

Abstract
Centromeres and their surrounding pericentric heterochromatic regions remain enigmatic and poorly understood despite critical roles in chromosome segregation1,2 and disease3,4. Their repetitive structure, vast size, low recombination rates and paucity of reliable markers and genes have impeded genetic and genomic interrogations. The potentially large selective impact of recurrent meiotic drive in female meiosis5,6 has been proffered as the cause of evolutionarily rapid genomic turnover of centromere-associated satellite DNAs, rapid divergence of centromeric chromatin proteins7, reduced polymorphisms in flanking regions8 and high levels of aneuploidy9. Addressing these challenges, we report here the identification large-scale haplotypic variation in humans10 that spans the complete centromere, centromere-proximal regions (CPR) of metacentric chromosomes, including the annotated ‘CEN’ modeled arrays comprised of Mbps of highly repeated (171 bp) α-satellites11,12. The dynamics inferred by the apparent descent of cenhaps are complex and inconsistent with the model of recurrent fixation of newly arising, strongly favored variants. The surprisingly deep diversity includes introgressed Neanderthal centromeres in the Out-of-Africa (OoA) populations, as well as ancient lineages among Africans. The high resolution of cenhaps can provide great power for detecting associations with other structural and functional variants in the CPRs. We demonstrate this with two examples of strong associations of cenhaps with α-satellite DNA content13 on chromosomes X and 11. The discovery of cenhaps offers a new opportunity to investigate phenotypic variation in meiosis and mitosis, as well as more precise models of evolutionary dynamics in these unique and challenging genomic regions.
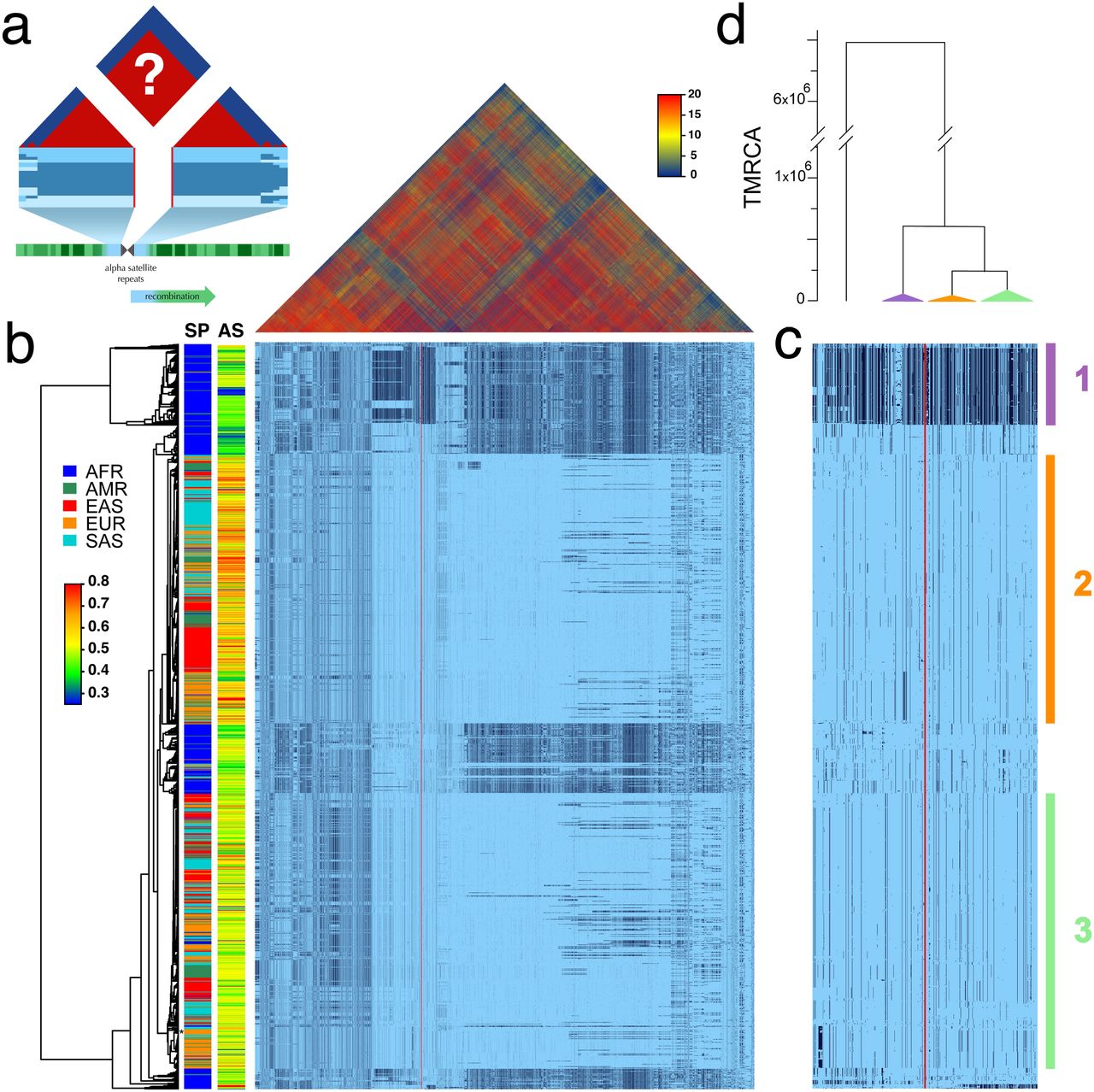
Recognizing the potential research value of well-genotyped diversity across human CPRs, we hypothesized that the low rates of meiotic exchange in these regions2 might result in large, diverse haplotypes in populations, perhaps spanning both the α-satellite arrays on which centromeres typically form and their flanking heterochromatic segments. Therefore, we examined the Single Nucleotide Polymorphism (SNP) linkage disequilibrium (LD) and haplotype variation surrounding the centromeres among the diverse collection of genotyped individuals in Phase 3 of the 1000 Genomes Project10. Figure 1a depicts the predicted patterns of strong LD (red) and associated unbroken haplotypic