

Abstract
The C. elegans Piwi Argonaute protein PRG-1 and associated piRNAs protect the genomes of germ cells by suppressing the expression of transposons and potentially deleterious foreign nucleic acids. Deficiency for prg-1 compromises germ cell immortality, resulting in normal fertility for many generations followed by progressively reduced fertility and ultimately sterility. The sterility phenotype of prg-1 mutants was recently shown to be a form of reproductive arrest, which implies that prg-1 mutants may become sterile in response to a form of heritable stress. The DAF-16 stress resistance and longevity factor can promote germ cell immortality of prg-1 mutants by activating a systemic RNAi pathway. We found that this RNAi pathway was not required for the somatic longevity function of DAF-16. Given that prg-1 mutant germ cells may transmit a form of heritable stress, we studied the somatic longevity of prg-1 mutant adults. We found that early generation prg-1 mutants had normal lifespans, but that late-generation adults that displayed reduced fertility or sterility were long lived. Germ cells of long-lived late-generation prg-1 mutants gave rise to F1 cross progeny that were heterozygous for prg-1, fertile and also long lived. However, in the absence of DAF-16, the heritable stress transmitted by prg-1 mutant germ cells was deleterious and caused lifespan to shorten. We conclude that deficiency for the genomic surveillance factor PRG-1/Piwi results in germ cells that transmit a heritable stress that promotes somatic longevity via DAF-16/Foxo, which could be relevant transgenerational regulationof aging.
Introduction
In principle, aging can be divided into two categories: the aging of post-mitotic cells, such as neurons or muscle cells, and the aging of cells that proliferate, such as hematopoetic stems cells. Primary human fibroblasts are deficient for telomerase and can divide for a limited number of cells divisions (40-60) due to telomere erosion, termed the Hayflick limit (1). Telomere erosion induces senescence, a state of permanent cell cycle arrest, but senescence can also be induced by other stresses that include aberrant cellular proliferation conditions (2). In many organisms, somatic aging occurs as a consequence of aging of proliferating cells and of cells that are post-mitotic, but the relationship between these two forms of aging is not well understood (3). The somatic cells of C. elegans adults are post-mitotic, whereas mitotic germ cells of C. elegans adults proliferate for several days. It has been shown that sterility induced by either physical or genetic ablation of the germline can elicit an increase in adult longevity (4), which implies that there may be a trade-off between reproduction and aging (5).
Germ cells are an effectively immortal cell lineage that can be maintained indefinitely. We study germ cells of the nematode C. elegans as a transgenerational model of aging of cells that proliferate. We identified mortal germline (mrt) mutants that initially display normal levels of fertility but after a number of generations these mutants succumb to complete sterility, which can be thought of as transgenerational senescence of the germ line (6). Some mrt mutants are deficient for telomerase-mediated telomere maintenance and their germ cells transmit chromosome termini that shorten with each generation, ultimately eliciting end-to-end chromosome fusions and genome catastrophe (7), (8). Given that germ cells of human families with telomerase deficiency transmit shortened telomeres that primarily elicit the lethal disorders aplastic anemia and pulmonary fibrosis that shorten lifespan (9), we previouslyexamined post-mitotic aging of C. elegans adults that were deficientfor the telomerase reverse transcriptase, trt-1. We found that early-,middle- and late-generation trt-1 mutants had wildtype lifespans, even though late-generation strains possessed telomere fusions and had lost segments of subtelomeric genomic DNA from some chromosome termini (8). These results indicate that telomere attrition, which is a known cause of proliferative senescence, does not impact the aging of post-mitotic C. elegans adult somatic cells.
A second pathway that promotes germ cell immortality is a genomic silencing pathway where small RNAs associate with Argonaute proteins to promote heterochromatin formation (10–12). This pathway initiates with the perinuclear Argonaute protein PRG-1, the C. elegans ortholog of Piwi, which localizes to P granules of germ cells and interacts with thousands of 21 nucleotide piRNAs that scan the genome for viruses, transposons or foreign nucleic acids such as man-made transgenes (13–19). When a genomic intruder is identified, possibly via a process that can detect genomic incongruities that occur when homologous chromosomes are paired during meiosis (20, 21), PRG-1 and associated piRNAs interact with RNA made from the foreign genetic element and recruit RNA-dependent RNA polymerases that act in P-granule associated Mutator bodies to promote biogenesis of 22 nucelotide effector siRNAs named 22G RNAs (22). 22G RNAs generated in response to PRG-1/piRNAs can interact with the P granule Argonaute protein WAGO-1 or the nuclear Argonaute protein HRDE-1, which promote genomic silencing via nuclear RNAi factors and histone modifying proteins (10, 13, 17). The PRG-1/Piwi pathway therefore represents a genomic immune system for germ cells that silences foreign genetic elements (23, 24).
