

Abstract
Despite the success of channel blocking drugs, only a single class of agent targeting virus-encoded ion channels, or “viroporins”, has been licensed since the 1960s1–3. Although resistance to adamantane inhibitors of the influenza A virus (IAV) M2 proton channel arose, a growing number of clinically important and emerging viruses encode essential viroporins4–6, providing targets for new interventions. Here, we describe rational antiviral development targeting the p7 viroporin from hepatitis C virus (HCV) and reveal a second biological function for its channel activity. Lead-like oxindole inhibitors potently blocked p7 function during virion secretion, but were also active during virus entry, supporting the presence of channel complexes within infectious HCV particles. Hence, p7 inhibitors (p7i) represent dual-acting HCV antivirals targeting both the spread and establishment of infection, which may be relevant to future antiviral prophylaxis.
HCV continues to pose a global clinical challenge as a major cause of chronic liver disease, with severe complications including cirrhosis and primary liver cancers. New HCV direct-acting antivirals (DAA) are an unprecedented drug development success. However, availability is limited by cost and resistant viral variants are a concern7. DAAs do not prevent re-infection and compliance within high-risk populations is low. In the absence of an HCV vaccine, ~6 million new infections occur annually, coincident with poor diagnostic rates and rapidly increasing burden in low/middle income countries (LMIC). Growing controversy also surrounds the efficacy of DAA in the prevention of HCV-associated liver cancer8.
In contrast to DAAs targeting viral RNA replication, we focused upon the p7 viroporin, whose channel function plays an essential role during virion secretion9–11. Our hairpin-conformation p7 monomer solution structure12 (PDB: 3ZD0) and defined heptameric stoichiometry13 enabled creation of genotype 1b channel ensembles as templates for in silico screening12. Whilst hexameric structures were determined for genotype 2a p714 (electron microscopy) and a mutated genotype 5a protein15 (solution NMR, PDB: 2M6X), molecular dynamics simulations favour 3ZD0-based channel models16. Moreover, functionality of the mutated 2M6X protein was not demonstrable15 and its unusual architecture is incompatible with p7i structure-activity relationships (SAR) herein (data not shown); this may relate to potential NMR artefacts resulting from the use of alkyl-phosphocholine detergents as membrane mimetics17.
Here we present second generation compounds targeting a membrane-exposed site containing several invariant residues (e.g. Gly46, Trp48, Leu52) and an L20F rimantadine resistance polymorphism12,18,19. SAR-focused chemical modification (Table 1) of an oxindole core scaffold identified “JK3/32” as a series lead (Figure 1a, S2) with excellent potency against chimaeric genotype 1b HCV (J4/JFH-1) secretion (IC50~184 nM) (Figure 1b, S1). The oxindole scaffold of JK3/32 is shared with licensed kinase inhibitor drugs (Sunitinib, Nintedanib). However, the JK series of inhibitors is chemically distinguished from these compounds by an N-alkyl substituent which was essential for anti-HCV activity (Table 1); accordingly, JK3/32 displayed no off-target activity against a panel of human kinases (Figure S2a). Reminiscent of first generation compounds12, JK3/32 retained cross-genotype activity, including HCV genotype 3a (IC50~738 nM) and more genetically distant 2a viruses (IC50~1900 nM) (Figure 1c). The compound retained a toxicity-based selectivity index of >500 (CC50 > 100 000 nM) in Huh7 cells, and had no effect against HCV subgenomic replicons (Figure S2b, c).

A. Determination of appropriate single dilution of infectious supernatants within linear range for secreted titre determination (red box). Appropriate dilutions for untreated virus were repeated alongside every inhibitor IC50 experiment to ensure that titres could be back-calculated accurately; each condition repeated in quadruplicate across multiple experiments, error bars show standard deviation. Example IncuCyte images are shown for comparison. B. Example IncuCyte images and calculated IC50 for inhibition of secreted titres using the licensed nucleotide analogue NS5B inhibitor, Sofosbuvir, error bars show standard deviation. C. Parallel determination of cytotoxic effects by imaging of producer cell confluency for a Huh7 cytotoxic agent, Hsp90 inhibitor Radicicol, lead p7i JK3-32 and control DAA Daclatasvir (DCV), error bars show standard deviation for quadruplicate samples in each representative experiment.
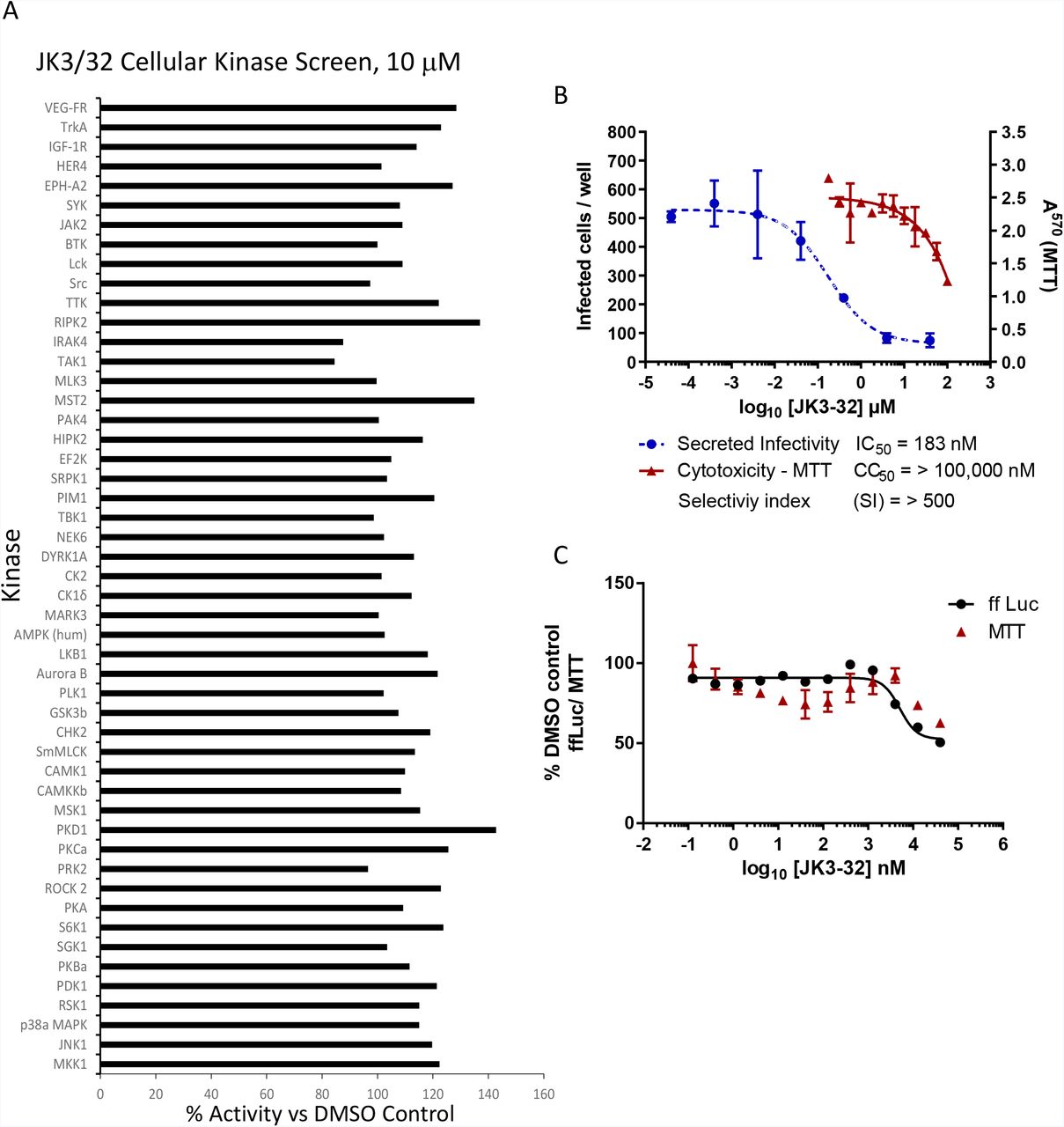
A. JK3/32 was tested for off-target in vitro effects vs. cellular kinases at the MRC Protein Phosphorylation Unit, Dundee. Reassuringly, no significant defects were observed upon incubating with 10 μM JK3/32, ~20x the IC50 concentration for treating particle secretion. B. JK3/32 IC50 vs chimaeric GT1b HCV (NS5A immunofluorescence) and CC50 determined by alternative method (MTT). C. Lack of JK3/32 activity against replication (firefly luciferase activity) of HCV subgenomic replicon (JFH-1). Each condition in quadruplicate across multiple experiments (see Table S2), error bars show standard deviation.

A. Summary of the structure activity relationships for the JK3/32 series of inhibitors (Table 1) for their effects upon chimaeric GT1b HCV (J4/JFH-1) secreted infectivity following electroporation. B. Comparison of JK3/32 potency vs. virion secretion of J4/JFH-1 with licensed HCV DAAs Sofosbuvir and Daclatasvir, as well as the prototypic adamantane viroporin inhibitor, rimantadine. Curves are representative of at least four experimental repeats for JK3/32 (see table S2), multiple for Sofosbuvir and Daclatasvir, and two for rimantadine, where each condition is carried out in quadruplicate and error bars represent standard deviations. C. Comparative IC50 curves for JK3/32 effects upon GT1b, 2a and 3a chimaeric HCV (J4, JFH-1, S52/JFH-1) secreted infectivity post-electroporation. Curves are again representative of multiple experiments and error bars show standard deviations between quadruplicate repeats. Secreted infectivity measurements were conducted following optimisation (figure S1) of a published rapid-throughput method for counting Huh7 cells fluorescently stained for NS5A.
JK3/32 SAR was consistent with its predicted binding pose (Figure 2a) following in silico docking within the peripheral binding site, defining key determinants of its activity. JK3/32 binds into a predominantly hydrophobic cleft created between helices on the membrane exposed site. Polar interactions are predicted between the side-chains of Tyr45 and Trp48 side and the carbonyl oxygen atom at the indole core (Figure 2a). Predicted close contacts included residues experimentally defined by NMR to interact with rimantadine12: Leu20, Tyr45, Gly46, Trp48, Leu50 and Leu52, and additional interactions with Ala11, Trp32 and Tyr42. A library of JK3/32 analogues was developed to explore SAR for inhibition of J4/JFH-1 secretion (Table 1). The observed SAR was largely consistent with the energetically preferred docking predictions (using Glide, Schrodinger). For instance, substitution of the N1 position of the oxindole core demonstrated a preference for benzyl substitution (e.g. JK3/32) consistent with the group occupying a relatively large hydrophobic pocket between helices created by Leu and Ala residues. Incorporation of a longer, more hydrophobic group (N-ethylphenyl, 1191-137), was less well tolerated. Introduction of a NH (JK3-38), N-Ph (JK3-42) and a N-heterocyclic substituent (2,5-dimethylisoxazolylmethyl, 1191-106) abrogated antiviral effects. Attempts at substitution at the ‘northern’ phenyl ring was not well tolerated, with 4-OMe (e.g. JK3/32) or H (21-RS-8) preferred over 4-cyano (1191-112) and 2-methoxy (1191-104). 4-alkynyloxy was only moderately less active than 4-OMe (compare 1191-146 (IC50 2.48 μM) to 1191-140 (IC50 0.46 μM)), suggesting that further synthetic expansion from this site was possible, consistent with the modelling which directed this vector outwards from the binding pocket into the membrane. The linker at the 3-position of the oxindole core was sensitive to modification. The hydrazone analogue (21-RS-7) was much less active whilst replacement of the NH for a carbonyl group (21-RS-17) was also not well tolerated. This is consistent with the enamine linker adopting an important bridging unit for correct placement of the N1 substituent into the deep hydrophobic pocket. Substitution of the oxindole core at the 5- and 6- positions with F atoms (1191-124 and 1191-121 respectively) was generally well tolerated.