structures surrounding the centromere of a metacentric chromosome. Unweighted Pair Group Method with Arithmetic Mean (UMPGA) clustering on 800 SNPs immediately flanking the chrX centromeric gap in males (Fig. 1c) reveals a clear haplotypic structure that, in many cases, extends to a much larger region (≈8 Mbp), Fig. 1b). Similar clustering of the imputed genotypes of females also falls into the same distinct high-level haplotypes (Extended Data Fig. 1). This discovery of the predicted haplotypes spanning CPRs, or cenhaps, opens a new window into their evolutionary history and functional potential.

a. The predicted patterns of the magnitude LD (triangle at top) and genotypes in CPR clustered into haplotypes surrounding the centromeric region of a metacentric chromosome in a large outbreeding population (central blue bands), if crossing over declines to zero in and around the highly repeated DNA where the centromere is typically found in the chromosome (blue and green at bottom of a). b. Above, the linkage disequilibria between pairs of 17702 SNPs (Left: chrX:55623011-58563685, Right: chrX: 61725513-68381787; hg19) flanking the centromere and α-satellite assembly gap (red vertical line) from 1231 human male X chromosomes from the 1000 Genomes Project. The color maps (see the adjacent legend) to the - log10(p)×10−3, where the p value derives from the 2×2 χ2 for each pair of SNPs. Below, broad haplotypic representation of these same data. SNPs were filtered for minor allele count (MAC)≤60. Minor alleles shown in black. Poorly genotyped SNPs near edges of the gap (red line) were masked. Superpopulation (SP; AFRica, AMeRicas, East ASia, EURope, South ASia) and scaled estimate of chrX-specific α-satellite array size (AS) indicated at left side. Approximate position of HuRef chrX indicated by black asterisk at right of the tree. Dendrogram represents UPGMA clustering based on the hamming distance between haplotypes comprised of 800 filtered SNPs immediately flanking the centromere (Left: chrX:58374895-58563685, Right: chrX:61725513-61921419; hg19), shown in detail in c. The three most common X cenhaps highlighted with colored vertical bars. d. A UPGMA tree based on the synonymous divergence in 17 genes (see Table S1) in the 3 major chrX cenhaps (indicated in c), assuming the TMRCA of humans and chimps is 6.5MY. Widths of the triangles are proportional to the log10 of number of members of each cenhap, and the height is proportional to the average divergence within each cenhap.
