

Abstract
Adeno-associated virus (AAV)-mediated gene replacement is emerging as a safe and effective means of correcting single-gene mutations, and use of AAV vectors for treatment of diseases of the CNS is increasing. AAV-mediated progranulin gene (GRN) delivery has been proposed as a treatment for GRN-deficient frontotemporal dementia (FTD) and neuronal ceroid lipofuscinosis (NCL), and two recent studies using focal intraparenchymal AAV-Grn delivery to brain have shown moderate success in histopathologic and behavioral rescue in mouse FTD models. Here, we used AAV9 to deliver GRN to the lateral ventricle to achieve widespread expression in the Grn null mouse brain. We found that despite a global increase in progranulin throughout many brain regions, overexpression of GRN resulted in dramatic and selective hippocampal toxicity and degeneration affecting both neurons and glia. Histologically, hippocampal degeneration was preceded by T cell infiltration and perivascular cuffing, suggesting an inflammatory component to the ensuing neuronal loss. GRN delivery with an ependymal-targeting AAV for selective secretion of progranulin into the cerebrospinal fluid (CSF) similarly resulted in T cell infiltration as well as ependymal hypertrophy. Interestingly, overexpression of GRN in wild-type animals also provoked T cell infiltration. These results call into question the safety of GRN overexpression in the CNS, with evidence for both a region-selective immune response and cellular proliferative response following GRN gene delivery. Our results highlight the importance of careful consideration of target gene biology and cellular response to overexpression in relevant animal models prior to progressing to the clinic.
Significance Statement Gene therapies using adeno-associated viral (AAV) vectors show great promise for many human diseases, including diseases that affect the central nervous system (CNS). Frontotemporal dementia (FTD) and neuronal ceroid lipofuscinosis (NCL) are neurodegenerative diseases resulting from loss of one or both copies of the gene encoding progranulin (GRN), and gene replacement has been proposed for these currently untreatable disorders. Here, we used two different AAV vectors to induce widespread brain GRN expression in mice lacking the gene, as well as in wild-type mice. Unexpectedly, GRN overexpression resulted in T cell infiltration, followed by marked hippocampal neurodegeneration. Our results call into question the safety of GRN overexpression in the CNS, with wider implications for development of CNS gene therapies.
Introduction
Frontotemporal dementia (FTD) (1, 2) and neuronal ceroid lipofuscinosis (NCL) (3) are neurodegenerative diseases resulting from haploinsufficiency or complete deficiency of progranulin (GRN), which is encoded by the gene GRN. FTD manifests in late middle age, with symptoms ranging from behavioral changes to deterioration of language and with death ensuing in a mean of 3-5 years after diagnosis (4). Mutations in GRN are a highly penetrant cause of FTD and account for up to 25% of inherited FTD cases (1, 2). Nearly 70 GRN mutations have been identified that cause FTD, and >90% are nonsense mutations that lead to GRN haploinsufficiency (1, 2, 5), with the remainder resulting in functional haploinsufficiency through other means (e.g. defects in GRN secretion). For reasons that are still poorly understood, GRN-deficient states result in accumulation of Tar-DNA binding protein of 43kD (TDP-43) (1, 2), in characteristic inclusion bodies, with subsequent neuronal loss and atrophy of the frontal and temporal lobes.
In the case of NCL, complete GRN deficiency leads to lysosomal dysfunction and accumulation of lipofuscin in neurons and other cell types and a clinical syndrome consisting of generalized seizures, mild cognitive dysfunction, vision loss, cerebellar degeneration, and palinopsia (6–8). Strategies to boost GRN in the FTD and NCL brain have been under development since its discovery as a major causal mutation for these diseases (9-11).
GRN is a secreted growth factor involved in embryonic development, wound healing, and immune modulation (12, 13). In the mouse brain, Grn is expressed highly in mature neurons and microglia and is upregulated in activated microglia following injury (14). In human postmortem brain tissue, GRN expression is widespread in both normal and FTD subjects (15). Both in vitro and in vivo, GRN has been shown to play a role in neuronal survival and neurite outgrowth (16-18), and a neuronal receptor for GRN, sortilin, has been identified (19). Based on its growth promoting properties, augmentation of GRN has been considered for the treatment of a wide range of neurodegenerative diseases. Indeed, lentivirus- and AAV-mediated GRN delivery to the central nervous system (CNS) have been investigated in preclinical models of Alzheimer’s disease (20, 21), Parkinson’s disease (22), motor neuron disease (23, 24), and Huntington’s disease (25).
Methods to augment or replace GRN expression include activating transcription (10), enhancing translation (9), increasing the levels of extracellular GRN (11), or using gene therapy based approaches. Among the latter, gene delivery using adeno-associated viral (AAV) vectors has risen to the forefront of gene therapy on the basis of its excellent safety and efficacy profile. AAV-mediated gene delivery has been used successfully in preclinical models for several decades and has recently shown success in treating a diverse range of diseases in humans, including hemophilia, Leber’s Congenital Amaurosis, and spinal muscular atrophy (26-28). To achieve CNS transduction, AAV can be delivered to the brain parenchyma or the cerebrospinal fluid (CSF), either via intrathecal delivery or by injection into the lateral ventricle, with therapeutic benefit in preclinical models with both gain-of-function and loss-of-function diseases (29-34). Interestingly, and in contrast to peripherally administered AAV (35, 36), numerous studies using different AAV vectors with various gene targets have shown minimal innate or adaptive immune response to AAV-mediated gene delivery in the CNS, even in animals with no prior exposure to the gene in question.
A recent study using direct bilateral injection of AAV1.Grn into the medial prefrontal cortex (mPFC) of Grn null mice demonstrated improvements in lipofuscinosis and microgliosis, with focal improvements in lysosomal function (37). This group had previously used this approach in Grn haploinsufficient mice and demonstrated improvement in lysosomal readouts and social dominance deficits (38). Notably, AAV GRN gene therapy in Grn null mice displayed robust microglial activation at the injection site, with induction of anti-GRN antibodies (37). No other immunologic phenotypes were reported in this short-term study.