A. Energetically preferred binding pose (in Glide) for JK3/32 within membrane-exposed binding site upon heptameric genotype 1b p7 channel complexes based upon pdb: 3ZD0. A single site is shown for simplicity; it is unclear how many sites need to be occupied in order to prevent channel activity. Left panel shows side view with orientation of JK3/32 and space-filling model of the channel complex to show membrane-exposed peripheral binding pocket. Insets show zoomed in spatial arrangements of binding pocket and key interacting residues including Tyr45 and Trp48. B. 100 ns atomistic molecular dynamics simulation of JK3/32 starting from a minimised pose derived from (A) in a hydrated lipid bilayer environment. C. Overlay of 0 ns and 100 ns protein structures and JK3/32 orientation.
Consistent with JK3/32 acting as an effective inhibitor targeting the peripheral site, atomistic MD simulations (100 ns) in hydrated lipid bilayers revealed marked stability of its binding pose, despite significant structural dynamics of the p7 bundle (Figure 2b, c). Structural dynamics of JK3/32-bound p7 fluctuated within tolerable values calculated as the root mean square deviation (RMSD) of the protein backbone Cα atoms and were indistinguishable from unbound protein (data not shown). Simulations of both wild type (figure 2b, c) and L20F (figure S4a) mutant p7 complexes resulted in the JK3/32 remaining within the binding pocket. The intramolecular H-bond between the NH and carbonyl oxygen was lost on simulation although the bifurcated H-bond to the side-chains of conserved residues Tyr45 and Trp48 was maintained. Consistent with 1st generation compounds12, L20F mutant p7 complexes also formed a stable interaction with JK3/32 (figure S4a), although Phe20 caused reorganisation and crowding of the binding pocket stabilized by π-π stacking interactions between Phe20 and Tyr45. JK3/32 carbonyl formed H-bonds with Tyr45, with subsequent bifurcation (after ~50 ns) with Trp48. The JK3/32 amino group made further interactions with various side chains over the course of the simulation. This tight interaction contrasts with the prototypic p7i rimantadine, binding of which was severely disrupted by L20F (figure S4b). We infer that efficient H-bond formation is highly contributory to JK3/32 potency. Furthermore, interactions with invariant p7 residues such as Tyr45 and Trp48 might generate a higher genetic barrier to resistance compared with rimantadine; several attempts to select JK/32 resistance failed (data not shown).

The amiloride derivative BIT225 was synthesised in-house and tested for effects against secretion of infectious HCV. A. No appreciable antiviral effects were determinable above inherent and significant cytotoxicity. Conditions in quadruplicate, error bars show standard deviation. B. Effects of BIT225 compared with selected current p7i series compounds upon Huh7 cell proliferation in real time (Incucyte).
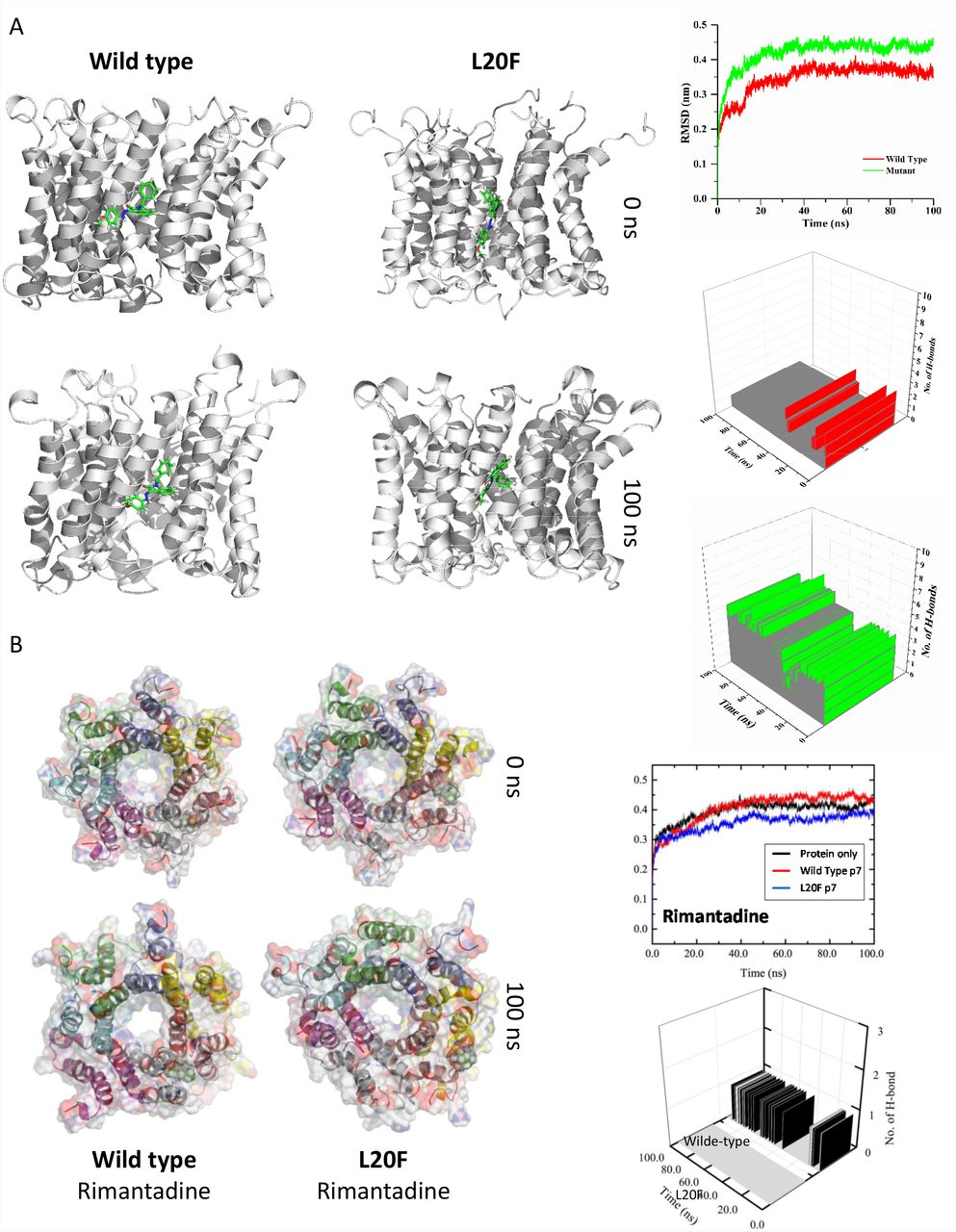
An L20F polymorphism was introduced into the genotype 1b p7 sequence in the context of the 3ZD0 heptamer, then subjected to energy minimisation within a hydrated lipid bilayer. A. JK3/32 was located into the peripheral binding pocket and the simulation allowed to proceed for 100 ns. The ligand remained stable within the pocket and in fact formed increased H-bonding and close contact interactions with the mutant protein compared to the wild type. B. Location of rimantadine into the peripheral binding pocket let to a loss of binding interactions for the L20F mutant compared with stable, but weak interactions with the wild type.
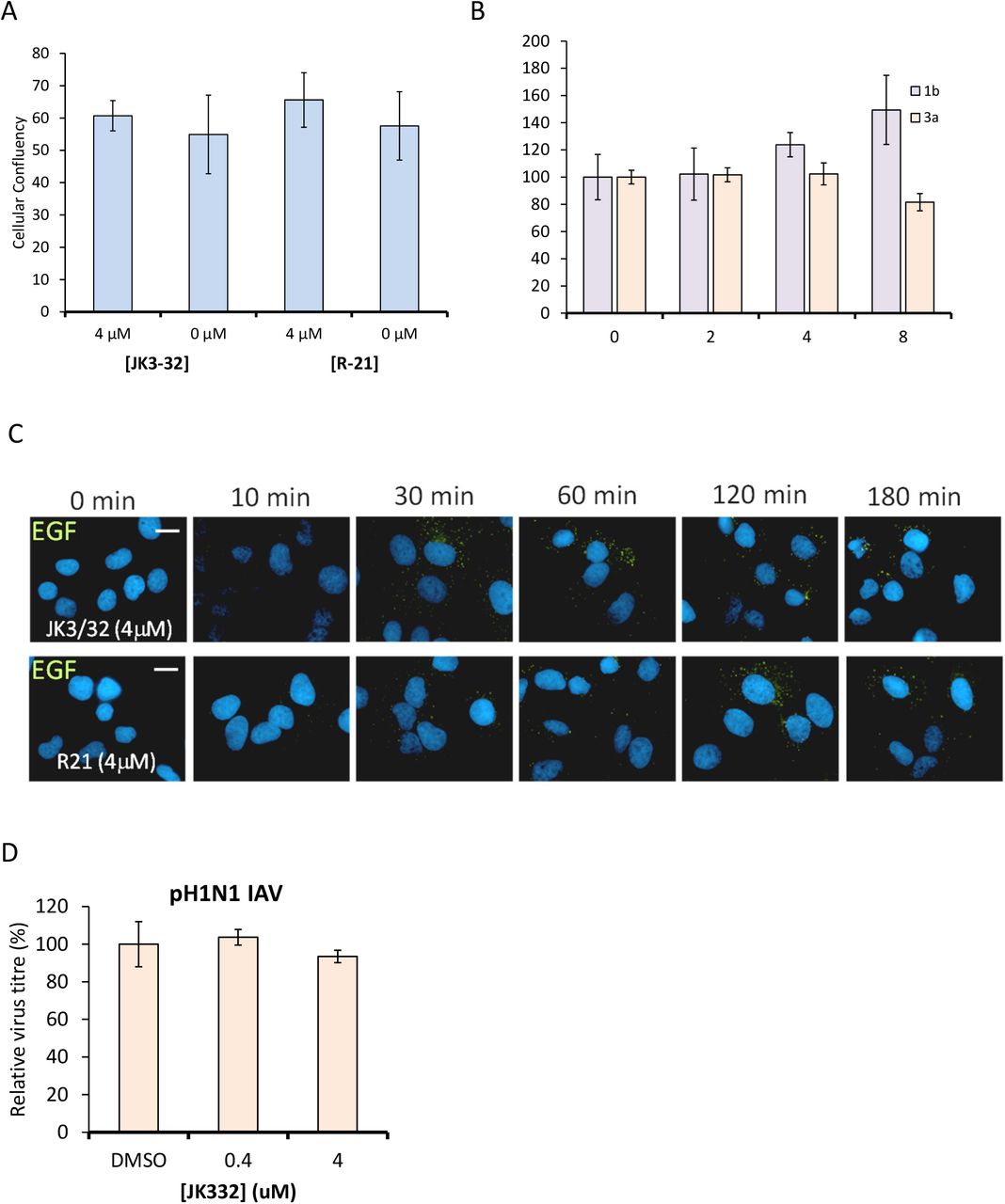
A link between p7 and the acid stability of secreted HCV particles20 prompted us to speculate that virion-resident channel complexes might influence virus entry in addition to p7 acting during secretion. Accordingly, JK3/32, but not an inactive analogue from a closely related series, compound R21, reduced infectivity (figure 3a), albeit at higher concentrations than those effective against virion secretion. Genotype-dependent variation in JK3/32 potency during entry supported virus-specific effects; genotype 3a chimaeric viruses required 2-4 fold higher concentration than genotype 1b (figure 3b). Neither JK3/32, nor R21 affected clathrin-mediated uptake of fluorescent epidermal growth factor (EGF) or infectivity of H1N1 IAV (figure S5), which itself retains particle-associated M2 proton channels21. Thus, JK3/32 effects during HCV entry were both virus- and viroporin-specific.