The pattern of geographic differentiation across the inferred cenhaps exhibits higher diversity in African samples, as observed throughout the genome10. Despite being fairly common among Africans today, a distinctly diverged cenhap at the top of Fig. 1b,c is rare in OoA populations. Examination of the haplotypic clustering and estimated synonymous and nonsynonymous divergence in the coding regions of 21 genes included in the chrX cenhap region (see Extended Data Table 1) yields a parallel relationship among the three major cenhaps and an estimated Time of the Most Recent Common Ancestor (TMRCA) of ≈700 KYA (Fig. 1d) for this most diverged example. While ancient, putatively introgressed archaic segments have been inferred in African genomes14,15, this cenhap stands out as genomically (if not genetically) large. The persistence of such ancient cenhaps is inconsistent with the simplest explanations of the rapid turnover of genetic variation in CPRs and may be connected to the atypically high conservation of α-satellite on chrX16,17,18. Further, the detection of near-ancient segments spanning the centromere contrasts with the observation of substantially more recent ancestry across the remainder of chrX and with the expectation of reduced archaic sequences on chrX19. A large block on the right in Fig. 1b, where recombination has substantially degraded the haplotypic structure, is comprised of SNPs in exceptionally high frequency in Africans. Its history in “anatomically modern humans” (AMH) may be shared with the apparently archaic cenhap in Africa. Many putative, distal recombinants are observed OoA that likely contribute to associations of SNPs in this region with diverse set of phenotypes, including male pattern hair loss20 and prostate cancer21.
This unexpected deep history of the chrX CPR region raises the possibility of even more ancient cenhap lineages, either derived by admixture with archaic hominins or maintained by balancing forces. A survey of the other chromosomes uncovered several interesting examples (see Extended Data Fig. 2), two of which we examined in detail. To identify Neanderthal and Denisovan admixture we looked for highly diverged alleles OoA that shared a strong excess of derived alleles with archaic hominids and not with AMH genomes22. Applying this approach to CPR of chr11 we find it represents a compelling example of Neanderthal admixture23. Fig. 2a illustrates this in the context of the seven most common chr11 cenhaps. The most diverged lineage contains a small basal group of OoA genomes (highlighted in green). Members of this cenhap carry a large proportion of the derived alleles assigned to the Neanderthal lineage, DM/(DM+DN) = 0.98, where DM is the cenhap mean number of shared Neanderthal Derived Matches, and DN is the cenhap mean number of Neanderthal Derived Non-matches (Fig2a, at left). AN is the number of Neanderthal-cenhap Non-matches that are Ancestral in the Neanderthal and derived in the cenhap. The ratio DM/(DM+AN) = 0.91 is a measure of the proportion of the cenhap lineage shared with Neanderthals, supporting the conclusion that this chr11 cenhap is an introgressed archaic centromere. Fig. 2b shows these mean counts for each SNP class by cenhap group, confirming that the affinity to Neanderthals is slightly stronger than to Denisovans. A second basal African lineage separates shortly after the Neanderthal (highlighted in purple). It is unclear if this cenhap represents an introgression from a distinct archaic hominin in Africa or a surviving ancient lineage within the population that gave rise to AMHs. The relatively large expanses of these cenhaps and unexpectedly sparse evidence of recombination could be explained either by relatively recent introgressions or by cenhap-specific suppression of crossing over (e.g., an inversion) with other AMH genomes in this CPR. As with chrX above, the clustering of cenhaps based on coding SNPs (Fig. 2d) yields a congruent topology and estimates of TMRCAs of the two basal cenhaps of 1.1 and 0.8 MYA, consistent with relatively ancient origins. Among the 37 genes ‘captured’ in this apparent Neanderthal introgressed chr11 cenhap are 34 odorant receptors (ORs) reported to be associated with variation in human chemical perception24. 52 amino acid replacements among 20 of these ORs are associated with the Neanderthal cenhap (Extended Data Table 2). Similarly eight of these ORs harbor 12 distinct amino acid replacements associated with the second basal cenhap found primarily within Africa. These two ancient lineages share only two nonsynonymous substitutions. Given relatively large number of substitutions24, this introgressed chr11 archaic cenhap likely determines Neanderthal-specific determinants of smell and taste with significant impacts on variation in perception.
