

ABSTRACT
Cilia and extracellular vesicles (EVs) are signaling organelles that play important roles in human health and disease. In C. elegans and mammals, the Autosomal Dominant Polycystic Kidney Disease (ADPKD) gene products polycystin-1 and polycystin-2 localize to both cilia and EVs, act in the same genetic pathway, and function in a sensory capacity, suggesting ancient conservation. Hence, the nematode offers an excellent system in which to address central questions regarding the biology of cilia, EVs, and the polycystins. We discovered an unexpected role of the mec-1, mec-5, and mec-9 genes encoding extracellular matrix (ECM) components. We determined that these ECM encoding genes regulate polycystin localization and function, ciliary EV release, cilia length, dendritic morphology, and neuron-glia interactions. Abnormal ECM and fibrosis are observed in ciliopathies such as ADPKD, nephronophthisis, and Bardet-Biedl Syndrome. Our studies reveal multifaceted roles for ECM proteins in the ciliated nervous system of the worm and provide a powerful new in vivo model to study the relationship between ECM, the polycystins, and ciliopathies.
INTRODUCTION
Cilia are antenna-like structures that project from many eukaryotic cells (Wood and Rosenbaum 2015). Cilia play essential roles in human development and health, with ciliary defects resulting in syndromic ciliopathies (Reiter and Leroux 2017). Cilia are endowed with receptors, channels, and signaling components, enabling cilia to act as cellular sensors. In addition to their sensory abilities, cilia may transmit signals via submicroscopic extracellular vesicles (Wang and Barr 2018; Wood and Rosenbaum 2015). The mechanisms that enable a cilium to simultaneously send and receive information remain mysterious.
Cilia and the extracellular matrix (ECM) share an intimate association, with cilia projecting into and being surrounded by ECM (Seeger-Nukpezah and Golemis 2012). ECM is made up of a network of interacting proteins that surround and support cells for adhesive, structural and signaling functions (Hynes 2009). ECM is necessary for tissue morphogenesis and homeostasis throughout the lifespan of an organism and dynamically interacts with and regulates body systems, organs, and tissues. In the brain and nervous system, ECM is important for neuronal development, anatomy, and synaptic transmission. Dysregulation of ECM contributes to pathological conditions such as invasive cancer and fibrosis (Bonnanset al. 2014). Fibrosis and abnormal ECM are observed in ciliopathies such as autosomal dominant polycystic kidney disease (ADPKD), nephronophthisis, and Bardet-Biedl Syndrome (Songet al. 2017).
ADPKD is a genetic disorder characterized by the presence of fluid filled cysts which form on and in the kidney epithelia lining renal tubules that replace normally functioning renal tissue and result in enlarged, polycystic kidneys and end stage renal failure (Ghata and Cowley 2017; Ong and Harris 2015). ADPKD affects 1 in ~500 persons regardless of race or gender and can be caused by a mutation in either of the polycystin encoding genes PKD1 or PKD2. The large extracellular domain of polycystin-1 extends into the ECM and contains domains that may mediate protein-protein or protein-ECM interactions. PKD2 has an often-mutated extracellular domain that is necessary for polycystin channel assembly, stimulation, and gating (Shenet al. 2016). These two polycystins can be cleaved and have been found to act individually and synergistically, performing myriad functions including acting as ion channels and regulation of ECM components (Hanaokaet al. 2000; Mangoset al. 2010). Polycystins and ECM seem to mutually regulate each other, suggesting dynamic feedback.
The polycystins localize to cilia and extracellular vesicles, and this subcellular localization is evolutionarily conserved and observed in Chlamydomonas, C. elegans, and mammals (Hoganet al. 2009; O’Haganet al. 2014; Semmoet al. 2014; Wanget al. 2014; Woodet al. 2013; Wood and Rosenbaum 2015). EVs carry many ECM proteins such as fibronectin and laminin, which provide communication necessary for altering ECM composition, signaling between ECM and the cells it surrounds, tumor proliferation, and inflammation (Rillaet al. 2017). In Chlamydomonas, ciliary EVs carry a proteolytic enzyme that degrades ECM required for hatching (Woodet al. 2013). In C. elegans, ciliary proteins such as polycystins are EV cargo that function in animal-to-animal communication (Wanget al. 2014).