Cell viability (Incucyte cell confluency) during treatment of Huh7 cells with JK3/32 or R21 during virus entry experiments. C. Uptake of fluorescent EGF by Huh7 cells over time following cold synchronisation (40 °C) and exposure to either R21 or JK3/32. D. Lack of effect for JK3/32 during cell entry of pH1N1 IAV following infection at an MOI of 0.001 and incubation for 24 hr. Titre was determined by plaque assay, error bars show standard deviation across triplicate samples.

10 μL of each gradient fraction was added to 10 000 naïve Huh7 cells within each well of a black-walled 96-well plate. 48 hr post-infection, cells were stained for HCV NS5A protein and infectious units calculated using the IncuCyte.

A. Infectivity following application of JK3/32, or the inactive R21 analogue, during entry of GT1b chimaeric HCV into Huh7 cells. Virus innoculae were pre-treated with either compounds or DMSO for 20 min at room temperature prior to application to Huh7 cells overnight. Cells were then washed extensively and assessed for infectivity 48 hr post infection by NS5A immunofluorescence staining and quantitation on the Incucyte Zoom. Representative images from Incucyte analysis of NS5A-stained Huh7 cells are shown for comparison (**p=0.00707, Student T-Test, n=2). B. Effects of titrating JK3/32 concentrations were assessed during entry as described in A, comparing chimaeric GT1b (J4/JFH-1) and GT3a (S52/JFH-1) viruses (**p≤0.01, *p≤0.05, Student T-Test, n=2; 2 μM p=0.007893452, 4 μM p=0.014655631, 8 μM, p=0.032708636). C. Effects of prototypic p7 channel blockers against wild type and rimantadine resistant GT2a HCV (JFH-1) during entry into Huh7 cells (**p≤0.01, *p≤0.05, Student T-Test, n=2; JFH-1: DMSO vs DNJ40 p=0.028522916, DMSO vs DNJ80 p=0.013759902, DMSO vs Rim40 p=0.052786405, DMSO vs Rim80 p=0.025954537, JFH-1(L20F): DMSO vs DNJ40 p=0.009901971, DMSO vs DNJ80 p=0.00606083). D. JK3/32 effects upon entry when added prior, during or post-infection with chimaeric GT1b HCV (J4/JFH-1), compared to Daclatasvir NS5Ai (**p=0.001890506, *p=0.037101137, Student T-Test, n=2). E. Inhibitory effects of p7-specific concentrated polyclonal antisera during HCV entry. Broadly neutralising AP33 monoclonal targeting E2 was used as a positive control. GT2a viruses (JFH-1 or J6/JFH-1) were exposed to sera either containing or lacking p7 cross-reactivity (% sera v/v as indicated) for 20 min prior to infection of Huh7 cells: JFH-1 N-terminus (2715/2716), JFH-1 C-terminus (2717), GT1b (J4) C-terminus (1055). Note, JFH-1 and J6 p7 share N-terminal epitopes (both NH2- ALEKLVVLHAAS-CO2H), but differ at C-terminus (NH2-PRQAYA-CO2H vs NH2-PQQAYA-CO2H in J6). Cell viability controls are shown in figure S5A, B. (***p≤0.001, **p≤0.01, *p≤0.05, Student T-Test, n=2).
As JK3/32-selected resistance was unachievable, we used previously defined strain- and polymorphism-dependent resistance to prototypic p7i18,22,23 to provide genetic evidence of p7 target engagement during HCV entry. Secretion of genotype 2a JFH-1 HCV is innately amantadine-resistant23, yet remains sensitive to rimantadine and the alkyl imino-sugar NN-DNJ. This pattern was recapitulated during virus entry with the latter two compounds displaying dose-dependent efficacy (figure 3c). Moreover, entry of JFH-1 L20F was resistant to both amantadine and rimantadine18,24, whilst remaining NN-DNJ-sensitive; NN-DNJ disrupts p7 oligomerisation rather than binding peripherally. Hence, p7 sequence dictated p7i-mediated blockade of HCV entry, providing genetic evidence for virion-resident channels. Consistently, JK3/32 only blocked infection when introduced during virus infection, concordant with a direct effect upon virion-resident channels (Figure 3d).
Next, p7-specific antisera22,23,25 were shown to neutralise HCV virions in a genotype/strain-specific manner (figure 3e). Dose-dependent inhibition of JFH-1 was observed for specific N-terminal and C-terminal sera, yet entry was insensitive to sera recognising the distinct C-terminus of genotype 1b p7 (figure 3e, left panel). Similarly, a shared epitope within both JFH-1 and J6 p7 (chimaeric J6/JFH-1) conferred sensitivity to N-terminal sera, whereas J6/JFH-1 was predictably insensitive to JFH-1 C-terminal sera. p7 sera were less potent than the anti-E2 monoclonal AP33, which is perhaps unsurprising given their poor reactivity and the predicted poor accessibility and low stoichiometry of virion-resident p7.
Elevated concentrations of JK3/32 required to block HCV entry compared with secretion made it critical to rule out off-target effects. JK3/32 SAR, docking, and MD simulations predicted that the northern 4-OMe group should tolerate the addition of a flexible linker without significant loss to binding affinity. Click chemistry was used to attach an azide reactive fluorophore (Alexa Fluor 488 nm) to an alkynyl group at the 4-position of the phenyl ring generating a chemical probe (JK3/32-488), allowing compound concentrations to be calculated by fluorimetry. We removed free ligand from virion-p7i complexes by centrifugation through iodixinol density gradients. Accordingly, following the incubation of compounds with purified J4/JFH-1 HCV particles, residual JK3/32-488 concentrations within peak infectivity fractions were <2nM (fractions 5 & 6), far lower than concentrations that blocked HCV secretion (figure 4a). However, treatment with both JK3/32 and JK3/32-488 virtually abrogated infectivity, whereas R21 again showed no effect (figure 4b, c, S7). The potency of JK3/32-488 thus supported the SAR around the core JK3/32 scaffold, as well as the binding hypothesis predicted by docking/MD. p7i blockade of HCV entry therefore involves irreversible effects upon particles, rather than cellular exposure to significant levels of compound, supporting the presence of channel complexes within particles.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Concentrated, purified high titre chimaeric GT1b HCV (J4/JFH-1) was incubated with DMSO, active JK3/32(−488) or control compounds (R21) at 10 μM for 20 min prior to separation on a continuous 10-40% iodixinol/PBS density gradient followed by fractionation. A. Incucyte quantitation of NS5A immunofluorescence staining within each fraction expressed as infectious units (IU) per mL. B. Immunofluorescence staining of HCV NS5A protein within naïve Huh7 cells 48 hr post-infection with 10 μl of fraction 6, corresponding to peak infectivity for untreated control gradients. C. Calculated concentration of JK3/32-488 based upon fluorimetry within each of 12 fractions taken from the top of the gradient.
JK3/32 activity against HCV virion secretion represents a distinct antiviral modality compared with replicase-targeted DAA. Supported by a robust SAR, JK3/32 demonstrated potency in line with licensed drugs against multiple HCV genotypes. We were unable to select resistance to this compound in culture (data not shown), likely reflecting predicted interactions with invariant residues (e.g. Tyr45, Trp48). JK3/32 was also superior to a clinically progressed compound, BIT22526,27, which, in our hands, showed no antiviral activity discernible from effects upon cellular viability (figure S3). Thus, we propose that the favourable potency and selectivity index demonstrated by JK3/32 could serve as a high quality starting point for a more comprehensive p7-targeted inhibitor drug development programme.
The potency and selectivity demonstrated by JK3/32 provided an excellent molecular tool for the study of p7 channel function, identifying a second role for channel activity during HCV entry. Whilst higher inhibitory concentrations were required compared with targeting secretion, evidence supported that p7i blockade was both virus- and indeed viroporin-specific. We speculate that differences in effective concentrations are due to dependency of respective biological processes upon p7 functionality, rather than altered drug binding; vesicle alkalinisation during secretion likely requires significantly more active p7 complexes than acidification of individual virions during entry. Hence, blocking entry may require saturating inhibition, whereas losing a mere fraction of vesicular channels might prevent p7 from effectively counteracting vATPase. Thus, whilst blocking both entry and secretion may require higher plasma concentrations compared with targeting secretion alone, the favourable selectivity index for JK3/32 suggests this may be feasible. Hence, p7i could be ideal for delivering effective antiviral prophylaxis against de novo exposure or transplanted graft re-infection, in addition to use in conventional combination therapies treating chronic infection.
In summary, viroporins represent an untapped reservoir of antiviral drug targets that has historically been under-explored following the shortcomings of adamantane M2 inhibitors. Our work shows that it is possible to take a step-change in ability to target such proteins, providing a new approach to HCV antiviral prophylaxis and generating novel research tools with which to dissect viroporin function.
Materials and Methods
Virus secretion inhibition assays
Huh7 cells were cultured and propagated as described previously. Secreted infectivity was measured as described28. Briefly, 1 μg of linearised HCV constructs pJFH129, pJ4JFH130, or pS52-JFH131 was used to perform in vitro transcription (RiboMax express, Promega) following the manufacturer’s protocol. Following phenol/ chloroform extraction, 4 × 106 Huh7 cells were electroporated with 10 μg HCV RNA. Electroporated cells were seeded at 2.5 × 104 cells/ well in 100 μL volume in 96 well plates and incubated 4 h. Compound dose responses were prepared at 400x in DMSO, diluted 1:20 into media in an intermediate plate, and 1:20 into the final cell plate to yield final 0.25% (v/v) DMSO. All compound treatments were dosed in duplicate. Dosed cells were incubated 72 h before performing 1:4 dilution (50 μL) of virus-containing supernatant onto a plate of naïve Huh7 cells (150 μL), seeded at 8 × 103 cells/well 6 h previously. For cytotoxicity analysis, producer plates were washed in PBS and fixed in 4% PFA prior to imaging cellular confluency using an IncuCyte ZOOM (Essen BioSciences). Infected Huh7 cells were incubated 48 h before washing 3x in PBS and fixing in 4% PFA. Fixed cells were washed in PBS and permeabilised using 0.1% Triton X-100 (v/v) in PBS for 10 min, RT. Following PBS wash, cells were immuno-stained for NS5A to quantify infected cells. Anti-NS5A antibody32 was used at 1:2000 in PBS supplemented with 10% FBS, 16 h, 4°C. Following 3x PBS washes, AlexaFuor594 Donkey anti-Sheep antibody was added at 1:500, 2 h, RT under subdued light. Cells were washed in PBS and imaged using phase and red channels (IncuCyte ZOOM). Infected cells positive for NS5A expression were quantified using IncuCyte parameters previously described28, normalised to DMSO control and non-linear regression fitted using Prism 6 (GraphPad) to determine EC50/CC50.