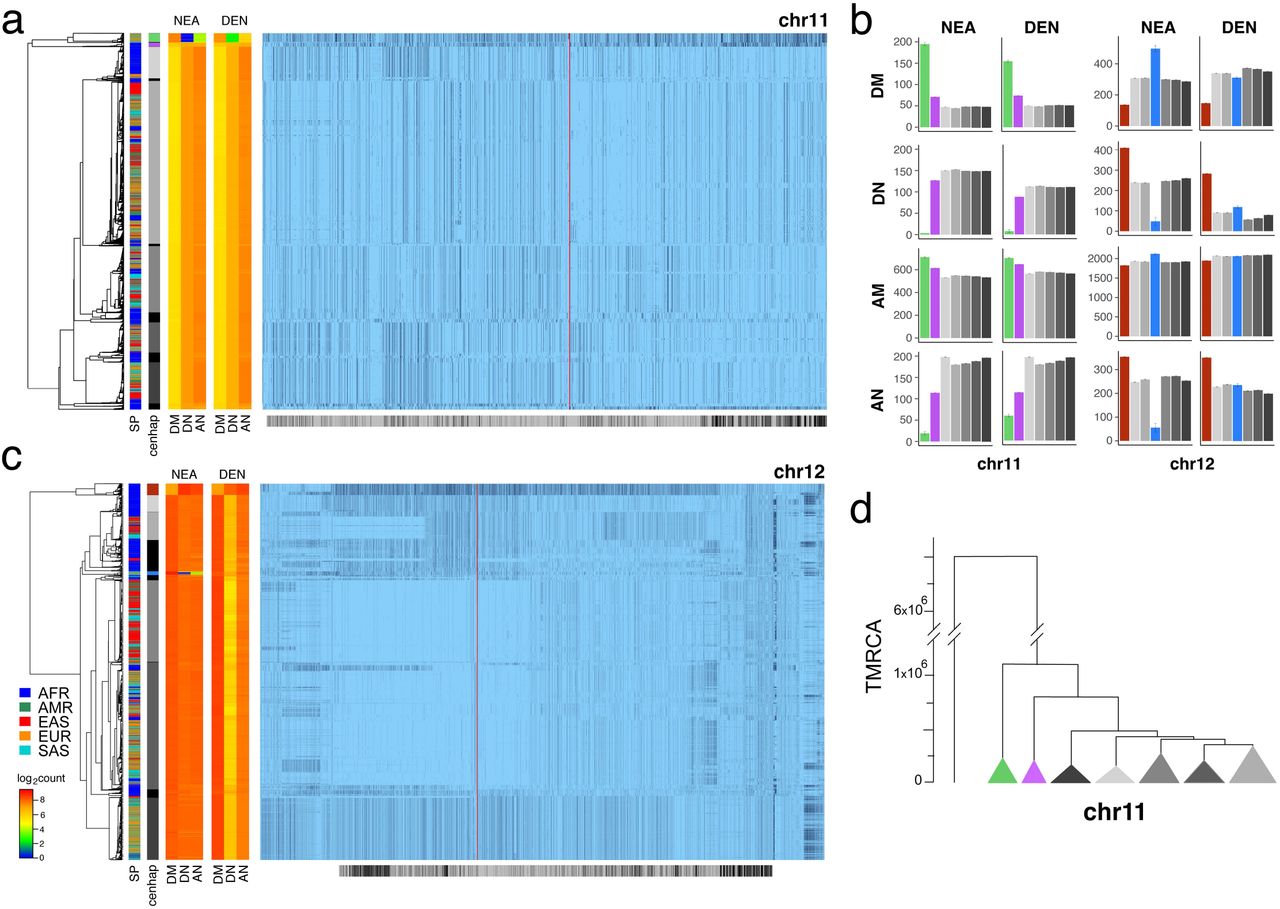
a. Haplotypic representation of 9151 SNPs from 5008 imputed chr11 genotypes from the 1000 Genomes Project (Left: chr11:50509493-51594084, Right: chr11:54697078-55326684; hg19). SNPs were filtered for MAC≤35 and passing the 4gt_dco with a tolerance of three (see Methods). Minor alleles shown in black, assembly gap indicated by red line. Haplotypes were clustered with UPGMA based on the hamming distance between haplotypes comprised of 1000 SNPs surrounding the gap (Left: chr11: 51532172-51594084, Right: chr11:54697078-54845667; hg19). Superpopulation and cenhap partitioning indicated in bars at far left. Log2 counts of DM (derived in archaic, shared by haplotype), DN (derived in archaic, not shared by haplotype) and AN (ancestral in archaic, not shared by haplotype) for each cenhap relative to Altai Neanderthal (NEA) and Denisovan (DEN) at left. Grey horizontal bar (bottom) indicates region included in analysis of archaic content; black bars indicate SNPs with data for archaic and ancestral states. b. Bar plots indicating the mean and 95% confidence intervals of DM, DN, AM (ancestral in archaic, shared by cenhap) and AN counts for cenhap groups (as partitioned in a. and c.) relative to Altai Neanderthal and Denisovan genomes, using chimpanzee as an outgroup1. c. Haplotypic representation of 21950 SNPs from 5008 imputed chr12 genotypes from the 1000 Genomes Project (Left: chr12:33939700-34856380, Right: chr12:37856765-39471374; hg19). SNPs were filtered for MAC≤35. Minor alleles shown in black. Centromeric gap indicated by red line. Haplotypes were clustered with UPGMA based on 1000 SNPs surrounding the gap (Left: chr12: 3482173834856670, Right: chr12:37856765-37923684; hg19). Bars at side and bottom same as in a. d. A UPGMA tree based on the synonymous divergence in 30 genes in the 7 major chr11 cenhaps (see Table S2), assuming the TMRCA of humans and chimpanzee is 6.5MY (see Methods and legend for Fig 1d).
