA was isolated and reverse transcribed as described, and the results of qRT-PCR were normalized against hk11, the expression of which does not change under the experimental test conditions.
Acknowledgements
This research was supported by NIH Grant #DE014711 to G.A.S., the T. Ragan Ryan Summer Research Fund to P.M., and by the Middlebury College Department of Biology. We acknowledge Mr. Gary Nelson for figure preparation and Dr. Indranil Biswas for providing plasmid pIB611. Many thanks to Dr. Robert Haney for his bioinformatics expertise and assistance with the microarray data, to Dr. Jessica Kajfasz for her guidance with the IVT experiments, and to Mr. Frank Spatafora for general technical assistance. We declare no conflicts of interest for this work.
References