Freshly outcrossed prg-1/piRNA mutants display normal levels of fertility for several generations (12). However, in later generations prg-1 mutants show a decline in fertility such that very few progeny are produced prior to the onset of reproductive delay and sterility (25). Some late-generation animals display pronounced effects on reproduction, such that sterility occurs on day 1 of adulthood, followed by fertility on days 2-4 adulthood. However, some prg-1 mutants that are sterile on day 1 of adulthood remain in an indefinite state of reproductive quiescence termed Adult Reproductive Diapause (ARD) that can be reversed if environmental conditions change (25). States of developmental arrest, termed diapause, occur in many species in response to environmental stresses such as nutritional or seasonal cues, and ARD has been shown to occur in C. elegans in response to starvation (26–28). We therefore hypothesized that prg-1 mutant germ cells may transmit a form of Heritable Stress that eventually becomes severe enough to induce a state of reproductive arrest (25).
We previously found that the number of generations that prg-1 mutant strains can grow, termed transgenerational lifespan, can be significantly extended by weekly bouts of starvation (12). This effect was mediated by the DAF-16/Foxo transcription factor that responds to a variety of stresses and can promote adult longevity (3, 29). Constitutive activation of DAF-16 (by mutation of the upstream daf-2 insulin/IGF-1 receptor) restores germ cell immortality to prg-1 mutants (12). This effect requires 22G RNAs created by the Mutator body protein MUT-7 as well as the H3K4 demethylase SPR-5, both of which promote genomic silencing, implying that DAF-16 activity represses the transgenerational sterility phenotype of prg-1 mutants by activating a compensatory small RNA-mediated silencing pathway (12). In the accompanying paper, by Simon, Spichal, Heestand and colleagues, we demonstrate that DAF-16 acts in somatic cells to upregulate a systemic small RNA pathway that requires Dicer and the double-stranded RNA (dsRNA) binding protein RDE-4, which function together to promote primary siRNA biogenesis in response to exogenous or endogenous dsRNA triggers (22).
microRNAs are produced from hairpin precursor molecules that processed by the ribonucleases Dicer and Drosha in conjunction with DGCR8/pash-1 (30). A conditional allele of pash-1 has revealed that loss of miRNAs shortens lifespan, although miRNAs were not required for the long life of daf-2 mutants whose longevity depends on the DAF-16 stress response transcription factor (31). Specific microRNAs have also been demonstrated to regulate aging in C. elegans (32–35), including some microRNAs whose levels change with age (36–38).
Here we address the relationship between aging of post-mitotic cells with transgenerational aging of germ cells by studying the adult longevity of prg-1 mutants. Our results suggest that prg-1 mutants transmit an epigenetic factor, termed heritable stress, that is intimately related to the function of DAF-16/Foxo and that can promote adult longevity.
Results
The systemic small RNA pathway activated by DAF-16 does not affect adult longevity
daf-2 mutants have been reported to be hypersensitive to RNA interference (39), and daf-2 mutations suppress the transgenerational sterility phenotype of prg-1 mutants by activating a systemic RNAi pathway that requires Dicer and the dsRNA binding protein RDE-4 (see related manuscript by Simon, Spichal, Heestand and colleagues). Because RDE-4 promotes the biogenesis of endogenous small RNAs and rde-4 mutants have relatively minor overt phenotypes (11, 40, 41), we asked whether deficiency for rde-4 affects the lifespan of daf-2 mutants. We found that daf-2; rde-4 double mutant lifespan was not significantly different than that of daf-2 single mutant controls (Fig 1A, S1 Table, p=1), indicating that the small RNA pathway that can be activated by DAF-16 in prg-1 mutants is dispensable for its function in adult longevity.

