

Abstract
Animal venoms are considered sterile sources of antimicrobial compounds with strong membrane disrupting activity against multi-drug resistant bacteria. However, bite wound infections are common in developing nations. Investigating the oral and venom microbiome of five snake and two spider species, we evidence viable microorganisms potentially unique to venom for black-necked spitting cobras (Naja nigricollis). Among these are two novel sequence types of Enterococcus faecalis misidentified by commonly used clinical biochemistry procedures as Staphylococcus; the genome sequence data of venom-specific isolates feature an additional 45 genes, at least 11 of which improve membrane integrity. Our findings challenge the dogma of venom sterility and indicate an increased primary infection risk in the clinical management of venomous animal bite wounds.
One Sentence Summary Independent bacterial colonization of cobra venom drives acquisition of genes antagonistic to venom antimicrobial peptides.
Introduction
The rise of single and multi-drug resistant (MDR) bacterial infections suggests the end of the antibiotic golden era might be approaching fast. Discovery of novel antimicrobials is therefore an urgent priority of exceptional socioeconomic value. Crude preparations of animal venoms exhibit strong antibiotic potencies, including against clinical MDR bacterial isolates such as Mycobacterium tuberculosis (Xie et al. 2003). With antimicrobial properties described for crotalid (pit viper) venom as early as 1948 (Glaser 1948), relevant compounds have been isolated from most animal venoms, including those of spiders, scorpions, and insects, as well as aquatic species. Examples include phospholipase-A2 enzymes, L-amino acid oxidases, cathelicidins, C-type lectins, and hydrophobic/cationic peptides, as well as venom toxin domains (Samy et al. 2013), which may act by physically disrupting bacterial cell membranes through pore formation (Stocker and Traynor 1986; Perumal Samy et al. 2017). Accordingly, venomous animal bite or sting (envenomation) wound infections are considered rare (Talan et al. 1991) and are attributed to secondary infection (Powers 2005). Yet over three quarters of snake bite victims may develop mono- or polymicrobial envenomation wound infections, characterized by Bacteroides, Morganella, Proteus, and Enterococcus (Wagener et al. 2017; Mao et al. 2016) – bacterial taxa commonly found in the gut. Indeed, Enterococcus faecalis and Morganella morganii have been independently reported as the most common Gram positive and Gram negative infections across several countries (Mao et al. 2016; Lam et al. 2011; Wagener et al. 2017). Historically associated with the oral snake microbiome (Jorge et al.), these bacteria are thought to originate from prey faeces (Garg et al. 2009) persisting in the snake oral cavity (Lam et al. 2011) with a diversity similar to that of the snake gut (Iqbal et al. 2014). Yet no ‘fixed’ oral microbiome was observed in early systematic studies, beyond a seasonal variation of diversity (Blaylock 2001). Curiously, non-venomous snake mouths were reportedly more sterile than those of venomous snakes (Blaylock 2001), a counterintuitive finding independently reproduced elsewhere (Lam et al. 2011). More recently, the oral microbiome of the non-venomous Burmese python (Python bivittatus) has also been reported to be native and not derived from prey guts (Costello et al. 2010).
As venom glands are connected to the tip of envenomation apparatus via a persistently open duct which is continuously exposed to the environment (Trim and Trim 2013), envenomation apparatus could be compared to clinical catheterisation assemblies: a transcutaneous needle resting on a non-sterile environment, connected to a continually open duct, leading to a liquid vessel. Such devices develop biofilms within a few days, making weekly catheter replacement necessary (Loveday et al. 2014). Unlike the high flow rates of catheters, however, envenomation apparatus normally ejects venom only sporadically. Captive snakes are often fed weekly and can fast for months whereas large arachnids are fed typically on a monthly basis. Wild animals may also undergo hibernation for several months when venom expulsion frequency can be assumed to be zero. We therefore postulated that the anatomy of envenomation apparatus allows it to be colonised by microbes and that their intermittent use may facilitate bacterial persistence, adaptation and establishment within antimicrobial venom.
Results
The snake venom microbiome varies on account of host species and not on account of the oral flora
Applying established culture-free methods (Muyzer et al. 1993) on commercially available Bothrops atrox venom (fer-de-lance; viperidae) and a venom sample from a captive Bitis arietans (African puff adder), we first optimised microbial DNA extraction for this unusual biological matrix (Fig. S1). Given animal availability, behavioural, and sampling limitations, we next focused our efforts on five snake, two spider and two scorpion species (Table 1). We collected a swab (O) of the oral cavity (snakes) or fang/aculeus surface (spiders/scorpions) and two consecutive envenomation samples (E1 and E2), expecting the second venom sample to have fewer contaminants from bacterial plugs possibly forming on the envenomation apparatus. In agreement with previous reports (Jorge et al.; Blaylock 2001), principle coordinate analysis and unsupervised clustering (Fig. S2) failed to discriminate the swab microbiomes by host species, suggesting the common diets and water sources in captivity would have the biggest impact on the swab data.
Animals sampled for the presence of microbiomes in venom.
This lead us to hypothesise that captive animals would feature more closely related microbiota in their venoms compared to commercial or wild samples. We therefore compared the venom microbiomes of all snakes using the same approach (Fig. 1). High Shannon-Weiner indices indicated considerably diversity in snake venom microbiomes, however, surprisingly closer relationships were observed between B. arietans and other Viperidae, despite samples spanning captive and wild animals; an exception was B. atrox venom, which was characterized principally by Gammaproteobacteria. Focusing on B. arietans also failed to cluster samples by origin (Fig. S2), despite the disparate locations across South Africa where wild B. arietans samples were collected (Fig. 1D). In contrast, N. nigricollis microbiomes largely formed a distinct cluster (Fig. 1) characterized bacteroidia (bacteroidaceae), a taxon less common among Viperidae. This could reflect anatomical differences in elapid (cobra) fang location at the front of the mouth compared to the sheathed nature of the longer, hinged viperid fangs, whose tips rest at the back of the oral cavity. In contrast, spider species did not seem to influence venom microbiome consistency and exhibited lower biodiversity (Fig. S4). These results likely reflected vertebrate/invertebrate anatomical differences and the limited venom yield from invertebrates (<1-30 μl) vs snakes (100-1,000 μl).