While these studies are promising, translation of intraparenchymal gene delivery to the human brain is a challenge based on the size of the target compared to the murine brain. The aim of our study was to deliver GRN globally using a method easily translatable to human subjects, namely a single intraventricular injection of AAV.GRN. As a first approach, we selected AAV9 on the basis of its ability to broadly disperse and infect neurons and glia after CSF delivery, as well as its track record of use in prior studies (28, 34, 39). We also tested AAV4 due to its selectivity for ependymal cells and excellent safety profile (30, 40, 41), to maximize CSF secretion with the goal of broad CNS uptake through the neuronal receptor sortilin. Regardless of serotype, our studies show that over-expression of GRN in brain for extended periods of time is deleterious, causing profound neurodegeneration and raising concern about excessive expression of GRN in mammalian brain as a therapy for FTD/NCL.
Results
Characterization of the Grn Null Phenotype
Mice lacking Grn have an age-dependent histopathologic phenotype consisting of habenular and hippocampal vacuolation and increased ubiquitination starting at 7 months of age, as well as diffusely increased astrogliosis and microgliosis starting at 12 months of age (42-44). In our Grn null animals, we confirmed the previously reported increase in vacuolation (42), which was most pronounced in the habenula and increased with age (Fig. 1A, arrowheads). Additionally, we noted astrocytosis in the Grn null striatum that is present as early as 6 months and progresses with age; this histopathological finding was not present even in 12-month-old WT mice (Fig. 1B) and has not been previously described. Hippocampal morphology was unaffected by genotype at any age (Fig. 1C).

Grn null mice recapitulate previously published histopathologic findings and exhibit previously undescribed abnormalities. (A) Grn null mice exhibit vacuolation that is most pronounced in the habenula and increases with age (arrowheads), and is absent from wildtype mice at all time points. (Scale bar: 50 μm.) (B) Grn null mice demonstrate an age-dependent increase in astrocytosis compared to wildtype mice, as seen by GFAP staining. Shown here is the striatum, an area in which astrocytosis in Grn null mice has not been previously described. (Scale bar: 100 μm.) (C) The hippocampus shows no gross morphological differences in Grn null mice compared to WT at 6 or 12 months. (Scale bar: 250 μm.)
AAV-Mediated Gene Transfer Results in Sustained GRN Expression in Grn Null Mice
Grn null mice were injected with AAV9 encoding human GRN (AAV9.hGRN) (Fig. S1) into the right posterior lateral ventricle at 6-7.5 months of age and sacrificed at time points ranging from 1 to 6 months post-injection as indicated (Fig. 2A). We observed the highest levels of GRN expression, as detected by enzyme-linked immunoassay (ELISA), in the ipsilateral periventricular region; GRN levels remained undiminished at 1-, 3-, 4.5- and 6 months post-injection compared to uninjected whole brain tissue from null mice (Fig. 2A, left). We also observed high GRN levels in the ipsilateral striatum and to a lesser extent, the ipsilateral frontal cortex, although cortical expression diminished over time. Similarly high levels of GRN were detected in the contralateral periventricular region (Fig. 2A, right), but with minimal increase in the left striatum or left frontal cortex. GRN was additionally detected at moderate levels in the periventricular region of the 3rd ventricle, the brainstem, and the spinal cord at all time points (Fig. S2). These data indicate broad, sustained hGRN expression.

AAV9 mediates sustained expression of GRN in Grn null mouse brain. Grn null mice were injected at 6-7.5 months of age with AAV9.hGRN or AAV9.eGFP in the right lateral ventricle and sacrificed 1, 3, 4.5, or 6 months post-injection. Brains were microdissected and GRN levels measured by ELISA. (A, left) GRN levels in the right (injected) peri-ventricular area (RV), right striatum (RStr), right frontal cortex (RFC) and uninjected homogenized whole brain (WB) are shown. (A, right) GRN levels in the left (uninjected) peri-ventricular area (LV), left striatum (LStr), left frontal cortex (LFC) and uninjected homogenized whole brain (WB) are shown. (B) Schematic illustrating the regions collected by microdissection in blue (striatum), red (cortex), and purple (peri-ventricular area). n = 3 mice/group at each time point.
Overexpression of GRN Results in Progressive Hippocampal
Having established sustained expression of GRN, we next performed detailed histological and immunohistochemical analyses to assess for rescue in our treated mice. In Grn null animals injected with AAV9.hGRN at 6-7.5 months of age and sacrificed 6 months after injection, we found striking morphological changes in the hemisphere ipsilateral to injection. Specifically, in the majority of injected animals, the hippocampus ipsilateral to the injection site demonstrated marked loss of structural integrity, while adjacent structures as well as the contralateral hippocampus appeared relatively unaffected (Fig. 3A). Across all affected mice, the most prominent and consistent changes involved the inferomedial hippocampus, and especially the dentate region (Fig. 3A, bottom, and Fig. 3B, right panels). Similar hippocampal degeneration occurred in Grn null mice injected with an AAV9 vector delivering mouse Grn (Fig. S3), indicating that the response was not specific to delivery of a human gene.

Overexpression of GRN is toxic to cells of the hippocampus. Mice were injected at 6-7.5 months of age and sacrificed 1-6 months post-injection, and brains were either embedded and stained for immunohistochemical analysis (A-D) or sectioned and imaged fluoroscopically (E). (A, left) Gross morphological differences were observed between the injected and uninjected hemispheres in 6 of 11 mice 6 months after injection of AAV9.hGRN, with marked atrophy of the hippocampus on the injected side. (A, right) H&E of AAV9.hGRN injected mouse brain reveals marked degeneration of the hippocampus ipsilateral to injection and a dense cellular infiltrate throughout the remaining hippocampal tissue, extending from anterior (upper panel) to posterior (lower panel). (B) The cellular infiltrate is observed along the length of the hippocampus as early as 1 month post-injection of AAV9.hGRN, both inferior to (arrows) and within the parenchyma of the hippocampus (left panels; n = 3 mice/group). By 6 months, marked morphological changes are observed (right panels). (Scale bars: 250 μm.) (C) High levels of GRN are detected throughout the hippocampus at all time points, with a diffuse cytoplasmic pattern of expression (inset). In some cases, cells expressing GRN exhibit pyknotic nuclei (arrow). Shown here is expression 6 months post injection. (Scale bars: 50 μm; inset: 25 μm.) (D) AAV9.eGFP injected brains appear normal at 1- and 6 months post injection (n = 1 and 4 mice/group respectively) and are similar in appearance to those of uninjected Grn null mice (n = 4, data not shown). (Scale bars: 250 μm.) (E) High levels of eGFP expression are detected by fluorescence microscopy throughout the hemisphere ipsilateral to injection, with some cross-over to the contralateral side. Shown here is expression 3 months post-injection (n = 2 mice).