The C. elegans polycystins LOV-1 and PKD-2 localize to cilia of male-specific sensory neurons (Barret al. 2001; Barr and Sternberg 1999). We previously performed a forward genetic screen for regulators of PKD-2::GFP ciliary localization (Baeet al. 2008). Here we identify a mutation in the collagen gene mec-5 that produced a PKD-2::GFP ciliary localization defect and discovered new functions for the mec-1, mec-5, and mec-9 ECM genes previously implicated in the function of non-ciliated touch receptor neurons (Kattaet al. 2015). MEC-1 and MEC-9 contain EGF/Kunitz domains and MEC-5 is a Type IV collagen (Emtageet al. 2004). MEC-1, MEC-5, and MEC-9 ECM proteins form the mantle surrounding non-ciliated touch receptor neurons, mediate touch neuron attachment to the hypodermal skin of the worm, and regulate the localization of the mechanosensitive DEG/ENaC (degenerin/sodium epithelial channel) complex MEC-4 and MEC-10 (Duet al. 1996; Guet al. 1996).
Here we demonstrate that mec-1, mec-5, and mec-9 are required for polycystin protein localization to the sensory cilia and discovered that proteins in the extracellular matrix regulate the movement and activity of proteins inside the cell. We find that these ECM components also regulate polycystin-mediated male mating behaviors, control neuron-glia interactions important for ciliary and dendritic integrity, and modulate the shedding and release of ciliary extracellular vesicles. While the polycystins have been implicated in sensing and regulating collagen in zebrafish models, roles for ECM proteins in regulating ciliary integrity, ciliary polycystin localization, and ciliary function have not been previously appreciated.
MATERIALS AND METHODS
Culture of C. elegans nematodes
Nematodes were maintained using standard conditions (Brenner 1974). Males and hermaphrodites were isolated at L4 stage d24hrs prior to experiments and kept at 20-22°C overnight. In C. elegans, the predominant sex is hermaphrodite and males spontaneously arise only rarely (less than 1%). Therefore, in all experiments in which males were tested, we used animals in either the him-5(e1490) or him-8(e1494) background. These backgrounds were considered wild type. him-5(e1490) and him-8(e1494) males exhibit normal mating behaviors and are used as wild-type controls for mating assays.
General molecular Biology
PCR amplification was used for genotyping and building transgenic constructs using the following templates: C. elegans genomic DNA, cDNA, or prebuilt constructs. High fidelity LA Taq (TaKaRa Bio Inc., Otsu, Shiga, Japan) or Phusion High Fidelity DNA Polymerase (Thermo Fisher Scientific, Vantaa, Finland) were used for amplification of DNA for constructs. Sequencing reactions were performed by Genewiz, (South Plainfield, NJ, USA). DNA and protein sequence analysis BLAST was used for identification of gene orthologs in C. elegans. Human and nematode protein sequence information was provided by NCBI smartBLAST, and C. elegans gene and protein sequence information was also provided by WormBase. Serial Analysis of Gene Expression (SAGE) data provided by WormBase (Release WS221). Whole genome sequencing was performed and analyzed by Richard Poole using CloudMap. ApE 1.17 was used for sequence manipulation.
Imaging
Nematodes were anaesthetized with 10 mM levamisole and mounted on agar pads for imaging at room temperature. Epifluorescence images were acquired using a Zeiss Axioplan2 microscope with 10x, 63x (NA 1.4), and 100x (NA 1.4) oil-immersion objectives with a Photometrics Cascade 512B CCD camera using Metamorph software (www.moleculardevices.com) or Zeiss Axio Imager.D1m microscope using a 63x and 100x objective with a Q imaging Regtiga-SRV camera. Optical Z-stack projections were stored as TIFF files and manipulated using ImageJ and Adobe Illustrator. Scale bars are 10 microns for head and tail images. EM scale bars are 200 or 500 nm as marked in image.
Transmission Electron Microscopy
mec-9(ok2853) and wild-type young adult males were fixed using high-pressure freeze fixation and freeze substitution in 2% OsO4 + 2% water in acetone as the primary fixative (Weimer 2006). Samples were slowly freeze substituted in an RMC freeze substitution device, before infiltration with Embed-812 plastic resin. For TEM, serial sections (70-75 nm thickness) of fixed animals were collected on copper slot grids coated with formvar and evaporated carbon and stained with 4% uranyl acetate in 70% methanol, followed by washing and incubating with aqueous lead citrate. Images were captured on a Philips CM10 transmission electron microscope at 80kV with a Morada 11-megapixel TEM CCD camera driven by iTEM software (Olympus Soft Imaging Solutions). Images were analyzed using ImageJ (FIJI) and manipulated with Adobe Illustrator.
Extracellular vesicle release
EV release was analyzed by counting all EVs from one-day old young adult males as described by Silva et al 2017. Individual animals were mounted on agar into four quadrants of agar slide. Free and newly released EVs that float to the cover slip were counted and reported as “number of EVs released.”
Transmission Electron Microscopy EV measurements
Using ImageJ (FIJI) software, WT and mutant electron micrographs were compared at and around the CEM transition zone. Qualitative examination was performed. EV diameter measurements were performed by drawing a line across the widest diameter of an EV and taking a measurement via FIJI measurement tool reporting diameter in nm.