Snake venom microbiomes cluster on account of host species. Viperid venom microbiomes cluster separately from N. nigricollis with the exception of B. atrox as determined by A) PCoA, B) UPGMA tree and C) class-level taxonomic profiling following 16S rRNA phylogenetic analysis. Dots in (A) and (B) are coloured by species (red: B. arietans; green: N. nigricollis; light blue: B. atrox; orange: C. atrox; dark blue: O. scutallatus), represent data of captivity individuals, are labelled with short species name, enumerated for individual number and identified for the envenomation number (E1 or E2) of the sample. The 8 wild (red dots B1-B8) and the commercially sourced, lyophilised (red L dot) B. arietans sample are independently labelled. Relative taxonomic diversity profiles in (C) are aligned to the UPGMA tree sample labels, with the Shannon-Weiner Index (SWI) of each sample indicated. The geographical origin of the wild B. areitans samples collected in South Africa are shown in D.
A fifth of the N. nigricollis venom microbiome is distinct to that of fangs
Encouraged by the distinctive bacterial taxonomies in N. nigricollis venom, the availability of animals under controlled conditions, and the paired nature of the fang swab and envenomation samples, we delved deeper into this dataset. Thus, fang microbiomes appeared to form a distinct cluster to that of venom microbiomes (Fig. 2A, B) suggesting the venom gland might be a distinct ecological niche (Fig. 2C). We therefore asked if any bacterial taxa were unique to subsets of these samples. Operational taxonomic unit (OTU) incidence analysis within each animal (Fig. 2D) suggested some 60% of OTUs were shared between corresponding venoms and fangs. Yet, importantly, up to 20% of these appeared to be unique to venom, and some 15% were unique to the fang (Fig. 2E), indicating an OTU continuum between the two microenvironments, with unique taxa in each site. Common sample types also featured a majority of common taxa, and OTUs unique to each site in each animal (Fig. 2F). However, taxa unique to each sample type (O, E1 or E2) were rarely found across all animals. These results suggested that although the microbiome between each snake fang and venom was largely common, venom contained distinct organisms.
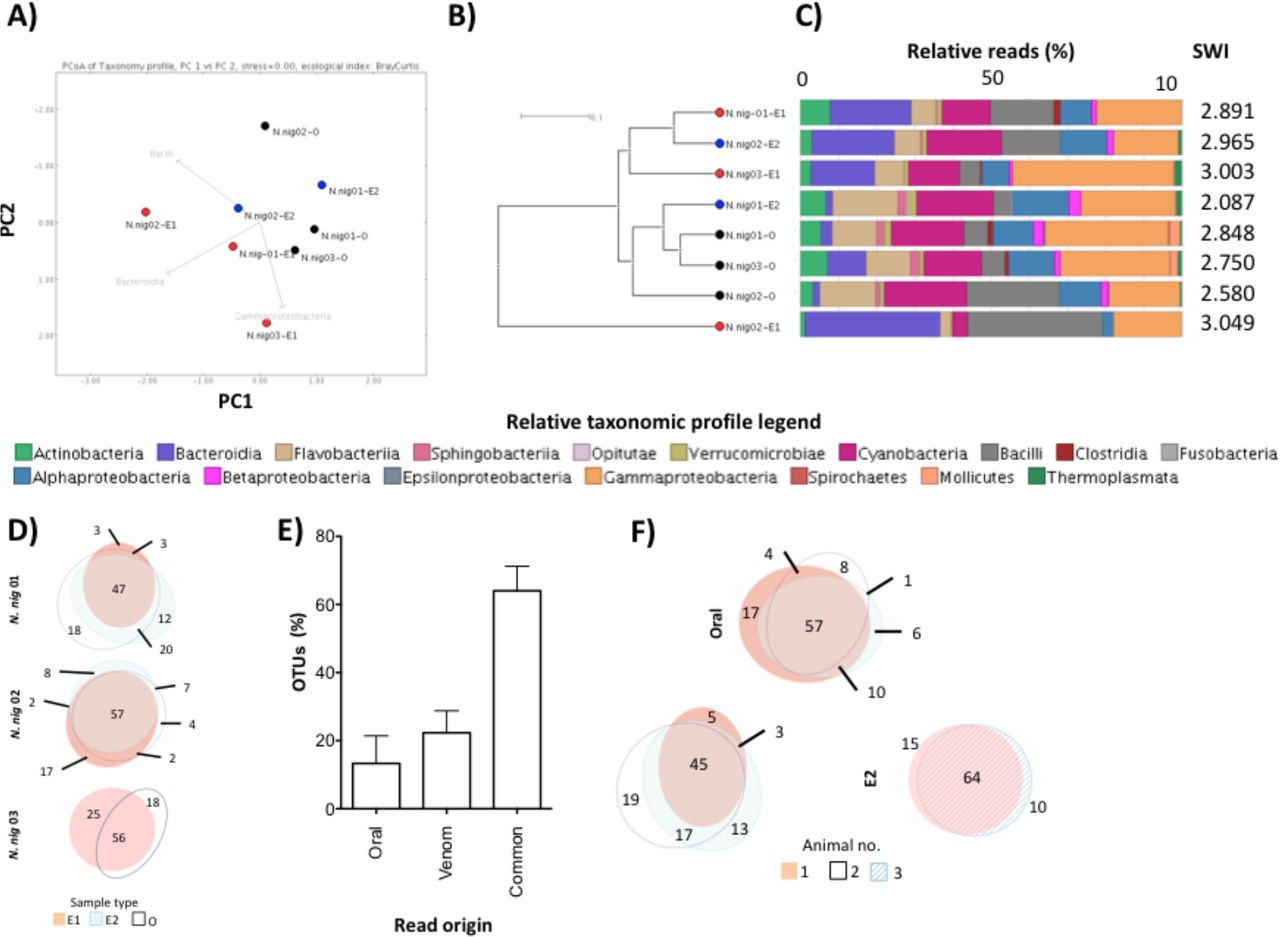
Comparison of the oral and venom microbiomes in three N. nigricollis individuals by A) PCoA, B) UPGMA tree and C) class-level taxonomic profiling following 16S rRNA phylogenetic analysis indicates separate clustering of the microbiotae in the two microenvironments. D) Within animal incidence comparisons of operational taxonomic units (OTUs) suggest E) unique taxa exist within the oral but also the venom microenvironments. F) Between animal comparisons per niche (E1, E2, Oral) indicate most OTUs are shared but some OTUs are unique to each animal for each site. Dots in (A) and (B) represent individual N. nigricollis (N.nig) animal data and are coloured/labelled by sample type (black: oral; red: envenomation 1 (E1); blue: envenomation 2 (E2)). Relative taxonomic diversity profiles in (C) are aligned to the UPGMA tree sample labels, with the Shannon-Weiner Index (SWI) of each sample indicated. The ‘venom’ histogram in E) represents the sum OTU fraction found in the two envenomation samples per individual (+/- standard deviation).
The venom flora in snakes and spiders is viable
Testing microbiome viability on identification agar (Supp. Table 1) yielded less growth with swab samples. Where this was significant, it was not usually matched by similar growth from the corresponding venom samples, further suggesting that the venom bacteria were probably not mouth contaminants. Strikingly, substantial and consistent growth was encountered amongst N. nigricollis (Fig. 3A) and P. regalis (Table S1) samples on blood agar. Unexpectedly, neither the wild (air dried) nor the commercial (lyophilised) venom samples yielded any growth, although colonies were obtained in blood agar from the captive B. arietans, underscoring the impact of venom handling on microbiome viability.