To better define the timeline of degenerative changes, animals were harvested soon after injection. At one month post-injection with AAV9.hGRN, a hypercellular infiltrate was noted to extend anteriorly and posteriorly along the entire hippocampus (Fig. 3B, left panels), most prominent inferior to and within the hippocampal parenchyma. By 6 months post-injection, prominent hypercellular infiltrates and perivascular cuffing accompanied loss of recognizable hippocampal structures (Fig. 3B, right panels). Staining confirmed strong GRN expression in these regions at all time points (Fig. 3C). Positive staining was noted in neurons that appeared to be healthy (Fig. 3C, lower inset) and in cells with pyknotic nuclei (Fig. 3C, arrow). In contrast, littermate control animals treated with AAV9-delivered eGFP (AAV9.eGFP) showed no pathology at 1- or 6 months post injection (Fig. 3D) and appeared similar to uninjected littermates (data not shown), despite high levels of eGFP expression (Fig. 3E).
Responses to hGRN Overexpression are Region and Cell-Type Specific
Gross morphological assessments were performed to determine if the degeneration observed after AAV9.hGRN delivery was present in other brain regions with high levels of expression. No gross morphological differences were seen between the cortex ipsilateral to the injection site and either the contralateral cortex or that of AAV9.eGFP-injected controls at 6 months post-injection (Fig. 4A-B), despite moderately high GRN levels in cortical brain isolates (Fig. 2). Indeed, cortical neuron organization remained intact ipsilateral to the site of AAV9.hGRN injection (Fig. 4C), and no obvious changes in architecture, neuronal number, or gliosis were found in the ipsilateral striatum (Fig. 4D-G), an area with high GRN levels (Fig. 2). Brain tissue from AAV9.eGFP-injected and uninjected animals was similar in appearance across all parameters (data not shown). These results suggest that hGRN overexpression selectively affects hippocampal brain regions.
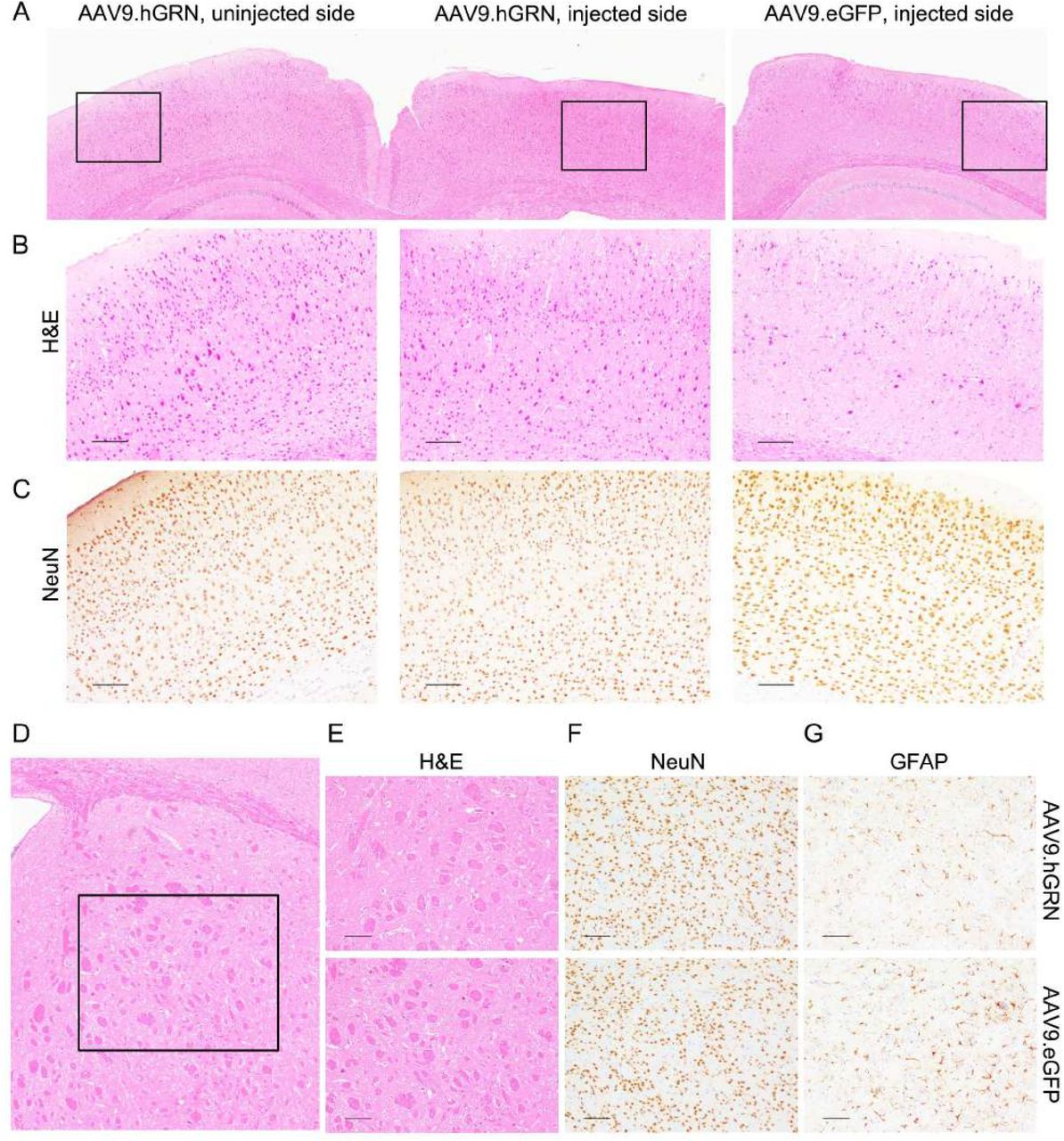
The cortex and striatum are unaffected by GRN overexpression. (A) The ipsilateral cortex immediately adjacent to the degenerated hippocampus appears unaffected by AAV9.hGRN overexpression when compared to the contralateral cortex and to cortex ipsilateral to AAV9.eGFP injected brain. Gross morphology, layer organization, and neuronal numbers appear unremarkable by H&E (B) and NeuN staining (C). (Scale bars: 100 μm.) Similarly, the ipsilateral striatum directly anterior to the degenerated hippocampus (D) appears unremarkable by H&E (E), NeuN (F), and GFAP (G) staining compared to AAV9.eGFP injected controls 6 months post-injection (scale bars: 100 μm), as well as to uninjected controls (data not shown). n = 11 AAV9.hGRN-injected mice/group, n = 4 AAV9.eGFP-injected mice/group.