Each experiment included a serial dilution of untreated virus confirming 1:4 dilution fell within a linear range and internal DAA EC50 controls (data not shown). Determined Z-factor was routinely > 0.45. In addition, the Incucyte cell confluence tool was used as a measure of cell viability in both transfected/inhibitor-treated producer cells and target cell populations. This was also undertaken over a time course for select compounds (see figure S3).
Virus entry inhibition assays
HCV entry experiments used virus supernatant stocks harvested 72 hours post-electroporation and stored at −80°C. Huh7 cells were seeded at 8 × 103 cells/ well in 100 μL in 96-well plates for 6 h. Indicated compound was added to virus-containing supernatant stocks immediately prior to infection at MOI of 0.6 and incubated 18 h. Cells were washed and incubated 48 h in media prior to quantifying infected cells via NS5A immunostaining as described above. For antibody inhibition, virus was mixed with increasing % v/v of p7-specific antisera as indicated (Figure 3E) for 20 min prior to infection of Huh7 cells as above. NS5A positive foci were counted at 48 hr post-infection.
Virus purification and ultracentrifugation
High titre J4/JFH-1 virus stocks were generated by sequential daily harvest of Huh7 culture supernatants over a 1 week period, comprising 20 mL of HEPES-buffered media in each of eight T150 cell culture flasks, seeded with 2 × 106 electroporated cells on day 1. Supernatants were clarified prior to addition of 1/3rd volume of 30% (w/v) polyethylene glycol (PEG)-8000 in PBS, thorough mixing and incubation at 4 °C overnight. The next day, precipitates were spun at 2000 rpm for 40 min at 4°C in a Hereas bench-top laboratory centrifuge, pellets harvested, and then resuspended in 1/100th the original culture volume of PBS. Stocks were titred using the IncuCyte and snap-frozen in dry ice/ethanol prior to storage at −80 0C.
Approximately 2×106 IU of virus were diluted into 200 uL PBS and treated with either DMSO or p7i at a final concentration of 10 μM. Suspensions were then layered over a pre-formed 10-40 % (v/v) iodixinol gradient in a 2.2 mL open-topped mini-ultracentrifuge tube. Gradients were then centrifuged at 150 000 x g for 3 h at 4°C in a S55S Sorvall rotor prior to harvesting into twelve equal fractions. 10 μL was removed for infectivity testing and 50 μL for fluorimetry after adjusting to 0.1 % (v/v) Triton-X100 to lyse virions.
Prototypic p7i
Rimantadine hydrochloride was purchased from Chembridge, amantadine hydrochloride from SIGMA and NN-DNJ from Toronto Biochemicals. BIT225 was synthesised via 5-bromo-2-naphthoic acid and 5-(1-methyl-1H-pyrazol-4-yl)-2-naphthoic acid according to the patent detail (US20150023921A1); analytical data was consistent with the expected structure.
Generation of optimised heptameric 3ZD0-based p7 channel structure
The initial heptameric channel model was constructed using the Maestro programme (Schrödinger) based upon the monomeric unit from the 3ZD0 NMR structure as described12. A heptameric bundle arrangement of protomers was oriented with His17 oriented towards the lumen equidistant from a centroid atom placed in the middle of the lumen to serve as a rotational centre with multiple energy minimisation steps. Iterative rotation of protomers in an octanol environment generated solutions. The preferred model was then refined using molecular dynamics simulations (methods described below)
Structure guided molecular dynamics simulations and compound docking
Molecular Docking
The p7 structure was energy minimized using the default energy minimization scheme in MOE software (Version 2015:1001, www.chemcomp.com), AMBER 94 force field was used with ɛ = 2. Hydrogen atoms were added and partial charges were assigned using MOE. The energy minimization was carried out by restraining protein backbone atoms as rigid. The pairwise alignment of the structures, the one before minimization and the one after minimization, gives an RMSD of 0.10 Å.
Ligands were prepared using MOE software. Hydrogens atoms were added using the protonate 3D option and then partial charges were assigned with MMFF94 force field default parameters. Energy minimizations were done using MMFF94 force field with a root mean square gradient of 0.1 kcal Mol-1.Å-1 and gas phase solvation model.
For docking, L20 or F20 residues were selected from each monomer of the wild type and mutated proteins respectively. Receptor sites with a radius of 5 Å is defined around L20 and F20 residue of each chain. The energy minimized ligands were loaded into the MOE graphical user interface. The ‘rigid receptor’ protocol of MOE was used, where side chains of the protein were kept ‘fixed’ during the forcefield base refinement. Ligand placement was performed using the Triangle Matcher protocol, where the active site was defined using the α-spheres and the centre of that spheres are triplets of atoms. These triplets represent the location of tight packing. Poses are generated by superposing the ligand atom triplets onto the centre of the α-spheres. The top 1000 poses received after the placement were then used to score using the London dG scoring function which is an energy term summing contributions from ligand entropy from its topology or ligand flexibility, geometric imperfections of protein-ligand and metal-ligand interactions and desolvation energy. The top 30 poses ranked accordingly based on the London dG scores are then taken for a forcefield based refinement within the rigid receptor. The resulting poses are then rescored using Generalized Born Volume Integral/Weighted Surface Area dG (GBVI/WSA dG) scoring function. At the end final 30 poses were ranked accordingly while removing the duplicate poses.
MD simulations
To simulate all the systems, Amber ff99SB-ILDN force field (ff) 33 with Amber/Berger combination of ff was used with Gromacs 4.5.5. The POPC lipid ((1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) topology was received from Cordomí et al.34 while for the calculation of ligand parameters the generalized amber force field (GAFF) was used. The partial charges (ESP) were estimated using a HF/6-31G* basis set and Antechamber package was used for restrained electrostatic potential (RESP) fitting.
Each of the bundles, (wildtype docked with JK3/32 and Rimantadine, L20F mutated bundle docked with JK3/32 and Rimantadine, and only protein) was inserted into patches of hydrated and pre-equilibrated POPC lipids. The overlapped lipids were removed and after steps of minimization (2000 steps of steepest decent and 5000 steps of conjugated gradient), it was equilibrated for a total of 1.9 ns by restraining the positions of the heavy atoms proteins and ligands. The systems were brought to equilibrium by gradually increasing the temperature of the systems from 100 K to 200 K and 310 K. During the initial equilibration, the peptides and ligands were fully restrained by applying a harmonic potential with a force constant, k = 1000 kJ mol-1 nm-2. The simulations at temperatures 100 K and 200 K were run for 200 ps each followed by simulation at 310 K for 500ps. Once the systems were equilibrated at 310K, the restraints were released by running 3 short (500 ps) simulations, one with k = 500 kJ mol-1 nm-2 the consecutive two simulations with k = 250 kJmol-1 nm-2 and finally 0 kJmol-1 nm-2.
For all the simulations SPC/E water model 35 was employed and ion parameters proposed by Joung et al. were adopted 36. A Nosé-Hoover thermostat with a coupling time of 0.1 ps coupling separately to the temperature of the peptide, lipid, and the water molecules and Berendsen barostat with a coupling time of 2.0 ps were used during the MD simulations.
For the systems without any docked ligand and with JK3/32 docked, 14 Cl- ions were added to neutralize the simulation box while for the systems with rimantadine docked it was 15 Cl-ions. All the systems were consisted of 449 lipids which were hydrated with 14605-14606 water molecules (with 14 and 15 Cl-ions respectively).
Compound synthesis and purification
Reagents and solvents were obtained from commercial supplier and used without further purification. Thin layer chromatography (TLC) analyses were conducted using silica gel (aluminium foil backing) plates and visualised under UV radiation in a dark-box. Compound purification was effected using gradient elution on a Biotage Isolera-4 running SiO2 cartridges. HPLC-MS was performed on a Bruker Daltronics spectrometer running a gradient of increasing acetonitrile (0 to 100%) in water containing 0.1% TFA at 1 ml.min-1 on a short path 18C reverse phase column detecting compounds with both a diode array detector and a Bruker Mass spectrum analyser. HRMS experiments were conducted on a Bruker MaxisImpact time-of-flight spectrometer operating in a positive ion mode with sodium formate as an internal standard. 1H, 13C experiments were recorded using either a Bruker DRX 500 instrument or a Bruker DPX 300 operating at 298K, by solubilising the sample in deuterated chloroform (CDCl3) with internal standard tetramethylsilane (TMS), CD3OD, d6-DMSO, or d6-acetone as the NMR solvent. Chemical shifts were expressed as parts per million (ppm) and the splitting signals of NMR assigned as s (singlet), d (doublet), t (triplet), dd (doublet of doublet), br (broad) or m (multiplet).
General experimental methods
Synthesis of alexafluor-JK3/32 adduct (JK3/32-488)
3-[(dimethylamino)methylidene)]-2,3-dihydro-1H-indol-2-one