Well-established clinical microbial biochemistry tests identified the multiple, punctate white colonies from N. nigricollis almost universally as Staphylococcus spp., albeit with assay confidence intervals (CI; TABLE S2) below 50%. In contrast, Stenotrophomonas maltophila (80.4% CI) was present in five out of six P. regalis (all animals positive) and two Lasiodora parahybana (salmon pink tarantula) venom samples, but not on any fang swabs. Perplexed by the N. nigricollis results we sequenced the isolates on the Ion Torrent PGM.

(A) White punctate colonies were recovered in blood agar (BA) and MacConkey agar (MAC) blinded cultures of individual oral swab (O) and two consecutive envenomation samples (E1 and E2) obtained from three captivity N. nigricollis snakes. N/D: none detected. (B) Blinded multiple sequence alignment (ClustalO followed by ClustalW phylogeny) of homologous sequences across the de novo assembled genomes against the E. faecalis V583 KatA gene (distance to V583 KatA indicated in V583 track) suggests two sequence groups reflecting the history and housing of the host animals. (C) Blinded MST construction based on the MLST of the N. nigricollis-derived isolates against nine E. faecalis reference genomes again separate samples into two distinct clusters that reflect the history and housing of the host animals. Partially available allele data are included in this analysis and allelic difference instances between nearest neighbours are annotated in white boxes. (D) Blinded complete genome MLST against a custom schema generated using three closely related E. faecalis reference genomes cluster these isolates by animal of origin (animals 1 (light blue), animal 2 (dark blue) and animal 3 (orange)). The host animal colour scheme depicted in D) is also used in B) and C).
Viable bacteria in N. nigricollis venom are two new E. faecalis sequence types
Resequencing against putative reference genomes (Table S2) demonstrated less than 6% base alignment across all isolates. Instead, BLASTn analysis of the largest contig per isolate after de novo assembly identified E. faecalis V583 as the closest likely relative, resulting in >80% base alignment, at an average coverage of 51.2x. This was puzzling given the catalase positive isolate biochemistry vs. the generally accepted catalase negative nature of E. faecalis (Fisher and Phillips 2009). However, the E. faecalis V583 katA gene, previously reported to encode a haem-dependent cytoplasmic catalase (Frankenberg et al. 2002), was confirmed by BLASTn amongst all isolates at 99% identity, explaining the biochemical misclassification. Blinded multiple sequence alignment (MSA) further revealed two katA alleles: one shared between isolates from animals 1 and 2 (allele 1) vs. another found in animal 3 isolates (allele 2; Fig. 3B) varying by less than 20 single nucleotide polymorphisms to the V583 allele (Fig. S5A). Interestingly, these alleles grouped isolates according to the origin and joint housing histories of animals 1 and 2 vs animal 3.
To explore isolate relationships further we generated minimum spanning trees (MST; Fig. 3C) by multi-locus sequence typing (MLST; Table 2), including at core genome level (cgMLST; Fig. 3D and Fig. S5B-D) (Mellmann et al. 2011; de Been et al. 2015). Comparisons to five complete reference genomes of the closely related E. faecium succeeded only for the gyd (alelles 16, 19) and adk (allele 18) loci. In contrast, MLST succeeded for all E. faecalis loci (Table 2), grouping isolates in line with the katA allele observations (Fig. 3B), and identifying two novel sequence types featuring two new alleles for pstS and yqiL (Fig. S6) as confirmed by Sanger sequencing. MLST also indicated closer relationships to the E. faecalis strains OG1RF, D32, and DENG1, with 87.5% +/- 1.7 of OG1RF cgMLST targets accepted for distance calculations vs D32 (78.8% +/- 2.1) and DENG1 (77.4% +/- 1.5). Pairwise comparisons of the resulting custom cgMLST schema including 5041 loci found across all the N. nigricollis-derived isolates also grouped isolates by their host animal (Fig. 3C) in line with the katA and MLST locus observations. Collectively, these results indicated independent acquisition of two separate and novel E. faecalis strains across these three animals.