The most diverged cenhap on chr12 is a basal clade (Fig. 2c, indicated in brown) common in Africa, but, like the most diverged chrX cenhap, it is not represented among the descendants of the OoA migrations25. The great depth of the lineage of this cenhap is further supported by analysis of archaic variation. Consistent with the hypothesis that this branch split off before that of Neanderthals/Denisovans, members of this cenhap share fewer matches with derived SNPs on the Neanderthals and Denisovans lineages (DM) and exhibit strikingly more ancestral nonmatches (AN) than other chr12 cenhaps (see Fig. 2b). This putatively archaic chr12 cenhap represents a large and obvious example of the genome-wide introgressions into African populations inferred from model-dependent analyses of the distributions of sequence divergence.14,15 The small OoA cenhap nested within a mostly African subclade (indicated in blue in Fig. 2c) appears to be a typical Eurasian archaic introgression with high affinity to Neanderthals (DM/(DN+DM) = 0.91 and DM/(DM+AN) = 0.90) than to Denisovans (Fig. 2b). This bolsters the conclusion that the basal cenhap represents a distinct and more ancient lineage. Unfortunately, there are too few coding bases in this region to support confident estimation of the TMRCAs of these chr12 archaic cenhaps, but the basal cenhap is twice as diverged as the apparent introgressed Neanderthal cenhap, placing the TMRCA at ~1.1 MYA, assuming the Neanderthal TMRCA was 575KYA26. While there is no direct evidence of recent introgression, the large genomic scale of this most diverged cenhap (relative to apparent exchanges in other cenhaps) is consistent with recent admixture with an extinct archaic in Africa, although, again, suppression of crossing over is an alternative explanation.
The CPRs of chromosomes X, 11 and 12 harbor a diversity of large cenhaps including those representing archaic lineages. Notably, the CPRs of many chromosomes harbor diverged/basal lineages that are likely to be relatively old, if not archaic (Extended Data Fig. 2). For example, chromosome 8 contains a putative archaic cenhap limited to Africa with an estimated TMRCA of 817 KYA (Extended Data Fig. 3) and a basal chr10 cenhap appears to be another clear Neanderthal introgression (Extended Data Fig. 4).