Lifespan curves for (A) daf-2 (e1368) rde-4 (ne301) and (B) early,late, and sterile generation prg-1 mutants. (C) Percent worms at each developmental stage after 72 hours at 25°C. n.s. is not significant, *** p<0.001, p values determined using log rank analysis and reported in S1 Table.
Heritable Stress of prg-1 mutants induces longevity but not dauer formation
In C. elegans, various environmental stresses including temperature, crowding and starvation can induce an alternative developmental stage termed the Dauer Larva, which is a state of development arrest that is long-lived and highly stress resistant (42). Late-generation prg-1 mutants can enter a reversible state of reproductive arrest, suggesting that prg-1 mutant germ cells may transmit a heritable stress (25). We therefore asked whether late-generation prg-1 mutants that were close to sterility gave rise to dauer larvae. We scored late-generation prg-1 mutants that were poised to become sterile at the stressful temperature of 25°C, but did not observe any dauer larvae, similarto wild-type controls, and to early and fertile late-generation prg-1 mutant controls (Fig 1C). To verify that prg-1 mutants were not defective for inducing dauer larvae, we crossed a daf-2 mutation, which leads to high levels of dauers at 25°C, to create prg-1; daf-2 double mutants and found that there was no defect in entry into dauer arrest (Fig 1C). This indicates that the heritable stress that is transmitted by prg-1 mutant germ cells can specifically induce developmental arrest of the adult germ line and does not perturb larval development, even though dauer larvae are induced by a variety of stresses like heat, crowding and starvation (42).
C. elegans worms that are subjected to dietary restriction, acute starvation or other stresses live much longer than worms fed ad libitum (26, 43–46). These observations are consistent with the phenomenon of hormesis, in which adult longevity can be promoted by stresses that are strong but not debilitating (47, 48). We therefore asked whether prg-1 mutant lifespan is affected by the heritable stress transmitted by prg-1 mutant germ cells, by measuring longevity of early-generation, late-generation prg-1 mutants that were sterile and late-generation prg-1 mutants that were fertile. We found that early-generation prg-1 mutants, with low amounts of heritable stress, were not significantly different than wild-type animals (Fig 1B, S1 Table, p=1). However, fertile but stressed late-generation and sterile prg-1 mutant adults displayed significant 46.5% and 81.8% increases in median lifespan in comparison to wild-type controls, respectively (Fig 1B, S1 Table, p=0.0008 and p=1.55E-16) suggesting that increased levels of heritable stress in germ cells is sufficient to confer an increase in somatic longevity.
prg-1 mutant germ cells transmit longevity to heterozygous prg-1/+ progeny
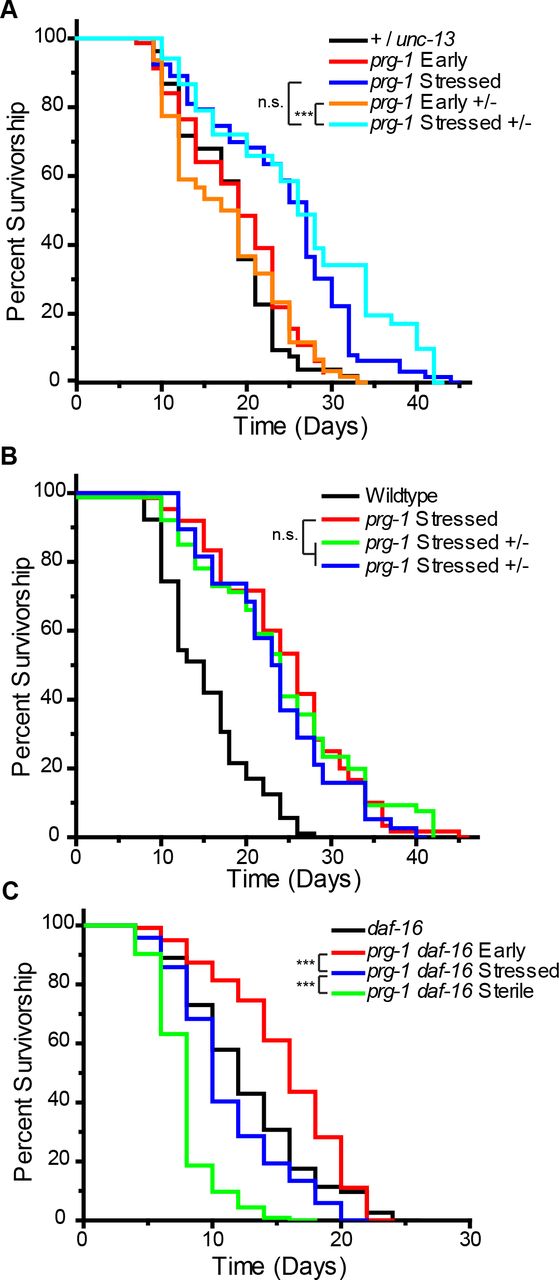
We tested the hypothesis that prg-1 mutant germ cells transmit a form of heritable stress that affects longevity by crossing late-generation prg-1 mutant hermaphrodites with wildtype males and measuring longevity of their F1 cross-progeny. We performed crosses of three independently derived lines of late-generation prg-1 hermaphrodites with males that were heterozygous for an unc-13 mutation and compared the lifespans of their prg-1 +/− F1 progeny with those of early- and late- generation prg-1 mutant controls. To ensure that the presence of the unc-13 balancer mutation did not confer an unexpected dominant effect on longevity, we also measured the lifespan of +/unc-13 heterozygous