Novel sequence types of E. faecalis recovered from fangs and venoms of N. nigricollis.
Genomic evidence of E. faecalis isolate adaptation to venom
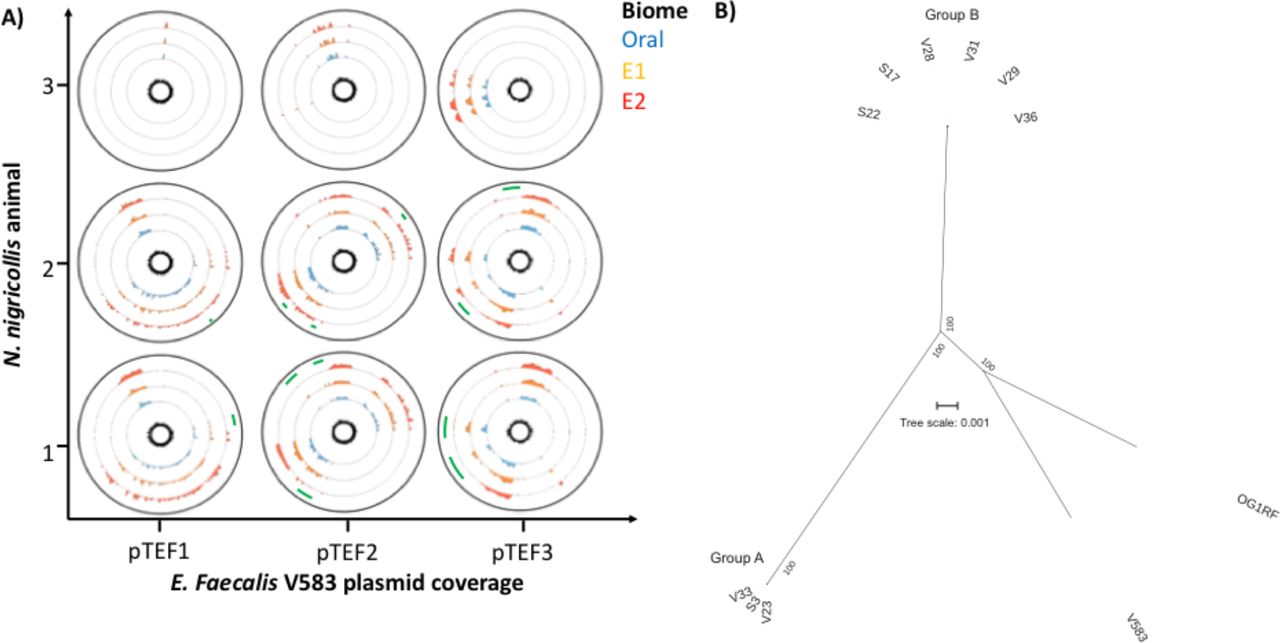
Including in cgMLST comparisons an additional 3060 loci found in some, but not all of the isolates (Fig. S5D) identified between 290 and 831 allelic differences occurring within each animal. Furthermore, whilst 80.9% of alleles varied between the two nearest neighbour isolates from the two strains, venom isolates from animals 1 and 2 were divergent by 7.15 – 10.3% to their oral isolate counterparts. Given the well-described plasticity of the E. faecalis genome (OG1RF: 2.74 Mb vs V583: 3.36 Mb), we next examined mobile genetic element divergence. Detecting the repA-2 gene from plasmid pTEF2 (Table S3) amongst all isolates in accordance to MLST-derived isolate groupings suggested only plasmid fragments were found in these genomes. However, as pTEF2 is one of three E. faecalis V583 plasmids associated to vancomycin resistance (Paulsen et al. 2003), we confirmed fragments from all three pTEF plasmids (Fig. 4A), consistent to MLST profiles (Table S4, Table 2), and with some pTEF sequence elements not found in secondary envenomation isolates from animals 1 and 2. Thus, the average per base read pTEF1/pTEF2 ratios for isolates from animal 3 were 0.25 (+/- 0.01), as elements with high (>95%) sequence identity to these plasmids were also known, highly mobile sequences common to other plasmids (e.g. the E. faecalis Bac41 bacteriocin locus) (Kurushima et al. 2016). These results therefore indicated the presence of highly mobile sequences in these compared to 0.90 (+/-0.15) and 0.81 (+/-0.05) for isolates from animal 1 and 2, respectively; similarly, the pTEF2/pTEF3 average per base read ratios were 0.68 (+/-0.02) for isolates from animal 3 vs. 1.57 (+/-0.04) for isolates from animal 1 and 2. Most importantly, however, many of the genomic isolates, either on plasmids or integrated in the bacterial chromosome, that appeared to participate in the observed genomic divergence of E. faecalis within each animal.

(A) Circos coverage plots of the vancomycin resistance-associated, V583 plasmids pTEF1, pTEF2 and pTEF3 in the E. faecalis isolates obtained from oral, envenomation 1 (E1) and envenomation 2 (E2) samples from three N. nigricollis individuals, reinforce the two sequence type groupings and highlight within animal variation (green arcs) indicative of sample-specific variation (lack of reads) across E2 samples in animals 1 and 2. The central plot for each plasmid and animal reflects GC content. All data is represented in 50 nt blocks. (B) Blinded maximum likelihood tree of the core genomic alignments for the 6 N. nigricollis- derived E. faecalis isolates against the V583 and OG1RF reference strains.