To determine what cell types were affected in the hippocampus anteriorly (Fig. 5A-D) and posteriorly (Fig. 5E-H), tissue sections were stained for the neuronal marker NeuN, the glial marker GFAP, and the microglial marker Iba-1. Across all affected mice, extensive hippocampal neuronal loss was noted, with the dentate gyrus most severely affected (Fig. 5B and 5F). Hippocampal astrocytes demonstrated fewer, less robust processes (Fig. 5C and 5G), and microglial infiltration ipsilateral to injection was prominent, particularly in the posterior hippocampus (Fig. 5D and 5H) (45). In all cases, the side contralateral to the injection (left panels) appeared similar to the hippocampus of both AAV9.eGFP-injected and uninjected control littermates (data not shown). These data indicate that AAV9-mediated hGRN overexpression is toxic to neurons and astrocytes in the hippocampus, and provokes a strong local microglial response.

AAV9.hGRN-overexpressing mice undergo cell-specific hippocampal degeneration. H&E-stained coronal sections show hippocampal degeneration 6 months post-injection on the injected side anteriorly (A) and posteriorly (E), with boxes indicating regions magnified below (B-D, F-H). On the injected side, NeuN staining shows striking neuronal loss throughout all regions of the hippocampus both anteriorly (B) and posteriorly (F); GFAP staining shows loss of astrocytic processes in anterior (C) and posterior (G) hippocampus; Iba-1 staining for microglia shows a dense microglial infiltrate in the hippocampal region that is present anteriorly (D) but is most prominent posteriorly (H). Throughout the posterior hippocampus there is a dense cellular infiltrate in the ependymal space underlying the hippocampus (F-H) ipsilateral to injection. (Scale bars: 100 μm.)
A T Cell Mediated Inflammatory Response Precedes Neuronal Loss and Occurs in Both Wild-Type and Grn Null Animals
The hypercellular infiltrate found in AAV9.hGRN-injected animals consists of cells with a high nucleus-to-cytoplasm ratio characteristic of lymphocytes. As such, sections were stained for the cell proliferation marker Ki-67, the B lymphocyte marker B220 (CD45), and the T lymphocyte marker CD3.
As shown in Fig. 6, abundant proliferative cells were noted (Fig. 6B and 6F), and the majority of these cells were positive for CD3 (Fig. 6C and 6G). In both anterior and posterior sections, there was extensive perivascular cuffing by CD3+ cells both within and adjacent to the hippocampus (Fig. 6G, arrowheads). These data collectively indicate a robust T cell infiltration of the hippocampal region with minimal contribution from B cells.
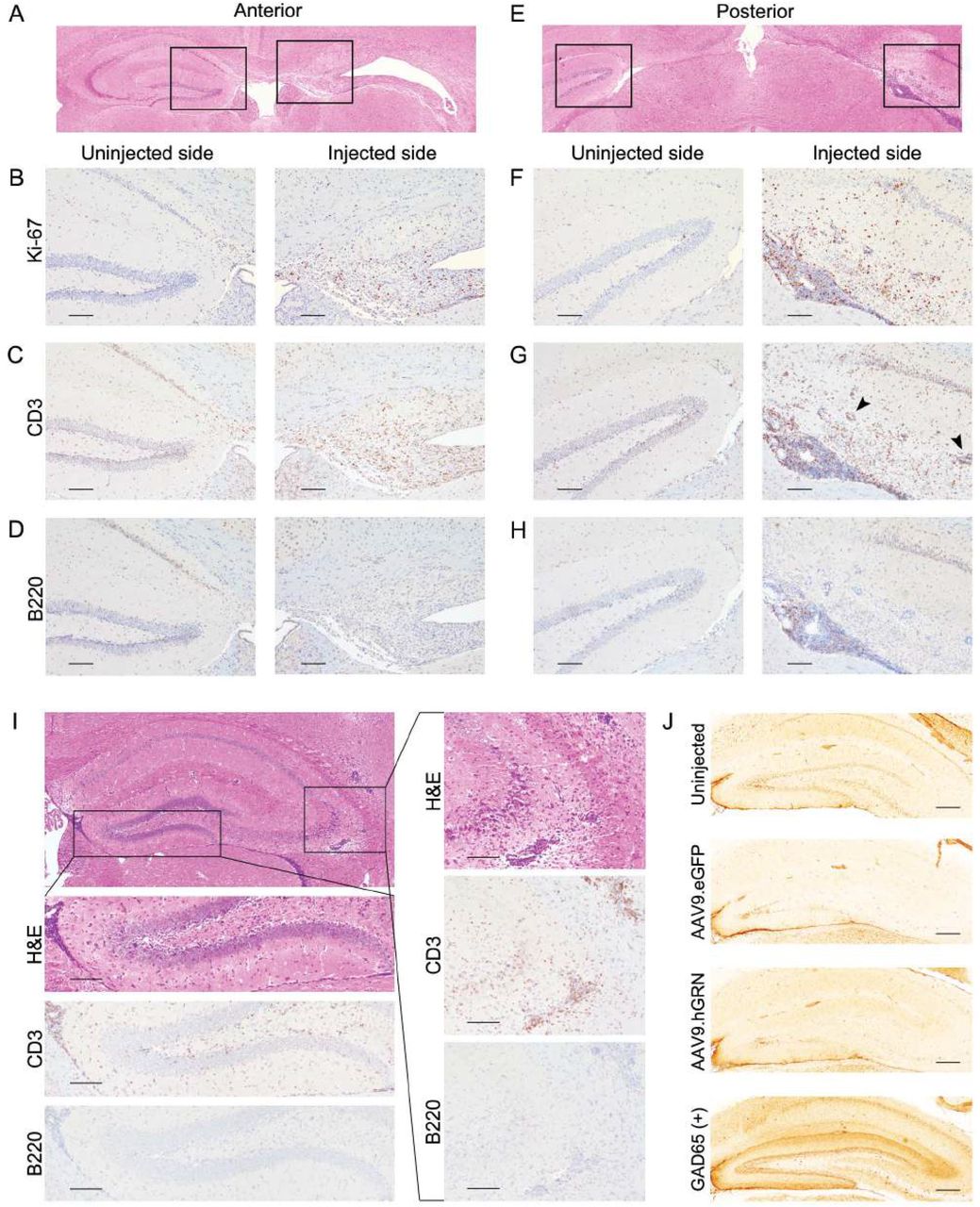
Hippocampal degeneration is characterized by a T cell inflammatory response that precedes cell death. Mice were injected with AAV9.hGRN at 6-8 months of age and sacrificed 6 months post-injection (A-H) or 1 month post-injection (I). H&E-stained coronal sections show hippocampal degeneration 6 months post-injection on the injected side anteriorly (A) and posteriorly (E), with boxes indicating regions magnified below (B-D, F-H). On the injected side, Ki-67 staining demonstrates proliferating cells throughout the hippocampus anteriorly (B) and more markedly posteriorly (F); CD3 staining identifies most of these proliferating cells as T cells (C, G), while B220 staining for B cells is largely negative (D, H), aside from some positively stained cells in the dense posterior sub-hippocampal infiltrate (H). (Scale bars: 100 μm.) Mice were then examined at 1 month post injection (I). H&E staining shows a dense infiltrate in the ependymal space inferior to the hippocampus that is CD3+ and B220− (left box and magnifications below; scale bars: 100 μm). In the CA2/3 region, there is also dense hypercellularity with perivascular cuffing that is CD3+ and B220- (right box and magnifications on right; scale bars: 100 μm, n = 3 mice). (J) Rat brain slices were incubated with mouse serum from uninjected (top, n = 3), AAV9.eGFP-injected (second, n = 2), or AAV9.hGRN-injected (third, n = 5) mice at 6 months post injection, with GAD65+ mouse serum used as a positive control for antibody-based hippocampal reactivity (bottom). There was no immune reactivity in serum collected from mice injected with AAV9.hGRN at 6 months, or at 1- (n = 3) or 3 (n = 2) months after AAV9.hGRN injection (data not shown). (Scale bars: 250 μm.)