controls and found that these were not significantly different from early-generation prg-1 mutant controls (Fig 2A, S1 Table, p=1). During the initial days of the longevity experiment (reproductive period), we transferred adults to new plates every day until they stopped laying progeny. Although late-generation prg-1 mutant control adults give rise to very few progeny, we observed that their prg-1 +/− F1 progeny had a marked increase in brood size, demonstrating that the strong fertility defect of late-generation prg-1 mutants is not transmitted to their heterozygous progeny. However, all three independent lines of prg-1 +/− F1 progeny displayed significant increases in lifespan compared to +/unc-13, early-generation prg-1, and early-generation prg-1 +/−controls (Fig 2A,B, S1 Table, p>0.05 in all comparisons). Notably, the lifespan of these prg-1 +/− F1 cross progeny was not statistically different than late-generation prg-1 mutant adults (Fig 2 A,B, S1 Table, p>0.72 in all cases).

(A) Lifespan curves for +/unc-13, early or late (stressed) generation prg-1, and early or late prg-1 +/− F1 progeny. (B) independent lines of heterozygous (prg-1 +/−) late generation F1 progeny compared to homozygous non-crossed sisters. p=1 (C) daf-16 (mu86) is required for somatic lifespan extension in late and sterile generation prg-1 (n4357) mutants. n.s. is not significant, *** p<0.001, p values determined using log rank analysis and reported in S1 Table.
Heritable stress is detrimental to longevity in the absence of daf-16
The DAF-16 transcription factor is well known to promote longevity (3, 29, 49, 50) and has been previously shown to interact in three distinct ways with deficiency for prg-1. First, when daf-2 mutations that activate DAF-16 are introducedinto early-generation prg-1 mutants, DAF-16 promotes germ cell immortality and suppresses the transgenerational sterility defect of prg-1 mutants (12). However, knockdown of daf-2 by mutation or RNAi does not suppress the sterility phenotype of late-generation prg-1 mutants (25), indicating that DAF-16 may lose its ability to promote germ cell immortality in late-generation prg-1 mutants. A second function of DAF-16 occurs when prg-1 mutants become sterile. Most of these animals display a pronounced germ cell atrophy phenotype as L4 larvae mature in to 1 day old adults, but deficiency for daf-16 significantly alters the germline profiles of sterile prg-1 mutant adults, leading to reduced levels of germ cell atrophy (25). This implies that daf-2 signaling promotes germ cell atrophy as sterile prg-1 mutant L4 larvae mature into 1 day old adults. A third function of DAF-16 occurs for 5 day old sterile prg-1 mutants, which are in a state of prolonged reproductive arrest, a small fraction of which can become fertile if moved to an alternative food source (25). This exit from reproductive arrest requires DAF-16. We therefore asked whether the longevity of late-generation prg-1 mutants depends on DAF-16. Fertile and sterile late-generation prg-1 daf-16 animals lived significantly shorter than either early-generation prg-1 daf-16 double mutants (late p=3.53E-12 and sterile p=1.55E-35) or daf-16 single mutant controls (late p=0.028 and sterile p=2.71E-19), (Fig 2C, S1 Table), indicating that DAF-16 is responsible for the longevity observed in late-generation prg-1 mutants. Furthermore, sterile late-generation prg-1 daf-16 double mutants were shorter lived than their fertile late-generation siblings (Fig 2C, S1 Table, p=5.41 E-13) implying that a detrimental form of heritable stress is transmitted by late-generation prg-1 mutants.