Closer examination of the draft genome assemblies indicated that the three E. faecalis isolates from animal 3 shared close genomes of ~2.9 Mb with 2,772 to 2,836 genes. The rest of the genomes varied from 3.04 to 3.24 Mb in size, and encompassed 3,128 to 3,282 genes (Table S3). The E. faecalis pan-genome, including the strains OG1RF and V583, contained a total of 5,130 genes of which 1,977 belonged to the core genome. As with other analyses, the core genomic diversity separated the snake-derived strains into two major groups (Fig. 4B): three isolates with smaller genome sizes corresponded to animal 3 forming group A, and the remaining six isolates with larger genome sizes forming group B. However, in line with cgMLST data (Fig. 3C) OG1RF and V583 were quite distinct from both of these groups. Comparison of the annotated genomes indicated that 235 genes that were specific to group A and were absent from group B isolates (Table S5). The majority of these genes (144/235) encoded hypothetical proteins. Among the rest, two genes were involved in capsule biosynthesis, and one pesticidal crystal protein cry22Aa, which may contribute to the virulence or fitness of these isolates. In addition, three genes belonged to a CRISPR-Cas type II system, a prokaryotic immune system that protects bacteria from invading nucleic acids, including bacteriophages and plasmids; these were absent from other isolates. However, a small proportion of the genes were mobile genetic elements that encoded phage associated proteins, integrases, and transposes (Table S5), further supporting the hypothesis that these group A-specific genes might have been acquired by recombination. Of the 321 genes specific to group B, again the largest fraction (203/321) encoded hypothetical proteins (Table S6). However, a number of genes encoded phage-associated proteins and transposases.
Most interestingly, examination of the nine genomes from the snake-derived isolates identified 45 genes unique to them and absent from the OG1RF and V583 reference genomes. In contrast to Group A- and Group B-specific genes, 21 (46.7%) of these 45 genes were of known function (Table S7). UniProt functional annotation indicated that 11 of these 21 (52.4%) genes were associated to cell wall/membrane integrity, with four additional genes (19.1%) associated to pathogen foreign protein and toxin defence. Pathway analysis via DAVID using B. subtilis orthologues identified significant enrichment (11.11 fold, p=0.00802) in the two component system pathway, specifically genes responsive to cationic antimicrobial peptides, cell wall active antimicrobials, bacitracin efflux, and salt stress.
Projecting primary infection clinical risk from venom-tolerant E. faecalis isolates
Given these viable E. faecalis strains could potentially infect envenomation wounds, we next examined the genomic data for known resistance determinants that might facilitate opportunistic primary infection. None of these strains had any acquired resistance genes to any antimicrobial classes (Table S8), although all isolates featured lsaA, which confers intrinsic streptogramin resistance to E. faecalis (Singh et al. 2002): MSA further reinforced isolate grouping based on host animals (Fig. S7). Since horizontally acquired genes are largely responsible for resistance to vancomycin, aminoglycosides, macrolides, and tetracycline in Enterococcus (Hollenbeck and Rice 2012), these strains were considered likely to be susceptible to drugs in each of these drug classes. In addition, the absence of known resistance-associated mutations in gyrA, parC, and the 23S rRNA genes also increased the likelihood that these isolates would be susceptible to oxazolidinones and fluoroquinolones. Thus, these data indicated that several available antimicrobials would likely be effective in treating infections caused by these strains of E. faecalis. However, a gene related to macrolide export (macB5) was detected in the venom microbiome strains (Table S7).
On the other hand, one additional concern was the potential misidentification of these isolates as Staphylococcus using standard diagnostic methods: this could potentially impact treatment decision-making. Although many of the same antibiotics, including vancomycin and linezolid, would be considered for treatment of both staphylococci and enterococci, there are some potential differences. For instance, oxacillin is often employed as a first-line agent to treat Staphylococcus (Andrews et al. 2000). This drug is not effective against enterococci, as the use of penicillins for E. faecalis infections would typically involve ampicillin, usually in combination with an aminoglycoside (Arias et al. 2010). In addition, cephalosporins such as cefotaxime are considered second-line therapies for coagulase-negative staphylococci such as S. epidermidis (Aldridge). However, enterococci are intrinsically resistant to this class of drugs, and their prevalence in the gut actually tends to increase in response to cephalosporin therapy (de Vries-Hospers et al. 1991). Thus, while the E. faecalis strains in this study did not have any known acquired resistance determinants, if they were to cause infections, ensuring their proper identification would be critical to issuing correct treatment and achieving positive clinical outcomes.
Discussion
In contrast to the generally held view that venoms are both antimicrobial (Xie et al. 2003; Glaser 1948; Samy et al. 2013; Stocker and Traynor 1986) and sterile (Talan et al. 1991; Palappallil 2015; Powers 2005), despite contrasting reports since the 1940’s (Pasricha and Abedin 1941), we show that microorganisms are common and viable in the venoms of both vertebrates and invertebrates. As with previous work (Jorge et al.; Garg et al. 2009; Iqbal et al. 2014), our data support the precept that prey faeces might seed the oral and the venom microbiome. However, significant adaptation takes place in these bacteria to persist in venom, occurring in parallel across distinct bacterial isolates and host animals probably through horizontal gene transfer, to contribute genes unique to these isolates. Of the genes with known function, an unusually large fraction (71.5%) is involved in maintaining bacterial membrane integrity or toxin defence. Notably, disruption of membrane integrity appears to be the most common mechanism of action for known, venom-derived antimicrobial peptides and enzymes (Stocker and Traynor 1986; Perumal Samy et al. 2017) of long-standing (Glaser 1948) and significant biotherapeutic interest, including MDR (Xie et al. 2003) and nociception (Trim and Trim 2013). It is unclear at present to which extent this form of parallel convergent evolution extends beyond Enterococcus or other antimicrobial resistance mechanisms, such as antibiotic resistance genes against last resort antibiotics found on mobile genetic elements (Liu et al. 2016; McGann et al. 2016) found in multiple continents.