To test if inflammatory infiltrates precede or follow the hippocampal degeneration, brain sections from animals sacrificed at earlier time points after AAV9.hGRN injection were characterized. At one month post-injection, hippocampal structures were maintained despite dense cellular infiltration ventral to the hippocampus, with widespread perivascular cuffing lateral to and within the hippocampus (Fig. 6I). Infiltrating cells were positive for CD3 and negative for B220, indicating that T cell infiltration precedes hippocampal neurodegeneration.
In human autoimmune encephalitides, hippocampal degeneration often ensues from autoantibodies specific for hippocampal antigens. Triggering events may be expression of an ectopic antigenic protein by a tumor, or unmasking of a native hippocampal antigen by an inflammatory process (46). We thus tested whether hGRN overexpression elicited a similar pathophysiological process in our Grn null animals using a previously-described rat hippocampal slice assay (47). As shown in Fig. 6J, serum from mice with the hippocampal degeneration phenotype screened negative for anti-hippocampal antibodies, regardless of whether serum was drawn 1-, 3-, or 6 months after AAV9.hGRN injection. Moreover, AAV9.hGRN intraventricular delivery into wild-type animals also elicited perivascular cuffing with infiltration of CD3 positive T cells as early as one month after gene delivery, which became prominent and was accompanied by loss of hippocampal structures by 3 months post-injection (Fig. 7). Taken together, these data support a role for T cell mediated hippocampal neurodegeneration following GRN overexpression.

Wildtype mice also mount a T cell response accompanied by hippocampal cellular loss after injection with AAV9.hGRN. Wildtype background-matched mice were injected in the right lateral ventricle at 6 months of age with AAV9.hGRN and sacrificed 1- (data not shown) or 3 months post-injection. A hypercellular infiltrate was observed most prominently adjacent to the third ventricle ipsilateral to injection (A) and extending posteriorly throughout the hippocampus (B, C). The infiltrate extended inferior to the hippocampus (arrows) as well as within the parenchyma, where we observed marked loss of cells in the CA2/3 region of the hippocampus on the injected side compared to the uninjected side (arrowheads; n = 3 mice).
hGRN Delivered by the AAV4 Ependymal-targeting Vector Elicits an Inflammatory Response and Ependymal Hypertrophy
AAV9 transduces multiple cell types, including neurons and glia (48). We next asked whether the observed inflammatory response and subsequent hippocampal degeneration was serotype specific. For this we used AAV4, an ependymal-targeting serotype (49) that allows secretion of transgene products into the CSF (30, 41, 50). In Grn null mice injected with AAV4.hGRN 1 month earlier into the right lateral ventricle, at the same dose and age as our AAV9.hGRN-injected animals, we observed a hypercellular infiltrate in the anterior (Fig. 8A-B) and posterior (Fig. 8C) hippocampus. Consistent with our previous findings, infiltrating cells were positive for the T cell marker CD3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Grn null mice expressing hGRN delivered by an ependymal-targeting vector (AAV4) show an inflammatory response and ependymal hypertrophy. Mice were injected at 6.5-8 months of age in the right lateral ventricle with AAV4.hGRN and sacrificed 1 month post-injection. AAV4.hGRN-injected mice show a dense cellular infiltrate in the far anterior hippocampus (A, middle panel), anterior hippocampus (B, middle panel), and posterior hippocampus (C, middle panel) that is not present in uninjected age-matched controls (A-C, left panels) or in an AAV4.eGFP-injected littermate (data not shown). (Scale bars: 250 μm.) The infiltrate is highly positive for the T cell marker CD3 in all regions (A-C, right panels; scale bars: 100 μm). When compared to age-matched AAV9.hGRN injected mice, AAV4.hGRN-injected mice show unique choroidal and ependymal hyperplasia and thickening and dense T cell infiltration in the ipsilateral (D) and contralateral (data not shown) lateral ventricles (scale bars: 100 μm). AAV4.hGRN-injected mice also demonstrate ependymal hyperplasia and T cell infiltration in the far anterior ventricles adjacent to the striatum (E; scale bars: 250 μm) that are not apparent in uninjected or in AAV9.hGRN-injected mice. (n = 3 mice/group. 2 of the 3 AAV4.hGRN-injected mice were affected.)