Dimethylformamide dimethylacetal (1.2 g, 10 mmol) was added with cooling to a suspension of oxindole (1.30 g, 9.80 mmol) in chloroform (15 ml) before the contents were stirred at room temperature for 15 mins, and heated under reflux for 4 hours. The contents were cooled to room temperature and concentrated under reduced pressure to afford an orange solid, which was recrystallized from ethanol-diethyl ether to afford a yellow solid which was used crude in the subsequent reaction.
3-[(dimethylamino)methylidene]-1-{(4-fluorophenyl)methyl]-2,3-dihydro-1H-indol-2-one

Cesium Carbonate (580 mg, 1.8 mmol) was added in a single portion with stirring to an acetonitrile (5 ml) suspension of 3-[(dimethylamino)methylidene)]-2,3-dihydro-1H-indol-2-one (200 mg, 1.1 mmol) at room temperature and the contents were stirred for a further 30 mins, prior to the addition of 4-fluorobenzylbromide (300 mg, 1.6 mmol) via a syringe, after which the contents were stirred for 4 hours at room temperature and then heated at 70°C for a further hour. The reaction mixture was cooled to room temperature, and transferred to a separating funnel, whereupon the reaction mixture was diluted with water and extracted into dichloromethane (2 × 15 ml). After drying over sodium sulfate, the contents were filtered and the organic phase concentrated under reduced pressure to yellow oil, which was chromatographed (SiO2; gradient elution; Hexane-EtOAc = 1:1 to 100 % EtOAc) to afford the desired product as pale yellow solid (210 mg, 67%) which was carried forward to the next step without additional characterisation.
(3Z)-1-[(4-fluorophenyl)methyl]-3-({[4-(prop-2-yn-1-yloxy)phenyl]amino}methylidene)-2,3-dihydro-1H-indol-2-one (1191-146)

An ethanol (3 ml) suspension of 3-[(dimethylamino)methylidene]-1-{(4-fluorophenyl)methyl]-2,3-dihydro-1H-indol-2-one (53 mg, 0.18 mmol) was treated with 4-aminophenylpropargylether (35 mg, 0.23 mmol) and p-toluenesulphonic acid (52 mg, 0.28 mmol), and the contents were heated under reflux with stirring for 18 hours until HPLC-MS had indicated consumption of the dimethylenamide. Upon gradual cooling to 60°C, together with intermittent scratching of the inside of the flask with a micro-spatula, there was obtained a precipitate that was filtered at 50°C, washed with cold ethanol and recrystallized from chloroform-diethyl ether to yield the desired product as a yellow solid (26 mg, 37%). IR: vmax/cm-1 (solid): 3080, 1633, 1610. HPLC-MS: 2.24 min, 399.5 [M+H]+. 1H NMR (500 MHz, CDCl3): 10.64 (d, J = 13 Hz, 1H), 7.87 (d, J = 13 Hz, 1H), 7.30 (d, J = 7 Hz, 1H), 7.21 (br, 3H), 7.06 (d, J = 8.5 Hz, 2H), 6.93 (m, 4H), 6.71 (d, J = 8.5 Hz, 1H), 4.96 (s, 2H), 4.63 (d, J = 2.5 Hz, 2H), 2.47 (d, J = 2.5 Hz, 1H); 13C (300 MHz, CDCl3): 157.6, 149.5, 137.7, 132.8, 132.3, 129.2, 124.0, 121.3, 117.7, 116.4, 116.0, 115.7, 108.5, 99.6, 98.6, 78.4, 75.7, 56.3, 42.5.
Alexafluor-JK3/32-triazole (JK3/32-488)

Alexafluor-488 (0.5 mg) was supplied in a light-proof centrifuge tube, to which was added a tertbutanol (0.2 mL) suspension of the (3Z)-1-[(4-fluorophenyl)methyl]-3-({[4-(prop-2-yn-1-yloxy)phenyl]amino}methylidene)-2,3-dihydro-1H-indol-2-one (1191-146) (0.25 mg), followed by a solution (0.1 mL) aqueous ascorbic acid (0.35 mg). The contents were vortexed for 30 mins, before an aqueous solution (0.05 mL) of copper sulfate (0.20 mg) was added, and the contents shaken overnight at 40°C. Following this, the contents concentrated on a Genevac to a residue which was re-suspended in DMSO (0.20 mL) and centrifuged to disperse the solutes.
Synthesis of JK3/32
1-benzyl-3-[(dimethylamino)methylidene]-2,3-dihydro-1H-indol-2-one

A DMF (4ml) suspension of (3Z)-3-[(dimethylamino)methylidene]-1,3-dihydro-2H-indol-2-one (300 mg, 1.6 mmol) was cooled to 0°C via an ice-bath with rapid stirring. Sodium hydride (125 mg, 60% dispersion) was added in three portions over 10 mins, and the resulting yellow suspension stirred for a further 20 mins at 5°C, prior to the addition of benzyl bromide (330 mg, 1.90 mmol), and the contents left to stir for a further 45 minutes and allowed to warm to 25°C over this period. Saturated ammonium chloride solution was added dropwise with cooling, and the contents transferred with ethyl acetate to a separating funnel whereupon the organic phase was removed, washed with water and brine, and dried over sodium sulfate. Evaporation and chromatography (SiO2; gradient elution; hexane: EtOAc = 2:1 to 100 % EtOAc) afforded the desired product (210 mg, 47%) as an oil which solidified upon standing. HPLC-MS: 1.89 mins, 279.1 [M+H]+. Spectral characteristics coincident with those reported37. IR: vmax/cm-1 (solid): 1625: 1H NMR (500 MHz, CDCl3): 3.34–3.37, (6H, m, 2CH3), 5.06 (2H, s, CH2Ph), 6.74–7.42 (8H, m, ArH), 7.49 (1H, d, H-4), 7.68 (1H, s, CH).
1-benzyl-3-[(4-methoxyphenyl)aminomethylidene]-2,3-dihydro-1H-indol-2-one (JK3/32)

An ethanol (3 ml) solution of p-toluenesulfonic acid (26 mg, 0.14 mmol) was combined with 4-anisidine (17 mg, 0.14 mmol) at room temperature, and sonicated briefly to disperse the contents. 1-benzyl-3-[(dimethylamino)methylidene]-2,3-dihydro-1H-indol-2-one (35 mg, 0.13 mmol) was added in a single portion and the contents heated under reflux with stirring for 12 hours to provide a precipitate, which was diluted with methanol (0.5 ml) and filtered at 50°C to afford the desired product as a yellow solid (18 mg, 39%). IR: vmax/cm-1 (solid): 3070, 1631, 1610. HPLC-MS: 2.21 mins, 357.1[M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.76 (d, J = 13 Hz, 1H), 7.97 (d, J = 13 Hz, 1H), 7.38 (d, J = 6.5 Hz, 1H), 7.31 (m, 3H), 7.24 (m, 2H), 7.12 (d, J = 8.5 Hz, 2H), 7.03 (m, 2H), 6.94 (d, J = 8.5 Hz, 2H), 6.82 (d, J = 8.0 Hz, 1H), 5.07 (s, 2H), 3.82 (s, 3H); 13C NMR (300 MHz, d6-DMSO) δ 167.8, 156.1, 139.4, 137.5, 137.1, 133.4, 128.6, 127.3, 127.0, 123.4, 120.7, 118.9, 117.8, 116.7, 115.1, 108.4, 97.4, 55.6, 42.3. HRMS (m/z): [M+H]+ calcd. for C23H20N2O2, 357.1598, found 357.1602.
(3Z)-3-[(4-methoxyanilino)methylidene]-1-phenyl-1,3-dihydro-2H-indol-2-one (JK3-42)

To N-phenyloxindole (0.9 mmol) was added dimethylformamide dimethyl acetal (5 mL) and the mixture heated at 70°C for 1 hour. The mixture was concentrated, dissolved in ethanol (3 mL) and then p-toluenesulfonic acid (48 mg, 0.24 mmol) and 4-anisidine (30 mg, 0.24 mmol) added at room temperature and the contents heated under reflux with stirring for 12 hours to provide a precipitate which was diluted with methanol (0.5 mL) and filtered at 50°C to afford the desired product as a pale yellow solid (19 mg, 42%). IR: vmax/cm-1 (solid): 3080, 1633, 1610. HPLC-MS: 2.13 mins, 345.1[M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.78 (d, J = 13 Hz, 1H), 7.94 (d, J = 13 Hz, 1H), 7.33 (d, J = 6.9 Hz, 1H), 7.25 (m, 3H), 7.18 (m, 2H), 7.12 (d, J = 8.3 Hz, 2H), 7.03 (m, 2H), 6.94 (d, J = 8.3 Hz, 2H), 6.78 (d, J = 8.3 Hz, 1H), 3.87 (s, 3H); 13C NMR (300 MHz, d6-DMSO) δ 165.8, 158.1, 141.4, 138.5, 137.3, 135.1, 128.6, 127.6, 127.0, 125.9, 124.8, 123.1, 117.9, 116.7, 116.1, 109.3, 96.4, 55.7. HRMS (m/z): [M+H]+ calcd. for C22H18N2O2, 345.1575, found 345.1601.
(3Z)-3-[(4-methoxyanilino)methylidene]-1,3-dihydro-2H-indol-2-one (JK3-38)

An ethanol (3 ml) solution of p-toluenesulfonic acid (26 mg, 0.14 mmol) was combined with 4-anisidine (17 mg, 0.14 mmol) at room temperature, and sonicated briefly to disperse the contents. (3Z)-3-[(dimethylamino)methylidene]-1,3-dihydro-2H-indol-2-one (0.14 mmol) was added in a single portion and the contents heated under reflux with stirring for 12 hours to provide a precipitate, which was diluted with methanol (0.5 ml) and filtered at 50°C to afford the desired product as a yellow solid (15 mg, 39%). IR: vmax/cm-1 (solid): 3100, 3050, 1640, 1610. HPLC-MS: 1.97 mins, 266.1[M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.50 (d, J = 11.0 Hz, 1H), 9.99 (s, 1H), 8.08 (d, J = 11.0 Hz, 1H), 7.55 (d, J = 6.9 Hz, 1H), 7.25 (m, 2H), 6.85-6.92 (4H, m), 6.78 (d, J = 8.3 Hz, 1H), 3.87 (s, 3H); 13C NMR (300 MHz, d6-DMSO) δ 165.8, 158.1, 141.4, 138.5, 135.1, 129.6, 126.0, 124.9, 124.0, 123.1, 117.5, 116.0, 115.1, 107.3, 93.4, 55.7. HRMS (m/z): [M+H]+ calcd. for C16H14N2O2, 266.1102, found 266.1197.
(Z)-1-benzyl-3-(2-(4-methoxyphenyl)hydrazono)indolin-2-one (21-RS-7)