From a clinical perspective, identification of E. faecalis as the most prevalent culturable microbe across our European N. nigricollis venom samples strikingly reflects three independent clinical reports across Africa and Asia that this non-sporulating microbe is the most common Gram positive infection cultured from infected envenomation wounds (Mao et al. 2016; Lam et al. 2011; Wagener et al. 2017). Further epidemiological data from countries with high envenomation incidence (Mao et al. 2016; Lam et al. 2011; Wagener et al. 2017; Johnston et al. 2017; Monteiro et al. 2002) challenge the consensus view in developed nations that venom is sterile, opportunistic primary infection upon envenomation is uncommon, and that venom wound infection is a consequence of poor hygiene or poor debridement practice (Talan et al. 1991; Powers 2005). Accordingly, in an experimental rabbit model of dermonecrosis (Monteiro et al. 2002) caused by Loxosceles intermedia (recluse spider) venom, Clostridium perfringens recovered from the spider fang and venom enhanced disease symptoms. Stenotrophomonas-like bacteria were also found to dominate cone snail venom microbiomes (Torres et al. 2017), indicating that microbial venom adaptation may extend well beyond snakes, spiders, scorpions, and snails. The cone snail study also reported comparable microbiomes in samples collected across the Pacific basin as well as Atlantic specimens. Furthermore, building upon the few instances of polymicrobial infection reported clinically (Mao et al. 2016; Lam et al. 2011; Wagener et al. 2017), the reports on L. intermedia (Monteiro et al. 2002), Conus (Torres et al. 2017), and herein suggest that diverse microbes effectively co-colonise venom glands in host species-specific manner, and thus envenomation wounds. Taken together, these studies support a review of the current standards of care for envenomation wound management (Palappallil 2015) beyond simply managing the severe tissue damage and necrosis that might be caused by venomous bites, to include clinical microbiology on envenomation wounds upon presentation. This would be particularly relevant to individuals immunocompromised through disease or malnutrition, e.g. in developing nations where envenomation incidence rates are high, or to children on a venom/colony forming unit dose per body weight basis.
Yet common diagnostic methods mistook E. faecalis for Staphylococcus, which could lead to unfavourable clinical decision making. It is unclear at present how common such misidentification events might be, contributing to MDR through inappropriate antibiotic use. At least one retrospective study reported higher incidence of Staphylococcus spp. in envenomation wounds (Garg et al. 2009), and Blaylock’s seminal snake oral flora studies also reported Proteus and Staphylococcus (Blaylock 2001): both relied on the same methods we used that resulted in pathogen misidentification. Our results therefore further support use of PCR/sequencing methods as they become more relevant to resource limited settings (Quick et al. 2016), and suited to the point of need (Shah et al. 2017), in line with World Health Organisation ASSURED criteria. Understanding the sensitivity of these methods will be crucial in their reliable implementation in envenomation care. It is therefore noteworthy that despite the limited biomass levels in these samples, species-level OTU analysis on MG-RAST (Wilke et al. 2016) correctly identified E. faecalis as one of the principle aerobic isolates in N. nigricollis venom. Thus, a simple phylogenetic or metagenomic approach, combined with local herpetogeography knowledge, could quickly and accurately inform clinical action regarding antivenom administration. This is because, unlike incidents involving venomous pets, envenomating animal capture is both rare and contraindicated to minimise further envenomation injuries (Mao et al. 2016). Culprit animals are also commonly misidentified through inadequate description (Mao et al. 2016), and antibody-based venom identification kits have so far proven unreliable (Johnston et al. 2017), generally complicating antivenom selection.
To conclude, we evidence that vertebrate and invertebrate animal venoms host diverse, viable microbiomes, with isolates genetically adapted to venom antimicrobials of medical interest against MDR. These results challenge perceptions on the sterility of venom and absence of primary infection risk upon envenomation, pointing to modern nucleic acid testing technologies to better inform envenomation care and antibiotic use.
Materials and Methods
Animals, sampling and microbial culture
All samples analyzed in this study were provided by Venomtech Ltd., with the exception of freeze-dried B. arietans venom (Latoxan, Portes les Valence, France) and field collected samples collected in South Africa. Briefly, captive animals were housed in 2 m by 1m wooden, glass fronted vivaria with large hide, thermal gradient and water ad-libitum. All procedures for venom collection and swabbing were approved as unregulated under the Animals (Scientific Procedures) Act 1976. Venom was collected by standard techniques; briefly snakes were restrained behind the head and presented to a collection vessel. Snakes freely bit into the vessel until envenomation was observed. Each snake was presented to two sterile collection vessels in succession, one for the first envenomation with potential fang plug, and the other for the second flow (labelled E1 and E2, respectively). While the snake was positioned over the second vessel, the oral cavity was swabbed with a sterile swab with individual collection tubes (invasive sterile swab with transport media, DeltaLab, VWR, Lutterworth, UK). The venom collection vessels were clear, sterile 125 ml polypropylene containers (ThermoFisher Scientific Ltd., Paisley, UK) covered by 2 x 9 cm2 pieces of parafilm stretched to fit (ThermoFisher Scientific Ltd.). The collection vessel was secured to a bench during collection. Samples collected in the field were from wild puff adders sampled as part of a previous phylogeographic study (Barlow et al. 2013). Venom samples were collected using a similar method to that described for captive animals, except that the entire venom sample was collected in a single collection vessel. Samples were lyophilised by storing <100 μl venom aliquots in a vacuum-sealed container that was half-filled with silica gel. Following drying, venom samples were stored in a refrigerator at 5°C.