We additionally observed marked ependymal and choroidal hypertrophy in the lateral ventricles adjacent to the hippocampus (Fig. 8D) as well as the anterior 3rd ventricle (Fig. 8A) and the ventricles adjacent to the striatum (Fig. 8E), suggesting a direct effect on cellular proliferation by GRN. This hypertrophy was not observed with AAV9-mediated gene delivery (Fig. 8D and Fig. 8E, second panels). As AAV9 does not efficiently transduce ependymal cells, these data indicate a hypertrophic effect on cells directly expressing the hGRN transgene. Cumulatively, our data suggest that GRN gene delivery may trigger harmful responses in the CNS, irrespective of the AAV serotype used for delivery.
Discussion
In these studies we sought to test the safety and efficacy of AAV-mediated GRN expression in a Grn null mouse model, towards development of a gene replacement strategy for treatment of GRN-deficient FTD and NCL. To our surprise we found that AAV9-mediated GRN overexpression led to severe degeneration of the hippocampus in Grn null mice. The observed degeneration was markedly selective, with sparing of the cortex above, striatum anterior to, and thalamic structures inferior to the hippocampus, despite high GRN levels in these tissues. We also observed a cellular infiltrate primarily composed of T cells as well as perivascular cuffing preceding the onset of hippocampal degeneration and persisting until late stages of degeneration. In addition, we detected a T cell mediated immune response irrespective of the genetic background of the mouse injected, and irrespective of the AAV serotype used, as well as a direct hypertrophic effect of GRN. These data emphasize the need for caution in pursuing GRN delivery in the human CNS.
Neuronal loss in our study was region-selective, with marked hippocampal neurodegeneration, preceded by T cell infiltration. We considered various explanations for our findings. First, the choice of the AAV9 vector – known to transduce both neurons and glia and to achieve high levels of expression – might have resulted in toxic levels of transgene expression. However, eGFP delivered by AAV9 under the same conditions did not elicit a similar response. Additionally, GRN delivered by the ependymally-targeting AAV4 serotype also provoked a marked inflammatory response. These data suggest that the choice of serotype is not sufficient to explain our findings.
Second, the intraventricular delivery route chosen here may have triggered immunogenicity and downstream tissue destruction. In this respect, the recent reports by Arrant et al. demonstrating rescue in mouse models of FTD and NCL after intraparenchymal delivery of Grn are noteworthy (37, 38). Specifically, whether GRN was expressed in mice lacking one or both copies of Grn, this group did not report the dramatic hippocampal degeneration we found in our study. It is possible that the intraventricular route of transgene delivery exposes particular antigen-presenting cells to GRN, thus provoking the T cell infiltration and inflammatory response observed in our animals. Our intraventricular delivery strategy was based on considerations of eventual clinical translation for GRN-deficient human diseases, for which intraparenchymal delivery poses safety and feasibility issues that are less concerning in preclinical models. Moreover, intraventricular CNS delivery of many different transgenes has been successfully achieved in preclinical models, including in animals null for the therapeutic gene, arguing that the choice of delivery route is not the sole explanation for our findings. In mice, at doses and injection routes similar to ours, gene replacement strategies have consistently been safely achieved in null models; for instance, one group safely delivered the ATP-binding cassette transporter (ABCD1) gene in a mouse model of X-linked adrenoleukodystrophy (39). Thus, a final consideration is the direct effect of GRN itself.
While much of the literature in the >10 years since GRN mutations were first linked to neurodegeneration concerns the role of GRN as a neurotrophic factor (14, 16-18), GRN is also widely expressed in cell types ranging from epithelial cells to hematopoietic cells, macrophages, and T cells (12, 13). Early studies described its role in wound healing and regulation of inflammation, which while poorly understood involves an interplay between GRN, which is itself active, and its cleavage products, the granulins, which have opposing effects on a number of immune cell-mediated processes (13). Thus, the existing literature on GRN suggests that tight regulation of its expression, both on the transcriptional side and with respect to the protein cleavage events that generate daughter peptides, may be needed to avoid untoward immunological effects.
There is also extensive literature on involvement of GRN in cell growth and proliferation, both in normal development and in cancer (12, 13). Indeed, GRN is overexpressed and promotes cell growth in many tumors, including glioblastoma (51, 52). In this respect, our findings using AAV4-mediated delivery of GRN are noteworthy. In mice, AAV4 selectively targets ependymal cells and astrocytes in the subventricular zone (53). We therefore reasoned that AAV4-mediated delivery of GRN might be safer, as we would avoid directly targeting neurons and glia, while allowing for endogenous mechanisms of GRN uptake through its neuronal receptor sortilin-1. We found, however, that AAV4-mediated GRN delivery resulted in marked hypertrophy of the ependyma, suggesting direct effects by GRN on the targeted cells, again accompanied by T cell infiltration.
Our data should be compared to recent studies by Arrant et al., who reported reversal of phenotypes associated with GRN deficiency using AAV mediated GRN replacement therapy in Grn null mice, and no evidence of T cell infiltration or hippocampal degeneration. Despite differences in our approach (intraventricular versus intraparenchymal), vector (AAV9 or AAV4 versus AAV1), dose (higher in our study), and transgene (our vector does not have a Myc tag and is able to interact with sortilin), both we and Arrant et al. observe an immune response. In the case of Arrant et al., the response consists of profound microglial activation and MHCII presentation at the injection site, as well as antibodies to GRN detected in plasma. In our studies, by contrast, we observed T cell infiltration and destruction of neurons and astrocytes in the hippocampus. It is possible that, given a longer period of time, evidence of hippocampal degeneration would emerge in their studies. Indeed, Arrant et al. remarked that the upregulation of MHCII is typically associated with a T cell response, and speculated that a longer exposure could result in T cell mediated neuronal degeneration and functional deficits.