Given that DAF-16 promotes the longevity of late-generation prg-1 mutants, we used a daf-16::GFP transgene that rescues the adult longevity defect of daf-16; daf-2 double mutants (51) to create prg-1; daf-16::GFP mutant lines. We found that early- and late-generation prg-1 mutant lines displayed DAF-16::GFP expression that was primarily cytoplasmic, similar to that of daf-16::GFP transgenic controls (Fig 3A,B). While DAF-16::GFP was only mildly nuclear in fertile late-generation prg-1 mutants (Fig 3C,E), we found that sterile late-generation prg-1 mutant strains displayed strong nuclear localization of DAF-16::GFP in somatic tissue, similar to that observed with daf-16::GFP; daf-2 mutant controls, where DAF-16 is constitutively activated (Fig 3D,E) (51).

(A) DAF-16::GFP remains primarily cytoplasmic in adult wild-type animals. (B) Mutant prg-1(n4357) animals display no nuclear localization of daf-16::GFP in early generation animals, but demonstrates partial nuclear localization in fertile late generation (C) and strong (D) nuclear localization in sterile generation adults. White arrows indicate nuclear localization. Scales bars = 50 μm. (E) P body aggregates visualized using dcap-11::GFP in wild-type, (F) early prg-1(n4357), (G) and stressed late generation prg-1(n4357) mutants. Scale bars = 50 μm. (H) Percent of wild-type, early generation prg-1, fertile late (stressed) generation prg-1, and sterile late generation prg-1 worms with no, partial, or full daf-16::GFP nuclear localization phenotypes. (n=25, 22, 22, 20 respectively). (I) Number of P body aggregates from (F-H). * is p=0.002 Kruskal Wallace test.
Transgenerational activation of P bodies in prg-1 mutants
Processing bodies (P bodies) and stress granules are typically cytoplasmic aggregates that are composed of proteins and RNA that function to protect RNAs under stressful conditions. P bodies appear under conditions of stress, including many types of environmental stress such as heat shock, radiation, osmotic stress, oxidative stress and ER stress (52). Given that prg-1 mutant germ cells may transmit a form of heritable stress that promotes somatic longevity, we crossed GFP-tagged component of P bodies and stress granules DCAP-1 (53) into prg-1 mutant backgrounds and monitored GFP expression in early- and late-generation strains. We observed that DCAP-1::GFP aggregates were rarely present in wild-type and early-generation prg-1 mutant strains but were significantly induced in late-generation sterile prg-1 mutants (Fig 3 F-I), similar to what has been previously observed under stressful conditions (53). These results are consistent the possibility that late-generation prg-1 mutants may experience a form of stress.
Discussion
daf-2 mutants have been previously reported to be hypersensitive to the exogenous RNAi pathway that normally targets foreign viruses and transposons (48). The PRG-1/Piwi silencing pathway also represents a form of immunity that protects the germ line from foreign genetic elements (54, 55). We found that deficiency for the prg-1-mediated genomic silencing pathway leads to a transgenerational sterility phenotype that can be suppressed by mutation of daf-2, via an endogenous systemic small RNA pathway that is activated by DAF-16 (see accompanying manuscript by Simon, Spichal, Heestand and colleagues). Given that DAF-16 can act cell-non-autonomously in distinct somatic tissues to promote longevity of daf-2 adults (56), we asked whether this systemic RNAi pathway was relevant to somatic longevity of daf-2 mutants and found it was not. This indicates that DAF-16 does not simply respond to stress by upregulating pathways that promote somatic stress resistance and longevity, but that it possesses a distinct function that promotes germ cell immortality when PRG-1/Piwi is deficient (12). Consistently, DAF-16 has been previously linked to the innate immune response to bacterial or fungal pathogens (57–60), an activity that is separable from its function in somatic longevity (57).