To a solution of N-benzyl isatin (0.05 g, 0.21 mmol) in ethanol (5 mL) was added p-methoxyphenylhydrazine hydrochloride (0.3 mL, 0.21 mmol) and the mixture heated at reflux for 2 hours. The reaction mixture was cooled to ambient temperature and the resultant precipitate filtered and dried to give the title compound as a yellow powder (0.04 g, 0.11 mmol, 50%). IR: vmax/cm-1 (solid): 1663, 1610. HPLC-MS (ES): 2.42 min, m/z = 737.6 (2M+Na)+ 1H NMR (300 MHz, DMSO-d6): δ 12.75 (s, 1H), 7.59 (dd, J = 7.2, 0.9 Hz 1H), 7.46 (d, J = 9.0 Hz, 2H), 7.38 – 7.25 (m, 5H), 7.22 (dd, J = 7.7, 1.3 Hz, 1H), 7.15 – 7.01 (m, 2H), 6.99 (d, J = 9.0 Hz, 2H), 5.03 (s, 2H), 3.76 (s, 3H) ppm; 13C NMR (300 MHz, CDCl3): δ 162.3, 156.2, 139.8, 136.4, 135.9, 128.8, 127.7, 127.4, 127.3, 125.6, 122.5, 121.6, 118.6, 115.7, 114.8, 109.2, 55.6, 43.2 ppm;; HRMS (m/z): [M+H]+ calcd for C22H19N3O2Na requires 380.1369 Found: 380.1374 (M+Na)+: vmax/cm-1 (solid): 1663, 1610; M.pt: 145-147 °C.
(Z)-1-benzyl-3-(2-phenylhydrazono)indolin-2-one (21-RS-8)

To a solution of N-benzyl isatin (0.05 g, 0.21 mmol) in ethanol (5 mL) was added phenylhydrazine hydrochloride (0.03 mg, 0.21 mmol) and the mixture heated at reflux for 2 hours. The reaction mixture was cooled to ambient temperature and the resultant precipitate filtered and dried to give the title compound as a yellow powder (0.03 g, 0.09 mmol, 42%).
IR: vmax/cm-1 (solid): 3060, 1663. 1H NMR (300 MHz, DMSO-d6): δ 12.72 (s, 1H), 7.64 – 7.59 (m, 1H), 7.52 – 7.46 (m, 2H), 7.43 – 7.30 (m, 6H), 7.30 – 7.22 (m, 2H), 7.16 – 7.07 (m, 1H), 7.10 – 7.01 (m, 2H), 5.03 (s, 2H) ppm; 13C NMR (300 MHz, DMSO-d6): 161.1, 142.4, 140.0, 136.3, 129.5, 128.7, 128.3, 127.5, 127.4, 126.5, 123.1, 122.5, 120.5, 118.5, 114.3, 109.8 ppm; HPLC-MS (ES): 2.07 min, m/z=677.7 (2M+Na)+; HRMS (m/z): [M+H]+ calcd for C21H17N3ONa requires 350.1264, Found: 350.1267 (M+Na)+,.; HPLC: RT = 4.51 min (100%); M.pt: 134-136 °C.
(Z)-3-(2-(4-methoxyphenyl)hydrazono)-1-phenylindolin-2-one (21-RS-9)

To a solution of N-phenyl isatin (0.05 g, 0.22 mmol) in ethanol (5 mL) was added p-methoxyphenylhydrazine hydrochloride (0.04 mg, 0.22 mmol) and the mixture heated at reflux for 2 hours. The reaction mixture was cooled to ambient temperature and the resultant precipitate filtered and dried to give the title compound as a yellow powder (0.06 g, 0.17 mmol, 77%).
IR: vmax/cm-1 (solid): 3186, 1673. 1H NMR (300 MHz, CDCl3): δ 12.88 (s, 1H), 7.69 – 7.60 (m, 1H), 7.52 – 7.45 (m, 2H), 7.43 – 7.33 (m, 4H), 7.26 (d, J = 9.0 Hz, 2H), 7.17 – 7.05 (m, 2H), 6.86 (d, J = 8.9 Hz, 2H), 3.75 (s, 3H) ppm; 13C NMR (300 MHz, CDCl3): δ 161.6, 156.2, 140.3, 136.3, 133.9, 129.6, 128.1, 127.4, 126.4, 125.3, 123.0, 121.6, 118.6, 115.7, 114.8, 109.7, 55.6 ppm; HPLC-MS (ES):: RT = 2.46 min, m/z= 709.9 (2M+Na)+; HRMS (m/z): [M+H]+ calcd for C21H17N3O2Na requires 366.1209: 366.1209 (M+Na)+,;;; M.pt: 195-197 °C.
1-benzyl-3-(2-(3,4-dimethoxyphenyl)-2-oxoethyl)-3-hydroxyindolin-2-one (21-RS-11 (aka “R21”))

To a microwave vial was charged N-benzyl isatin (0.20 g, 0.84 mmol), 3,4-dimethoxyacetophenone (0.17 g, 0.93 mmol), diethylamine (2 drops) and ethanol (2 mL). The reaction was heated at 100 °C and 300 W within a microwave reactor for 1-2 hours. The crude product was purified via column chromatography (50:50 hexane:ethyl acetate) to yield the title compound as a cream powder (0.24 g, 0.58 mmol, 68%). IR: vmax/cm-1 (solid): 3329, 1668, 1598. 1H NMR (300 MHz, CDCl3): δ 7.54 (d, J = 8.4 Hz, 1H), 7.48 (s, 1H), 7.43 (d, J = 7.4 Hz), 7.40 – 7.29 (m, 5H), 7.21 (t, J = 7.8 Hz, 1H), 7.01 (t, J = 7.5 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 6.74 (d, J = 7.8 Hz, 1H), 4.95 (s, 2H), 4.83 (br s, 1H), 3.95 (s, 3H), 3.91 (s, 2H), 3.86 (d, J = 17.6 Hz, 1H), 3.59 (d, J = 17.1 Hz, 1H) ppm; 13C NMR (300 MHz, CDCl3): δ 196.8, 176.6, 153.9, 149.0, 142.8, 135.5, 130.2, 129.8, 129.6, 128.8, 127.7, 127.3, 124.1, 123.3, 123.1, 110.0, 109.9, 109.7, 80.5, 74.7, 56.2, 56.0, 43.9 ppm; HPLC-MS (ES): RT = 1.87 min, m/z = 857.6 (2M+Na)+; HRMS (m/z): [M+H]+ calcd for C25H23NO5Na requires 440.1468, Found: 440.1488 (M+Na)+. M.pt: 157-159 °C.
1-benzyl-3-(2-(3,4-dimethoxyphenyl)-2-oxoethylidene)indolin-2-one (21-RS-17)

To a stirred solution of 1-benzyl-3-(2-(3,4-dimethoxyphenyl)-2-oxoethyl)-3-hydroxyindolin-2-one (21-RS-11) (0.04 g, 0.08 mmol) in acetic acid (5 mL) was added 12M aqueous hydrochloric acid (2 mL). The mixture was heated at 80 °C for 0.5 hours then stirred at ambient temperature for 2 days. The mixture was quenched with cold aqueous sodium bicarbonate (20 mL), extracted with ethyl acetate (3×20 mL) and concentrated to dryness. The crude product was purified via column chromatography (50:50 hexane:ethyl acetate) to give the title compound as an orange powder (0.02 g, 0.05 mmol, 57%). IR: vmax/cm-1 (solid): 1705, 1599. 1H NMR (300 MHz, CDCl3): δ 8.19 (d, J = 7.6 Hz, 1H), 7.88 (s, 1H), 7.72 (dd, J = 8.4, 2.0 Hz, 1H), 7.61 (d, J = 2.0 Hz, 1H), 7.31 – 7.12 (m, 6H), 6.96 – 6.83 (m, 2H), 6.64 (d, J = 7.8 Hz, 1H), 4.91 (s, 2H), 3.92 (d, J = 2.4 Hz, 6H) ppm; 13C NMR (300 MHz, CDCl3): δ 189.6, 168.2, 154.2, 149.5, 144.9, 135.6, 135.5, 132.2, 130.8, 128.9, 127.8, 127.6, 127.3, 127.2, 124.4, 122.8, 110.1, 109.2, 105.0, 80.2, 56.2, 56.1, 43.9 ppm; HPLC-MS (ES): RT = 2.18 min, m/z = 00.1 (M+H)+; HRMS (m/z): [M+H]+ calcd for C25H21NO4Na requires 422.1363 Found: 422.1356 (M+Na)+,; M.pt: 146-148 °C.
(3Z)-1-benzyl-3-[(2-methoxyanilino)methylidene]-1,3-dihydro-2H-indol-2-one (1191-104)

An ethanol (3 mL) solution of p-toluenesulfonic acid (52 mg, 0.27 mmol) was combined with 4-anisidine (34 mg, 0.27 mmol) at room temperature, and sonicated briefly to disperse the contents. 1-benzyl-3-[(dimethylamino)methylidene]-2,3-dihydro-1H-indol-2-one (68 mg, 0.27 mmol) was added in a single portion and the contents heated under reflux with stirring for 12 hours to provide a precipitate, which was diluted with methanol (0.5 mL) and filtered at 50°C to afford the desired product as a yellow solid (70 mg, 75%). IR: vmax/cm-1 (solid): 3079, 1630. HPLC-MS: 2.28 min, 357.1 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 11.02 (d, J = 12.5 Hz, 1H), 8.10 (d, J = 12.5 Hz, 1H), 7.43 (d, J = 8.5 Hz, 1H), 7.32 (m, 6H), 7.05 (m, 4H), 6.99 (d, J = 8.0 Hz, 1H), 6.80 (d, J = 7.8 Hz, 1 H), 5.12 (s, 2H), 4.09 (s, 3H); 13C NMR (125 MHz, CDCl3) 157.6, 149.1, 132.7, 131.7, 130.4, 125.8, 123.5, 122.0, 119.2, 118.3, 115.7, 111.8, 110.8, 107.5, 105.9, 103.8, 97.2, 50.7, 37.8.
4-{[(Z)-(1-benzyl-2-oxo-1,2-dihydro-3H-indol-3-ylidene)methyl]amino}benzonitrile (1191-112)