These findings raise concerns regarding how to safely translate these studies into humans. Specifically, our combined data suggest that GRN replacement could be highly immunogenic in NCL (GRN null) patients, while achieving the levels needed to reverse FTD will require use of vectors with high transduction efficiency. High levels of GRN expression delivered by transgene, in turn, may run risks of exposure to the tumorigenic growth effects of GRN suggested by our AAV4 data. In addition, these results suggest future pharmacological toxicity studies will be challenging, as over-expression of the human clinical product will be problematic in rodents.
In summary, while GRN-associated FTD and NCL may appear to be attractive targets for gene replacement therapy, our results suggest that concerns stemming from the identity and function of the transgene in question – GRN – are paramount in considerations of a path to human intervention. Specifically, work elucidating the molecular mechanisms by which GRN modulates inflammatory and growth responses, particularly in the CNS, are needed. In addition, our work highlights the potential for inflammatory and tumorigenic adverse effects with GRN overexpression, to which specific attention should be paid in preclinical models. More broadly, these findings call into question our current conception of the brain as an immune privileged organ, AAV as a safe vector for all CNS delivery, and maximization of gene expression as the goal of gene replacement therapy, as these are in tight interplay and should be carefully individualized to the specific therapy being developed.
Materials and Methods
Detailed materials and methods can be found in SI Materials and Methods. Procedures involving animals were performed in accordance with Institutional Animal Care and Use Committee of the University of Pennsylvania. Grn null mice were generated as previously described (54) and provided to the University of Pennsylvania by the Nishihara laboratory at the University of Tokyo. AAV vectors were produced by the CHOP Research Vector Core. Specific tests used are described in the figure legends. Transfections were performed with Lipofectamine 2000 (Invitrogen). For cells and mouse tissues, protein was obtained using RIPA buffer with 0.2% PMSF and 0.1% protease inhibitors. GRN was quantified by human progranulin ELISA kit (Adipogen). For IHC, the following antibodies were used: anti-human GRN rabbit polyclonal antibody (developed by CNDR as previously described (15)), anti-NeuN rabbit polyclonal antibody (ABN78; Sigma-Aldrich), anti-GFAP rabbit polyclonal antibody (Z0334; Dako/Agilent), anti-Iba1 rabbit polyclonal antibody (Saf5299), anti-Ki67 rabbit monoclonal antibody (Ab16667; Abcam), anti-CD45R rat monoclonal antibody (RA3-6B2; Invitrogen,), and anti-CD3e rabbit monoclonal antibody (MA1-90582; Invitrogen). Autoantibody studies were performed as previously described (55).
EXPANDED MATERIALS AND METHODS
Viral Vector Constructs
AAV serotype 9 and AAV serotype 4 were used for these studies. AAV9 vectors contained a cytomegalovirus (CMV) promoter, while AAV4 vectors contained a chicken ß actin (CBA) promoter. The human GRN cDNA (GenBank accession no. BC000324) was amplified from a Hek293 cell cDNA library and the mouse Grn cDNA (GenBank accession no. NM_008175) was cloned using gBlocks (IDT). The transgenes or an eGFP reporter gene were inserted into the G0347 pFB.AAV.CMV.bHGpA plasmid (Iowa Vector Core, Iowa City, IA) and were then used to produce AAV vectors by the Children’s Hospital of Philadelphia (CHOP) Research Vector Core (Philadelphia, PA). All constructs were verified by Sanger Sequencing (CHOP Napcore, Philadelphia, PA).
Mice
Generation of progranulin-null (Grn null) mice on a C57BL/6J background through targeted disruption of the Grn gene has been reported previously (1). Grn null mice were provided to the University of Pennsylvania from the Nishihara laboratory at the University of Tokyo, and the colony was expanded and maintained. Male and female wildtype C57BL/6J (WT; Jackson labs, Bar Harbor, ME) and Grn null mice ages 6-12 months were used in these studies, as well as age-matched wildtype C57BL/6J controls. Mice were housed in a controlled temperature environment on a 12-hour light/dark cycle and were given free access to food and water. All animal studies were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Stereotaxic Delivery
Mice were deeply anaesthetized with isoflurane and immobilized in a stereotaxic frame (David Kopf instruments, Tujunga, CA) installed with both a digital stereotaxic control panel (Leica Biosystems, Buffalo Grove, IL) and microinjection robot (KD Scientific, Holliston, MA) for motorized injections. Mice were injected unilaterally in the posterior right lateral ventricle via a Hamilton syringe using the following coordinates: AP, −2.18 mm; ML, −2.9 mm; and DV, −3.5 mm (from bone) relative to bregma. Each mouse received 10 μL of vector at a concentration of 5e+12 vg/ml for a total dose of 5e+10 VG, infused at a rate of 0.5 μL/min with a 3 minute wait time post-infusion prior to withdrawal of the trochanter. Mice were injected between 6-7.5 months of age and sacrificed between 1 and 6 months post-injection.
Mouse Brain Isolation
Mice were sacrificed at indicated ages by anesthetizing with a ketamine/xylazine/acepromazine mixture, followed by transcardial perfusion with 15mL of ice-cold 0.9% phosphate-buffered saline (PBS). Brains were quickly removed from the skull. Those used for fluorescent imaging studies were fixed whole in 4% paraformaldehyde overnight at 4°C followed by placement in a 30% sucrose/0.05% sodium azide solution for cryoprotection at 4°C. Those used for immunohistochemistry or for GRN quantification by ELISA were blocked into 2mm-thick coronal slices and then either fixed in 4% paraformaldehyde overnight at 4°C or microdissected and flash-frozen in liquid nitrogen, respectively.