{kind=link}
{kind=link}
{kind=link}
a. Empirical cumulative density (ecdf) of chrX α-satellite array size for cenhap, homozygotes and heterozygotes. 1_2 and 1_3 heterozygotes were excluded due to insufficient data. Female (F) values were normalized (x 0.5) to facilitate plotting with hemizygote male (M) data. b. Haplotypic representation of 1000 SNPs from 1640 imputed chr11 genotypes from 820 cenhap-homozygous individuals. SNPs were filtered for MAC≥35 and passing the 4gt_dco. Minor alleles shown in black. Assembly gap indicated by red line. Superpopulation (SP) and scaled chr11-specific α-satellite array size (AS) at left. Cenhap partitions at right; most common cenhap (“1”) and cenhap with larger mean array size (“2”) are highlighted. Most probable HuRef cenhap genotypes are indicated by black asterisks at right. c. Empirical cumulative density of array size for chr11 cenhap (from b) homozygotes (1_1 and 2_2) and heterozygotes (1_2).
These SNP-based cenhaps portray a rich view of the diversity in the unique segments flanking repetitive regions. While the divergence of satellites may be dynamic on a shorter time scale27, we reasoned that the paucity of evidence of exchange in or near regions known to contain satellite DNA arrays would create cenhap associations with satellite divergence in both sequence and array size. Miga, et al. 201413 generated chromosome-specific graphical models of the α-satellite arrays, which revealed a bimodal distribution in estimated chrX-specific α-satellite array (DXZ1) sizes28 for a subset of the 1000 Genomes males (Fig. 1b extends this to the entire data). Fig. 3a shows the substantial differences in the cumulative distributions of the three common chrX cenhaps designated in Fig. 1c. The distributions of α-satellite array size in cenhap-homozygous females are parallel to males, and imputed cenhap heterozygotes are intermediate, as expected. Similarly, Fig. 3b shows an even more striking example of variation in array size between cenhap homozygotes on chr11, and Fig. 3c demonstrates that heterozygotes of the two most common cenhaps are reliably intermediate in size. While we confirmed that reference bias does not explain the observed cenhaps with large array size on chrX and chr11 (see Methods, Fig. 1b, Fig. 3b and Extended Data Fig. 4), it is a potential explanation for particular instances of cenhaps with small array sizes, e.g., the relatively low chrX-specific α-satellite content in the highly diverged African cenhap (see Fig1b,c and Fig. 3a in purple). Importantly, our results demonstrate that cenhaps robustly tag a component of the genetic variation in array size.
The potential impact of sequence variation in CPRs and their associated satellites on the function of centromeres has been long recognized5,6 but difficult to study. The natural opportunity for meiotic drive in asymmetric female meioses has been cited as the likely explanation for the rapid turnover of satellite sequences and excess nonsynonymous divergence of several centromere proteins, some of which interact directly with the DNA7. The observed deep lineages and high levels of haplotypic diversity across the CPRs (Extended Data Fig. 2) conflict with the predictions of a naïve turnover model based on recurrent strong directional selection yielding sequential fixation of driven centromeric haplotypes. Models that maintain variation, including the inherent frequency-dependence of meiotic drive, the likely tradeoff with transmission fidelity9, and the expected impact of unlinked suppressors29, are plausible alternatives.
Our identification and characterization of human cenhaps raise new questions about the evolution of these unique genomic regions, but also provide a depth of diversity to quantitatively address them in the future. These results transform large, previously obscure and avoided genomic regions into genetically rich and tractable resources. Most importantly, cenhaps can now be investigated for associations with variation in evolutionarily important chromosome functions, such as meiotic drive30 and recombination2, as well as diseases arising from aneuploidy in the germline3 and in somatic cells during development31,32,33 and aging.4
Acknowledgements
Benjamin Vernot, Graham Coop, Yuh Chwen Grace Lee.
Literature Cited