The disposable soma theory of aging posits that trade-offs occur when deciding on activating processes that benefit either somatic longevity or reproduction (5, 61). However, our study implies that these choices are not mutually exclusive and that stresses that compromise germ cell function can be counteracted by a pathway that promotes somatic longevity in times of stress. Our data suggests that a trade-off may occur for the DAF-16 pathway in the context of deficiency for prg-1 /piRNAs and lead us to propose the following model (Fig 4): In early generation prg-1 mutants, DAF-16 activity is wildtype from the perspective of somatic longevity and DAF-16::GFP fluorescence. In this context, DAF-16 can be activated transiently or constitutively to suppress the sterility phenotype of prg-1 mutants (12). In a population of wild animals that experience a virus- or transposon-based attack that compromises the PRG-1/Piwi silencing system (54, 55), germlines of those animals that experience strong bouts of environmental stress may be better able to withstand a genomic parasite. Indeed, the feast or famine lifestyle of C. elegans populations that rely on ephemeral food sources such as rotten fruits or vegetables may experience significant levels DAF-16 activity on a consistent basis, which may suppress dysfunction of prg-1/Piwi (62, 63). If, however, prg-1 deficient strains are transferred regularly under laboratory conditions of replete food, these strains transmit a form of heritable stress that builds up to the point where it engages DAF-16 to promote somatic longevity (Fig 1). Fertile late-generation prg-1 mutant strains were long-lived and showed a mild and transient DAF-16::GFP nuclear localization pattern. Given that genetic or RNAi knockdown of daf-2 in late-generation prg-1 mutant strains no longer ameliorates their sterility defect (25), we propose that if DAF-16 is already engaged in responses such as heritable stress-induced adult longevity that may preclude DAF-16from activating the systemic RNAi pathway that suppresses the sterility of prg-1 mutants (12). An alternative explanation is that the heterochromatin defect of late-generation prg-1 mutants is so severe that it can no longer be suppressed by activation of DAF-16.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Loss of prg-1 leads to a transgenerational increase in heritable htress which promotes somatic longevity through DAF-16/Foxo.
Support for the concept that heritable stress engages specific facets of the DAF-16 pathway comes from our observation that late-generation prg-1 mutants do not display dauer larvae that are commonly produced under circumstances of stress (Fig 1C) (42). Instead, DAF-16 specifically promotes longevity and reproductive arrest in late-generation prg-1 mutant adults (25), both of which may represent adaptive responses to the heritable stress transmitted by prg-1 mutant germ cells. We found that F1 cross progeny of late-generation prg-1 mutants were also long-lived yet lacked the strong fertility defect of their prg-1 mutant mothers, which suggests a dilution of the stress, but also partial separation between the stress and the organismal response to it. In the absence of daf-16, late-generation prg-1 mutants lived shorter than daf-16 single mutants, which are themselves short-lived (64, 65). This is consistent with the longevity of late-generation prg-1 single mutants being a hormetic stress response (47, 48), where stressis beneficial unless the relevant stress response pathway is inactivated.
Additional support for the concept of heritable stress is provided by the observation that strong increases in P bodies occur in late-generation prg-1 mutants. As P bodies or stress granules are created in response to many forms of stress (52), their presence implies that the heritable stress transmitted by prg-1 mutant germ cells induces a stress response in adult somatic tissues. P bodies or stress granules can promote viral resistance and aspects of the innate immune response (52), which are also properties of perinuclear germ granules that house the PRG-1/Piwi/piRNA genome silencing proteins (66). Transgenerational dysfunction of the PRG-1/piRNA silencing pathway leads to desilencing of transposons (12, 67, 68), and expression of one or more transposon classes could facilitate stress granule formation in a manner that promotes lifespan extension. While the precise identity of the heritable stress transmitted by prg-1 mutant germ cells remains uncertain, our data provide strong evidence that germ cells can transmit factors other than DNA mutations that can have a significant impact on longevity. We suggest that desilencing of heterochromatin of prg-1 mutant germ cells over the generations may perturb an essential biochemical pathway that corresponds to the heritable stress that leads to increased longevity and P body formation in late-generation prg-1 mutants.