Lasiodora parahybana and Poecilotheria regalis were housed in 5 and 8 litre polypropylene boxes (Really Useful Products Ltd, Normanton, UK), respectively, with moist vermiculite (Peregrine Livefoods Ltd., Ongar, UK), plastic hide and 5 cm water bowl, as previously described.58 Arachnids were anaesthetised with a rising concentration of carbon dioxide, the fangs were swabbed with a sterile swab which was then placed in an individual 1 ml sterile, DNA free, polypropylene collection tube (FluidX Ltd, Nether Alderley, UK), and venom was subsequently collected from arachnids by electrical stimulation. All samples were stored at −80°C. The same transport swabs (VWR) as those used for snakes were also used for invertebrate oral / aculear swabbing. Samples were stored at 4°C and cultured within 24 hours of collection. Aerobic microbial viability was determined by plating swabs or aliquoting 10 μl volumes of venom samples onto oxalated whole horse blood agar, MacConkey agar, or mannitol salt agar (ThermoFisher Scientific) plates and incubating at 30°C for 72 hours. Biochemical isolate identification was undertaken using API® strips (20E, 20NE and Staph) interpreted via the APIWEB interface (BioMerieux, Basingstoke, UK). All isolates were stored on beads at −80°C at the University of Westminster microbial isolate library. Naja nigricollis sub-culture was performed by restoring cryogenically stored bacteria on lysogeny broth agar (ThermoFisher Scientific) grown for 48 hours at 30°C, and single colony overnight culture in lysogeny broth (ThermoFisher Scientific) using aerated culture (300 rpm).
DNA extraction
Neat venom samples or samples diluted in 18 megaohm water previously confirmed as bacterial DNA free by 16S PCR were subjected to DNA extraction using TRIzol™, PureLink Genomic DNA kits or MagMAX Cell-Free DNA kits (ThermoFisher Scientific) according to the manufacturer’s instructions. For combined extraction of Gram positive and Gram negative bacteria from liquid samples, diluted samples were split in equal volumes and processed according to the manufacturer’s Gram wall specific lysis protocols, with lysates combined prior to DNA binding onto columns by simple admixture. DNA content was then analyzed by Nanodrop (ThermoFisher Scientific) spectrophotometry and purified material was stored at −80°C until further analysis.
16S phylogenetic library preparation and sequencing
For short amplicon library preparation, the hypervariable V3 region of the 16S rDNA gene was amplified from 20 ng of DNA using the primers 5’-CCTACGGGAGGCAGCAG-3’ and 5’-ATTACCGCGGCTGCTGG-3’ (Integrated DNA Technologies BVBA, Leuven, Belgium),(Muyzer et al. 1993) 1U Platinum® PCR SuperMix High Fidelity (ThermoFisher Scientific) and 10 μM of primer-mix. The reaction mixes were incubated at 94 °C for 5 min followed by 30 cycles of 30 seconds at 94 °C, 30 seconds at 55 °C and 1 min at 72 °C and then final elongation at 72 °C for 10 min using a Techne Prime Thermal cycler (ColePalmer, Staffordshire, UK). PCR products (193 bp) were confirmed by 2% w/v agarose gel electrophoresis in TAE buffer (ThermoFisher Scientific).
NGS library preparation was carried out using the Ion Plus Fragment Library Kit according to the manufacturer’s instructions (Rev. 3, ThermoFisher Scientific), except that reactions were reduced to 1/5th volumes. Pooled libraries were diluted to ~26 pM for templating on the Ion OneTouch 2 system (ThermoFisher Scientific) using the Ion PGM Template OT2 200 v2 kit according to the manufacturer’s instructions (Rev. B, ThermoFisher Scientific). Templated samples were sequenced on the Ion Torrent Personal Genome Machine (PGM; ThermoFisher Scientific) system on a single 318 Ion Chip (ThermoFisher Scientific) using the Ion PGM™ 200 Sequencing kit according to the manufacturer’s instructions (Rev G., ThermoFisher Scientific).
Whole genome sequencing
DNA extracted from cultured isolates was mechanically sheared using the Covaris S220 Focused-ultrasonicator (Covaris, Brighton, UK). NGS libraries were generated using the NEBNext Fast DNA Library Prep Set for Ion Torrent (New England Biolabs, Hitchin, UK). Pooled samples were size selected with the LabChip XT (LabChip XT DNA 300 Assay Kit; PerkinElmer, Seer Green, UK) and diluted to 26 pM for templating with the Ion OneTouch 2 system using the Ion PGM Template OT2 200 kit. Templated samples were sequenced on the Ion PGM using the Ion PGM™ Sequencing 200 v2 reagent Kit (ThermoFisher Scientific) and Ion 318™ v2 Ion Chip (ThermoFisher Scientific).
Bioinformatic analyses
Raw Ion Torrent sequencing data reads were quality controlled and demultiplexed using the standard Ion Server v. 4.0 pipeline (ThermoFisher Scientific). Referenced and de novo assemblies were carried out using TMAP v.4.0 and the SPAdes plugin in the Ion Server. Phylogenetic data analyses were carried out after independent data deposition and curation on the MG-RAST v.3.0 pipeline (Wilke et al. 2016) (project IDs MGP5177 and MGP5617) which uses a BLAST approach and the EBI-METAGENOMICS v.1 (project ID ERP004004) pipeline (Hunter et al. 2014) which uses a Hidden Markov Model approach. Raw 16S sequencing reads were deposited in the European Nucleotide Archive (PRJEB4693). Quality control for both resources included length and quality filtering followed by a dereplication step where sequences with identical 50 nucleotides in 5′ positions were clustered together. MG-RAST taxonomy annotation involved RNA identification using VSearch, and assignments using a custom database generated by 90% identity clustering of SILVA, GreenGenes and RDP prokaryotic databases. EBI-METAGENOMICS identified rRNA using Hidden Markov Models present in the RDP databases and assigned taxonomy using Qiime and the GreenGenes prokaryotic database.