An ethanol (3 mL) solution of p-toluenesulfonic acid (52 mg, 0.27 mmol) was combined with 4-aminophenylnitrile (33 mg, 0.27 mmol) at room temperature. 1-benzyl-3-[(dimethylamino)methylidene]-2,3-dihydro-1H-indol-2-one (68 mg, 0.27 mmol) was added in a single portion and the contents heated under reflux with stirring for 12 hours to provide a precipitate, which was diluted with methanol (0.5 mL) and filtered at 50°C to afford the desired product as a yellow solid (50 mg, 52%). IR: vmax/cm-1 (solid): 3079, 2127, 1630. HPLC-MS: 2.20 min, 352.5 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.89 (d, J = 13.5 Hz, 1H), 7.89 (d, J = 12.8 Hz, 1H), 7.58 (d, J = 7.5 Hz, 2H), 7.35 (d, J = 9.0 Hz, 1H), 7.24 (m, 4H), 7.18 (m, 1H), 7.12 (d, J = 7.5 Hz, 2H), 7.04 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 6.0 Hz, 1H), 6.75 (d, J = 6.0 Hz, 1H), 4.92 (s, 2H); 13C NMR (125 MHz, CDCl3) 153.8, 143.6, 134.1, 130.8, 128.9, 127.7, 127.2, 125.7, 122.6, 121.8, 118.6, 116.9, 115.1, 109.16, 105.7, 104.6, 93.1, 43.4.
(3Z)-1-benzyl-6-fluoro-3-[(4-methoxyanilino)methylidene]-1,3-dihydro-2H-indol-2-one (1191-121)

A DMF (4ml) suspension of (3Z)-3-[(dimethylamino)methylidene]-6-fluoro-1,3-dihydro-2H-indol-2-one (300 mg, 1.60 mmol) was cooled to 0°C via an ice-bath with rapid stirring. Sodium hydride (125 mg, 60% dispersion) was added in three portions over 10 mins, and the resulting yellow suspension stirred for a further 20 mins at 5°C, prior to the addition of benzyl bromide (1.80 mmol) and the contents left to stir for a further 45 minutes and allowed to warm to 25°C over this period. Saturated ammonium chloride solution was added dropwise with cooling, and the contents transferred with ethyl acetate to a separating funnel whereupon the organic phase was removed, washed with water and brine, and dried over sodium sulfate. Evaporation and chromatography (SiO2; gradient elution; hexane:EtOAc = 2:1 to 100 % EtOAc) afforded the desired product as an oil which solidified upon standing.. This was then added to an ethanol (3 mL) solution of p-toluenesulfonic acid (52 mg, 0.27 mmol) and 4-anisidine (34 mg, 0.27 mmol) and the contents heated under reflux with stirring for 12 hours to provide a precipitate, which was diluted with methanol (0.5 mL) and filtered at 50°C to afford the desired product as a pale yellow solid (45 mg, 48%). IR: vmax/cm-1 (solid): 3099, 1630. HPLC-MS: 2.24 min, 375.3 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.79 (d, J = 13.5 Hz, 1H), 7.92 (d, J = 13.5 Hz, 1H), 7.28 (m, 5H), 7.12 (d, J = 10 Hz, 2H), 7.07 (m, 1H), 6.93 (d, J = 8.5 Hz, 2H), 6.70 (m, 2H), 5.05 (s, 2H), 3.82 (s, 3H); 13C NMR (300 MHz, CDCl3): 155.4, 148.1, 138.6, 133.5, 132.2, 128.7, 127.7, 127.4, 127.2, 126.0, 125.2, 118.1, 115.3, 109.9, 108.9, 103.3, 99.2, 55.8, 43.8.
4-({(3Z)-5-fluoro-3-[(4-methoxyanilino)methylidene]-2-oxo-2,3-dihydro-1H-indol-1-yl}methyl)benzonitrile (1191-120)

A DMF (4ml) suspension of (3Z)-3-[(dimethylamino)methylidene]-5-fluoro-1,3-dihydro-2H-indol-2-one (300 mg, 1.6 mmol) was cooled to 0°C via an ice-bath with rapid stirring. Sodium hydride (125 mg, 60% dispersion) was added in three portions over 10 mins, and the resulting yellow suspension stirred for a further 20 mins at 5°C, prior to the addition of 4-cyanophenylmethyl bromide (369 mg, 1.90 mmol) and the contents left to stir for a further 45 minutes and allowed to warm to 25°C over this period. Saturated ammonium chloride solution was added dropwise with cooling, and the contents transferred with ethyl acetate to a separating funnel whereupon the organic phase was removed, washed with water and brine, and dried over sodium sulfate. Evaporation and chromatography (SiO2; gradient elution; hexane:EtOAc = 2:1 to 100 % EtOAc) afforded the desired product as an oil. This was then added to an ethanol (3 mL) solution of p-toluenesulfonic acid (52 mg, 0.27 mmol) and 4-anisidine (34 mg, 0.27 mmol) at room temperature and the contents heated under reflux with stirring for 12 hours to provide a precipitate which was diluted with methanol (0.5 mL) and filtered at 50°C to afford the desired product as a yellow solid (51 mg, 48%). IR: vmax/cm-1 (solid): 3130, 2145, 1620. HPLC-MS: 2.02 min, 400.4 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.75 (d, J = 13 Hz, 1H), 7.95 (d, J = 13 Hz, 1H), 7.61 (d, J = 10 Hz, 2H), 7.38 (d, J = 8.5 Hz, 2H), 7.12 (d, J = 10 Hz, 2H), 7.09 (m, 1H), 6.94 (d, J = 8.5 Hz, 2H), 6.73 (t, J = 7.5 Hz, 1H), 6.60 (dd, J = 8.5 Hz, 4.2 Hz, 1H), 5.1 (s, 2H), 3.82 (s, 3H); 13C NMR (300 MHz, CDCl3) 169.1, 160.3, 157.0, 142.2, 139.1, 133.1, 132.9, 127.9, 118.7, 118.1, 115.2, 111.6, 110.4, 108.6, 55.9, 43.1.
(3Z)-5-fluoro-1-[(4-fluorophenyl)methyl]-3-[(4-methoxyanilino)methylidene]-1,3-dihydro-2H-indol-2-one (1191-124)

A DMF (4ml) suspension of (3Z)-3-[(dimethylamino)methylidene]-5-fluoro-1,3-dihydro-2H-indol-2-one (300 mg, 1.6 mmol) was cooled to 0°C. Sodium hydride (125 mg, 60% dispersion) was added in three portions over 10 mins, and the resulting yellow suspension stirred for a further 20 mins at 5°C, prior to the addition of 4-fluorophenylmethyl bromide (1.80 mmol), and the contents left to stir for a further 45 minutes. Saturated ammonium chloride solution was added dropwise with cooling, and the contents transferred with ethyl acetate to a separating funnel whereupon the organic phase was removed, washed with water and brine, and dried over sodium sulfate. Evaporation and chromatography (SiO2; gradient elution; hexane:EtOAc = 2:1 to 100 % EtOAc) afforded the desired intermediate as an oil. This was then added to an ethanol (3 mL) solution of p-toluenesulfonic acid (52 mg, 0.27 mmol) and 4-anisidine (34 mg, 0.27 mmol) at room temperature and the contents heated under reflux with stirring for 12 hours to provide a precipitate which was diluted with methanol (0.5 mL) and filtered at 50°C to afford the desired product as a yellow solid (51 mg, 53%). IR: vmax/cm-1 (solid): 3115, 1625. HPLC-MS: 2.26 min, 393.2 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.71 (d, J = 13.5 Hz, 1H), 7.86 (d, J = 13.0 Hz, 1H), 7.19 (br, 1H), 7.05 (d, J = 8.5 Hz, 2H), 6.99 (d, J = 8.5 Hz, 1H), 6.92 (m, 3H), 6.87 (d, J = 7.7 Hz, 2H), 6.66 (t, J = 7.7 Hz, 1H), 6.59 (br, 1H), 4.93 (s, 2H), 3.76 (s, 3H); 13C NMR (300 MHz, CDCl3): 157.9, 149.6, 140.2, 138.0, 133.4, 133.2, 132.9, 128.9, 122.2, 118.0, 115.8, 115.6, 115.2, 110.2, 108.8, 103.4, 98.1, 55.6, 42.6.
(3Z)-1-[(3,5-dimethyl-1,2-oxazol-4-yl)methyl]-3-[(4-methoxyanilino)methylidene]-1,3-dihydro-2H-indol-2-one (1191-106)

A DMF (4mL) suspension of (3Z)-3-[(dimethylamino)methylidene]-1,3-dihydro-2H-indol-2-one (300 mg, 1.60 mmol) was cooled to 0°C via an ice-bath with rapid stirring. Sodium hydride (125 mg, 60% dispersion) was added in three portions over 10 mins, and the resulting yellow suspension stirred for a further 20 mins at 5°C, prior to the addition of 2,5-dimethylisoxazole-4-methylchloride (1.80 mmol) and the contents left to stir for a further 45 minutes. Saturated ammonium chloride solution was added dropwise with cooling, and the contents transferred with ethyl acetate to a separating funnel whereupon the organic phase was removed, washed with water and brine, and dried over sodium sulfate. Evaporation and chromatography (SiO2; gradient elution; hexane:EtOAc = 2:1 to 100 % EtOAc) afforded an intermediate as an oil. This was then added to an ethanol (3 mL) solution of p-toluenesulfonic acid (48 mg, 0.24 mmol) and 4-anisidine (30 mg, 0.24 mmol) at room temperature and the contents heated under reflux with stirring for 12 hours to provide a precipitate which was diluted with methanol (0.5 mL) and filtered at 50°C to afford the desired product as a pale yellow solid (57 mg, 54%). IR: vmax/cm-1 (solid): 3120, 1625. HPLC-MS: 1.91 min, 375.3 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.67 (d, J = 13 Hz, 1H), 7.97 (d, J = 13 Hz, 1H), 7.72 (d, J = 10 Hz, 1H), 7.39 (d, J = 10 Hz, 1H), 7.15 (d, J = 10 Hz, 1H), 7.08 (m, 2H), 6.96 (d, J = 10 Hz, 2H), 6.74 (d, J = 8 Hz, 1H), 4.84 (s, 2H), 3.86 (s, 3H), 2.42 (s, 3H), 2.25 (s, 3H); 13C NMR (300 MHz, CDCl3) 168.9, 166.9, 159.5, 156.7, 138.4, 137.0, 133.6, 129.0, 126.3, 123.9, 121.7, 117.9, 116.1, 115.3, 109.7, 108.5, 55.8, 32.5, 11.4, 10.6.
(3Z)-3-[(4-methoxyanilino)methylidene]-1-(2-phenylethyl)-1,3-dihydro-2H-indol-2-one (1191-137)