Inactivation of members of the ASH-2 trithorax histone H3K4 methylation complex for a single generation promotes an adult longevity phenotype that can be transmitted transgenerationally in C. elegans (69). Functionally, ASH-2/trithorax transcriptional activation complex might be predicted to oppose the activity the PRG-1/piRNA genome silencing pathway, and it was demonstrated that the RBR-2 H3K4 demethylase that promotes genome silencing in germ cells is required for the longevity caused by inactivation of ASH-2/trithorax (69). RBR-2 is required for germ cell immortality at 25°C (70), likely because it acts downstream of PRG-1/piRNAs. Moreover, ASH-2 trithorax inactivation was capable of extending the lifespan of daf-16 mutants, whereas late-generation prg-1 mutants were very short-lived in the absence of daf-16 (Fig 2). Together, these results suggest that independent epigenetic defects can promote longevity in a manner that is heritable, such that knockdown of the ASH-2 trithorax transcriptional activation complex induces an immediate somatic longevity phenotype, whereas inactivation of the PRG-1/piRNA genomic silencing complex for many generations leads to a functionally distinct form of longevity.
Like RBR-2, the SPR-5 H3K4 demethylase is required for germ cell immortality (70). While this study was in progress it was shown that late- but not early-generation spr-5 mutants are long lived (71). However, daf-16 is not required for the longevity of late-generation spr-5 mutants (71), implying that deficiency for prg-1 and spr-5 lead to distinct transgenerational epigenetic defects that promote distinct mechanisms of longevity.
The transgenerational sterility of prg-1 mutants suggests a defect that accumulates as germ cells proliferate over the generations, which is potentially analogous to proliferative aging or senescence of mammalian somatic cells (72). Several links between aging and dysregulation of the epigenome have been documented, such as desilencing of retrotransposons in senescent human cells, and epigenetic silencing of retrotransposons by the anti-aging protein SIRT6 in the mouse (73–75). The induction of reproductive arrest in response to heritable stress therefore suggests a form of inheritance that is potentially relevant to epigenetic modulation of senescence of mammalian somatic cells.
Our study suggests that mutation of prg-1/Piwi can induce a form of heritable stress that is transmitted by gametes and can promote longevity, which could represent one facet of the evolutionary biology of aging in metazoans. An analogous form of non-Mendelian stress that is transmitted by germ cells could affect the rate of aging or adult onset disorders in mammals, potentially by modulating aging of post-mitotic somatic cells in a manner that depends on Foxo or by promoting replicative aging of proliferating cells.
Materials and Methods
Strains
Unless noted otherwise, all strains were cultured at 20°C on Nematode Growth Medium (NGM) plates seeded with E. coli OP50. Strains used include Bristol N2 wild type, unc-13(e450) I, prg-1(n4357) I, daf-16(mu86) I, kri-1 (ok1251) I, daf-2 (e1368) III, zIs356 [daf-16p::daf-16a/b::GFP + rol-6(su10060], Pdcap-1::dcap-11::GFP::3′ UTRdcap-1
prg-1 mutations were outcrossed versus an outcrossed stock of unc-13(e450), and freshly isolated homozygous F2 lines were established for analysis. To create the linked prg-1, daf-16 double mutant, prg-1, dpy-24 and unc-13, daf-16 double mutants were first created, then progeny of prg-1, dpy-24 / unc-13, daf-16 heterozygotes that lost unc-13 were identified, and the resulting putative prg-1, daf-16 recombinant chromosomes were made homozygous and PCR genotyped to verify the presence of prg-1 or daf-16 deletions.
Lifespan analysis
Animals were singled from non-starved plates and transferred every two days until the end of their reproductive cycles. Animals were kept uncrowded and free of contamination throughout experiment and transferred to new plates once per week. All animals were maintained at 20°C.
Statistical analysis
Statistical analysis was performed using the R statistical computing environment (76) and the survival R package (77). For lifespan experiments, pairwise log-rank tests were performed to detect significant differences between groups. For the P bodies experiment, a Kruskal-Wallace test was performed to detect the presence of any significant differences within the dataset. Pairwise Mann Whitney U tests were performed as a post-hoc test to detect the presence of significant differences between specific groups. All reported p-values were adjusted using Bonferroni correction to control the familywise error rate.
Competing interests
Authors declare no competing interests.
Author contributions
B.H. and M.S. performed experiments. S.F. analyzed the data. B. H. and S.A. wrote the manuscript.
Acknowledgements
We thank members of the Ahmed lab for critical reading of the manuscript. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This study was supported by NIH grant RO1 GM083048 (S.A).
References