For post-processing analyses, the EBI-curated dataset was analyzed using MEGAN v.5.5.3 (Huson et al. 2016). Classical multi-locus sequence typing (http://efaecalis.mlst.net/) and cgMLST (Mellmann et al. 2011; de Been et al. 2015) were carried out using Ridom SeqSphere+ v.4.0 running on a 2 core, 10 GB RAM, 500 GB hard disk Biolinux v.8.0 installation on a VirtualBox virtual machine instance on a 16GB RAM, 1TB hard disk Apple iMac. Extended cgMLST analysis to include partially detected loci, excluded loci annotated as ‘failed’ due to sequencing error suggesting genuine E. faecalis genomic divergence occurring within each animal. Plasmid detection was carried out using the PlasmidFinder v.1.3 server (Carattoli et al. 2014), followed by NCBI BLASTn analysis to detect shorter fragments, e.g. the same 398 nt fragment of repA-2 in animal 3 isolates (<40% of the full-length gene) at 90.1% identity to the plasmid-borne reference sequence. Single gene comparisons and multiple sequence analyses were carried out using TCoffe and MView on the EMBL-EBI server, with base conservation visualized by BoxShade v.3.3.1 on mobyle.pasteur.fr. Genome-level plasmid coverage analyses were carried out by NCBI BLASTn and comparisons were visualized using Circos v.0.69-4.
The sequencing reads were assembled using SPAdes v.3.9.0 (Bankevich et al. 2012), and the draft assemblies were annotated using Prokka (Seemann 2014) before NCBI deposition (BioProject No. PRJNA415175). The genome sequences of E. faecalis strains V583 and OG1RF (Accession numbers NC_004668.1 and NC_017316.1, respectively) were obtained from GenBank and were re-annotated using Prokka to have an equivalence of annotation for comparative analyses. The genomes were compared using the program Roary (Page et al. 2015; Tange). A maximum- likelihood tree was constructed from the core genomic alignment using IQ-Tree (Nguyen et al. 2015) with 100,000 ultra-fast bootstraps and 100,000 SH-aLRT tests. The tree was visualized using Interactive Tree Of Life (iTOL) (Letunic and Bork 2016). To identify acquired resistance genes, nucleotide BLAST analysis was performed on the ResFinder (Zankari et al. 2012) and NCBI (https://www.ncbi.nlm.nih.gov/pathogens/) resistance gene databases using cutoffs of 50% length and 85% identity to known resistance determinants. Additional BLAST analysis was performed to identify single nucleotide polymorphisms in the quinolone resistance determining region (QRDR) of gyrA and parC (Kanematsu et al. 1998). Additional mutational analysis was performed on region V of the 23S rRNA-encoding genes (Marshall et al. 2002).
BLASTP was performed in Ensembl Bacteria (release 38), against the E. faecalis V583 and E. faecalis (GCA_000763645) to obtain further geneID’s from significant matches. Bacillus subtilis orthologue gene ID’s were collated as this species is the closest relative to E. faecalis (VetBact.org) with the most comprehensive genome annotation required for gene ontolgy and KEGG pathway analysis. From the 45 genes unique to venom isolates, useable B. subtilis GeneID’s were obtained for 22, of which 12 of these successfully converted to ENTREZ Gene ID’s using the functional annotation tool (DAVID Bioinformatics resource 6.8)(Huang et al. 2009b, 2009a), selecting B. subtilis as the background species.
Funding
This work was funded by the University of Westminster, University of Northumbria, and Venomtech Ltd.;
Author contributions
A MMGL and TDL sampled, and CT and ST prepared the library of captive animal venoms. WW and AB collected and prepared the wild snake samples. EE, JT, PG and SAM optimized and performed the DNA extractions and 16S PCR. JT and EE performed the preliminary and main study library preps and next generation sequencing experiments, respectively. AD, HD, PK, LS, and SAM performed the phylogenetic data quality control, curation and analysis. KFR and SAM performed the microbial cultures and biochemical characterization. MKV and LU grew the E. faecalis isolates and performed the whole genome sequencing. MKV, KW and SAM performed the E. faecalis isolate genomic characterization and MLST+ analysis. GT performed E. faecalis resistome analysis. VS performed the E. faecalis isolate pangenome data reduction and ST identified the venom resistance gene ontology subset. SAM conceived the study and designed experiments together with ST. All authors contributed equally to the overall interpretation of the dataset and manuscript preparation;
Competing interests
Authors declare no competing interests; and
Data and materials availability
Phylogenetic data are deposited on MG-RAST (project IDs MGP5177 and MGP5617) and the EBI-METAGENOMICS servers (project ID ERP004004). Raw 16S sequencing reads were deposited in the European Nucleotide Archive (PRJEB4693). Annotated draft E. faecalis genome assemblies are deposited on NCBI (BioProject No. PRJNA415175).
Acknowledgments
We would like to thank: Drs. Pamela Greenwell and Caroline Smith for their invaluable input on non-standard DNA extraction methodology options suited to unusual samples; Dr. Patrick Kimmit for his input on microbial characterisation; Mr Peter Gibbens for housing and venom collection from captive N. nigricollis and B. areitans.





{kind=link}
{kind=link}
{kind=link}
{kind=link}