A DMF (4mL) suspension of (3Z)-3-[(dimethylamino)methylidene]-1,3-dihydro-2H-indol-2-one (300 mg, 1.6 mmol) was cooled to 0°C via an ice-bath with rapid stirring. Sodium hydride (125 mg, 60% dispersion) was added in three portions over 10 mins, and the resulting yellow suspension stirred for a further 20 mins at 5°C, prior to the addition of phenylethyl bromide (369 mg, 1.90 mmol), and the contents left to stir for a further 45 minutes and allowed to warm to 25°C over this period. Saturated ammonium chloride solution was added dropwise with cooling, and the contents transferred with ethyl acetate to a separating funnel whereupon the organic phase was removed, washed with water and brine, and dried over sodium sulfate. Evaporation and chromatography (SiO2; gradient elution; hexane:EtOAc = 2:1 to 100 % EtOAc) afforded an intermediate product as an oil which solidified upon standing. The material was used immediately in the subsequent reaction. The oil was added to an ethanol (3 mL) solution of p-toluenesulfonic acid (53 mg, 0.27 mmol) and 4-anisidine (35 mg, 0.27 mmol) at room temperature and the contents heated under reflux with stirring for 12 hours to provide a precipitate which was diluted with methanol (0.5 mL) and filtered at 50°C to afford the desired product as a pale yellow solid (49 mg, 44%). IR: vmax/cm-1 (solid): 3095, 1620.HPLC-MS: 2.29 min, 371.2 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.61 (d, J = 13 Hz, 1H), 7.86 (d, J = 13 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.22 (m, 3H), 7.17 (m, 2H), 7.03 (m, 3H), 6.99 (d, J = 7.0 Hz, 1H), 6.85 (d, J = 7.0 Hz, 2H), 6.81 (d, J = 8.0 Hz, 1H), 3.99 (t, J = 7.5 Hz, 2H), 3.77 (s, 3H), 2.95 (t, J = 7.5 Hz, 2H); 13C NMR (300 MHz, CDCl3) 166.2, 149.5, 137.6, 133.7, 133.1, 131.4, 129.6, 128.9, 128.6, 128.4, 127.7, 121.1, 117.6, 117.5, 115.1, 108.4, 98.5, 55.8, 34.7, 33.5.
(3Z)-1-[(4-fluorophenyl)methyl]-3-[(4-methoxyanilino)methylidene]-1,3-dihydro-2H-indol-2-one (1191-140)

A DMF (4mL) suspension of (3Z)-3-[(dimethylamino)methylidene]-1,3-dihydro-2H-indol-2-one (300 mg, 1.6 mmol) was cooled to 0°C via an ice-bath with rapid stirring. Sodium hydride (125 mg, 60% dispersion) was added in three portions over 10 mins, and the resulting yellow suspension stirred for a further 20 mins at 5°C, prior to the addition of 4-flurophenylbromide (1.65 mmol), and the contents left to stir for a further 45. Saturated ammonium chloride solution was added dropwise and the contents transferred with ethyl acetate to a separating funnel whereupon the organic phase was removed, washed with water and brine, and dried over sodium sulfate. Evaporation and chromatography (SiO2; gradient elution; hexane:EtOAc = 2:1 to 100 % EtOAc) afforded an oil which was added to ethanol (3 mL) and p-toluenesulfonic acid (53 mg, 0.27 mmol) and 4-anisidine (35 mg, 0.27 mmol) at room temperature and the contents heated under reflux with stirring for 12 hours to provide a precipitate which was diluted with methanol (0.5 mL) and filtered at 50°C to afford the desired product as a pale yellow solid (67 mg, 63%). IR: vmax/cm-1 (solid): 3105, 1635.HPLC-MS: 2.24 min, 375.3 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.63 (d, J = 13 Hz, 1H), 7.87 (d, J = 13 Hz, 1H), 7.31 (d, J = 7.5 Hz, 1H), 7.21 (m, 2H), 7.04 (d, J = 9 Hz, 2H), 6.94 (m, 3H), 6.85 (d, J = 8.5 Hz, 2H), 6.72 (d, J = 8.0 Hz, 1H), 4.97 (s, 2H), 3.75 (s, 3H); 13C NMR (300 MHz, CDCl3) 165.1, 156.5, 137.9, 137.4, 136.8, 135.3, 133.5, 132.6, 130.3, 128.8, 123.9, 123.8, 121.4, 117.1, 115.6, 115.1, 108.7, 98.1, 55.7, 42.7.
(3Z)-3-[(4-fluoroanilino)methylidene]-1-[(4-fluorophenyl)methyl]-1,3-dihydro-2H-indol-2-one (1191-141)

A DMF (4ml) suspension of (3Z)-3-[(dimethylamino)methylidene]-1,3-dihydro-2H-indol-2-one (300 mg, 1.6 mmol) was cooled to 0°C via an ice-bath with rapid stirring. Sodium hydride (125 mg, 60% dispersion) was added in three portions over 10 mins, and the resulting yellow suspension stirred for a further 20 mins at 5°C, prior to the addition of 4-flurophenylbromide (1.65 mmol), and the contents left to stir for a further 45. Saturated ammonium chloride solution was added dropwise and the contents transferred with ethyl acetate to a separating funnel whereupon the organic phase was removed, washed with water and brine, and dried over sodium sulfate. Evaporation and chromatography (SiO2; gradient elution; hexane:EtOAc = 2:1 to 100 % EtOAc) afforded the desired product as an oil which was used immediately in the next reaction. The oil was added to ethanol (3 mL) and p-toluenesulfonic acid (53 mg, 0.27 mmol) and 4-fluorophenylaniline (0.27 mmol) at room temperature and the contents heated under reflux with stirring for 12 hours to provide a precipitate which was diluted with methanol (0.5 mL) and filtered at 50°C to afford the desired product as a pale yellow solid (45 mg, 57%). IR: vmax/cm-1 (solid): 3130, 1615. HPLC-MS: 2.20 min, 362.4 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.60 (d, J = 12 Hz, 1H), 7.99 (d, J = 12 Hz, 1H), 7.23-7.39 (m, 3H), 7.15 (d, J = 9 Hz, 2H), 6.91 (m, 3H), 6.80 (d, J = 8.5 Hz, 2H), 6.77 (d, J = 7.9 Hz, 1H), 4.90 (s, 2H); 13C NMR (300 MHz, CDCl3) 165.1, 156.5, 139.1, 138.1, 136.7, 136.1, 133.5, 132.4, 131.1, 128.9, 124.0, 122.8, 121.8, 117.0, 114.1, 111.1, 104.6, 97.5, 58.9
(3Z)-5-fluoro-3-[(anilino)methylidene]-1-[(4-fluorophenyl)methyl]-1,3-dihydro-2H-indol-2-one (1191-125)

A DMF (4ml) suspension of (3Z)-3-[(dimethylamino)methylidene]-5-fluoro-1,3-dihydro-2H-indol-2-one (300 mg, 1.6 mmol) was cooled to 0°C via an ice-bath with rapid stirring. Sodium hydride (125 mg, 60% dispersion) was added in three portions over 10 mins, and the resulting yellow suspension stirred for a further 20 mins at 5°C, prior to the addition of 4-fluorobenzyl lbromide (1.65 mmol), and the contents left to stir for a further 45. Saturated ammonium chloride solution was added dropwise and the contents transferred with ethyl acetate to a separating funnel whereupon the organic phase was removed, washed with water and brine, and dried over sodium sulfate. Evaporation and chromatography (SiO2; gradient elution; hexane:EtOAc = 2:1 to 100 % EtOAc) afforded an oil which was used immediately in the next reaction. The oil was added to ethanol (3 mL) and p-toluenesulfonic acid (53 mg, 0.27 mmol) and aniline (0.27 mmol) at room temperature and the contents heated under reflux with stirring for 12 hours to provide a precipitate which was diluted with methanol (0.5 mL) and filtered at 50°C to afford the desired product as a pale yellow solid (43 mg). IR: vmax/cm-1 (solid): 3130, 1620. HPLC-MS: 2.30 min, 362.1 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.65 (d, J = 11.5 Hz, 1H), 7.99 (d, J = 11.5 Hz, 1H), 7.15-7.39 (m, 6H), 6.91-7.12 (m, 3H), 6.77 (m, 3H), 4.90 (s, 2H).
4-{[(Z)-(1-(4-fluoro)phenylmethyl-2-oxo-1,2-dihydro-5-fluoro-3H-indol-3-ylidene)methyl]amino}benzonitrile (1191-126)

A DMF (4ml) suspension of (3Z)-3-[(dimethylamino)methylidene]-5-fluoro-1,3-dihydro-2H-indol-2-one (300 mg, 1.6 mmol) was cooled to 0°C via an ice-bath with rapid stirring. Sodium hydride (125 mg, 60% dispersion) was added in three portions over 10 mins, and the resulting yellow suspension stirred for a further 20 mins at 5°C, prior to the addition of 4-fluorobenzylbromide (1.65 mmol), and the contents left to stir for a further 45. Saturated ammonium chloride solution was added dropwise and the contents transferred with ethyl acetate to a separating funnel whereupon the organic phase was removed, washed with water and brine, and dried over sodium sulfate. Evaporation and chromatography (SiO2; gradient elution; hexane:EtOAc = 2:1 to 100 % EtOAc) afforded an oil which was used immediately in the next reaction. The oil was added to ethanol (3 mL) and p-toluenesulfonic acid (53 mg, 0.27 mmol) and aniline (0.27 mmol) at room temperature and the contents heated under reflux with stirring overnight to provide a precipitate which was diluted with methanol (2 × 0.5 mL) and filtered at 50°C to afford the desired product as a yellow solid (57 mg). IR: vmax/cm-1 (solid): 3100, 2200, 1615. HPLC-MS: 2.00 min, 387.1 [M+H]+. 1H NMR (500 MHz, CDCl3): δ 10.71 (d, J = 12.0 Hz, 1H), 7.86 (d, J = 12.0 Hz, 1H), 7.45 (br, 1H), 7.35 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.5 Hz, 1H), 6.92-6.99 (m, 3H), 6.83 (m, 3H), 6.76 (t, J = 7.5 Hz, 1H), 4.90 (s, 2H).
Activity vs gt1b p7 (J4-JFH1 HCV), 72h treatment


Additional compounds:

Acknowledgements
This work was supported by Medical Research Council grant G0700124 (S.G.) and an MRC Confidence in Concepts Award (S.G. & R.F). We are grateful to Jens Bukh (Hvidovre University Hospital and University of Copenhagen, Hvidovre, Copenhagen, Denmark), Takaji Wakita (National Institute for Infectious Diseases, Tokyo, Japan) and Wendy Barclay (Imperial College, London) for the generous provision of reagents. We thank Adrian Whitehouse (Leeds) for useful discussion. We also thank Morgan Herod and Adam Davidson (Leeds) for technical advice regarding the Incucyte Zoom.
Footnotes
Conflict of interest: None
References