Protein Extraction
Frozen isolated brain tissues were weighed and transferred to a 500 μL Potter-Elvehjem dounce homogenizer (Sigma-Aldrich, Allentown, PA), and manually homogenized in 1% radioimmunoprecipitation buffer (RIPA; 50 mM Tris, 150 mM NaCl, 5mM EDTA, 0.5% sodium deoxycholate, 1% NP-40, 0.1% sodium dodecyl sulfate [SDS], pH 8.0) with 0.2% phenylmethane sulfonyl fluoride (PMSF) and 0.1% protease inhibitors (Penn Center for Neurodegenerative Research [CNDR], Philadelphia, PA). Homogenates were centrifuged at 21,380 RCF for 30 minutes and supernatant harvested, and protein concentration was measured by Pierce colorimetric BCA protein assay (ThermoFisher Scientific, Waltham, MA). GRN was quantified by human progranulin ELISA kit (Adipogen, San Diego, CA). 118 μg of total protein was plated per well, with samples run in duplicate. Plates were read on a Berthold LB941 TriStar vTI Multimode reader (Berthold Technologies, Bad Wildbad, Germany) at a wavelength of 450 nm and progranulin concentration calculated using the provided standard curve per kit instructions.
Antibodies
Primary antibodies used for immunohistochemistry included the following: anti-human GRN (anti-hGRN) rabbit polyclonal antibody (0.01mg/mL; developed by CNDR, Philadelphia, PA as previously described (2)), anti-NeuN rabbit polyclonal antibody (0.5μg/mL; ABN78; Sigma-Aldrich, St. Louis, MO), anti-GFAP rabbit polyclonal antibody (0.58μg/mL; Z0334; Dako/Agilent, Santa Clara, CA), anti-Iba1 rabbit polyclonal antibody (0.25μg/mL; Saf5299; Wako, Richmond, VA), anti-Ki67 rabbit monoclonal antibody (dilution 1:1000; Ab16667; Abcam, Cambridge, UK), anti-CD45R rat monoclonal antibody (5μg/mL; RA3-6B2; Invitrogen, Carlsbad, CA), and anti-CD3e rabbit monoclonal antibody (dilution 1:150; MA1-90582; Invitrogen, Carlsbad, CA). Biotinylated goat anti-rabbit and goat anti-rat IgG secondary antibodies (Vector laboratories, Burlingame, CA) were used at a concentration of 1.5μg/mL in all cases except for anti-hGRN, for which secondary antibody concentration was 7.5μg/mL. In the autoantibody studies, biotinylated goat anti-mouse IgG secondary antibody (Vector laboratories, Burlingame, CA) was used at a concentration of 0.75μg/mL.
Section preparation and immunohistochemistry
Fixed coronal brain slices were serially ethanol-dehydrated and paraffin-embedded. Blocks were sectioned coronally at 6 μm and incubated at 37-42°C overnight. Sections were deparaffinized in Xylene and rehydrated in serially dilute ethanol solutions, and were either stained with hematoxylin (ThermoScientific, Kalamazoo, MI) and eosin (Fisher Chemical, Waltham, MA) (H&E) or underwent immunohistochemical (IHC) staining, as follows: sections underwent deactivation of endogenous peroxidase with 5% hydrogen peroxide in methanol for 30 minutes as well as microwave antigen retrieval using antigen unmasking solution (Vector laboratories, Burlingame, CA), followed by washing in Tris buffer then blocking against nonspecific binding sites with 2% fetal bovine serum for 30 minutes at room temperature. Sections were then incubated in primary antibody overnight at 4°C in humidified chambers, blocked again, and incubated in biotinylated secondary antibody for one hour at room temperature. Sections were again blocked, followed by treatment for one hour with the VECTASTAIN ABC kit (Vector laboratories, Burlingame, CA) for avidin binding and peroxidation, then treated with Vector ImmPACT 3,3’-Diaminobenzidine (DAB) peroxidase substrate solution for detection (Vector laboratories, Burlingame, CA). Sections were counter-stained with hematoxylin and dehydrated prior to coverslipping. Brightfield images were taken on a Nikon 80i upright fluorescence microscope and analyzed with Nikon NIS-Elements AR Imaging software.
Section preparation and fluorescence imaging
Fixed, cryoprotected brains were sectioned at 60 μm on a freezing microtome and stored at −20°C in a cryoprotectant solution (30% ethylene glycol, 15% sucrose, 0.05% sodium azide in phosphate buffered saline) until use. Sections were imaged using a DM6000B Leica microscope equipped with a Hamamatsu ORCA-Flash4.0 camera.
Autoantibody detection
Studies were performed as previously described (3). Adult female Wistar rats were anesthetized and decapitated. Brains were removed and washed in 1x phosphate-buffered saline (PBS), then bisected sagitally and fixed in 4% paraformaldehyde in PBS at 4°C for one hour. Brains were then transferred to 40% sucrose in 0.1M PBS for 48 hours, followed by embedding in Tissue-Tek OCT compound embedding medium (Sakura Finitek, Torrance, CA), then snap-frozen in 2-methylbutane cooled with liquid nitrogen. Sections were cut sagitally at 7 μm, mounted on glass slides, and stored at −20°C until use. Endogenous peroxidase was quenched with 0.3% hydrogen peroxide in PBS for 15 minutes. Sections were blocked in 5% goat normal serum for one hour, after which serum samples were applied to sections at a dilution of 1:200 in 5% normal goat serum (Jackson ImmunoResearch Laboratories, West Grove, PA) and incubated overnight at 4°C in humidified chambers. Sections were incubated in biotinylated goat anti-mouse secondary antibody for 2 hours at room temperature, then treated for one hour with the VECTASTAIN Elite ABC kit (Vector laboratories, Burlingame, CA) for avidin binding and peroxidation. Slides were incubated for 30 seconds in 0.5% Triton X solution in PBS, then treated with Vector ImmPACT DAB peroxidase substrate kit (Vector laboratories, Burlingame, CA) for detection. Slides were counter-stained with 50% hematoxylin and dehydrated prior to coverslipping. Brightfield images were taken on a Nikon 80i upright fluorescence microscope and analyzed with Nikon NIS-Elements AR Imaging software.
ACKNOWLEDGEMENTS
We thank Masugi Nishihara for providing us with Grn null animals and Virginia Lee for providing GRN antibody reagents. Sources of support for this project include the Pechenik Montague Award Fund (to ACP), the Benaroya Award Fund (to ACP), and the Brody Foundation (to DA).
Footnotes
↵* co-first authors
REFERENCES