Transmission Electron Microscopy cilia length measurements
Electron micrographs were stacked using the TrakEM2 component of ImageJ(FIJI) software. The transition zone was identified by Y-links and used as the bottom-most measurement. The ciliary tip was used as the top measurement. The Z stack position was multiplied by the thickness of the cut (~70-75 nm) and length was reported in μm.
PKD-2 antibody generation and staining
Animals were staged young adults and washed off plates with M9. Antibodies were prepared using Abmart protocols and staining against PKD-2 was prepared and performed using a Finney Ruvkun protocol (Bettingeret al. 1996) [Wormatlas.org]. The monoclonal PKD-2 primary antibody was created by Abmart against the intracellular N-terminal domain (amino acids: DERWANPPQPVA) and an intracellular C-terminal domain (amino acids: KRGKRPDAPGED). The secondary antibody was α mouse Alexa Fluor ® 568 donkey anti-mouse IgG (H + L) (2 mg/ml) by Invitrogen TM. The following dilutions and incubation times were used: primary antibodies 1:200 overnight (18-24 hours); secondary antibody 1: 1000 for two hours.
Response behavior assay
Control Strains used: CB1490: him-5(e1490), CB1489: him-8(e1489), PT9: pkd-2(sy606) him-5(e1490), and CB169: unc-31(e169)IV. L4 larval males were moved to a fresh plate approximately 24 hours before mating. unc-31 mutant hermaphrodites were also picked as L4 larvae ~24 hours before experiments. Male mating assays were conducted on a fresh NGM agar plate with a small lawn of E. coli (OP50) containing 25 young-adult unc-31 hermaphrodites. One, two, or three males were placed in the center of the lawn and observed for four minutes. When a male began scanning a hermaphrodite and the male tail maintained contact with a mate for at least ten seconds, a response was scored and that male was removed from the test plate.
Location of vulva assay
was performed as described (Barr and Sternberg 1999). Location of vulva efficiency is calculated by successful vulva location divided by the total number of vulva encounters for each male. Total time measured was four minutes.
Male leaving assay
was performed as described (Barrioset al. 2008). L4 males were picked and isolated from hermaphrodites on plates, then assayed 24 hours later in 20 μl food on a 9 cm diameter plate. Animals are positive for leaving when males exhibit tracks that approach within 1 cm of the edge of the plate. Time points scored were 2, 5, 8, and 24 hours after the males were placed on the spot of food. A minimum of 20 animals per strain and three replicates were scored for each genotype assayed. Statistical significance was determined by R software.
Dye filling assays
Standard dye-filling assays (Perkinset al. 1986) were performed using Dil (Invitrogen). The number of amphid and phasmid cell bodies were counted, and the results reported as number of neurons out of 12 (amphid) or 4 (phasmid) that fill with DiI.
Dendritic trafficking velocity measurements
To directly measure in vivo velocity of PKD-2::GFP in CEM dendrites, we acquired time lapse image stacks, which were later converted to kymographs using the KymographClear V2.0 plugin in FIJI. Motile particles were automatically and indiscriminately detected and traced, and velocities analyzed with KymographDirect (Mangeolet al. 2016). We observed a reduction of overall observed velocities compared to our previous publication (Baeet al. 2006), likely due to differences between automatic versus manual particle tracing. This reduction does not affect our analysis or overall conclusions, as our model is not based on absolute velocities, but rather on their relative changes.
Ciliary localization and fluorescence intensity
Ciliary localization was performed by blind assay of a stack of images collected into a maximum intensity image using the “Z project” function in ImageJ software. Animals were scored as ciliary localization defective (Cil) if excess or misplaced PKD-2::GFP or endogenous PKD-2 (detected by α-PKD-2 antibodies) was detected (excess in cilium, ciliary base, or dendrite). Fluorescence intensity was measured using ImageJ by drawing a range of interest and using measurement tool. All measurements have background subtracted to create final value.
Strains used: (transgenic lines created by Knudra)
CB1066 mec-1(e1066)V
CB1292 mec-1(e1292)V
CB1490 him-5(e1490)
CB1494 mec-9(e1494)V
CB1503 mec-5(e1503)X
CB169 unc-31(e169)IV
COP1472 knuEx206[pNU1416-mec-9Sp:: GFP::tbb-2utr, unc-119(+)];unc-119(ed3)lll
COP1473 knuEx207[pNU1415-mec-9Lp::GFP::tbb-2utr,unc-119(+)];unc-119(ed3)III
KU25 pmk-1(km25)IV
PT3296 mec-9(ok2853)pmk-1(km25)IV;myIs4 him-5(e1490) V
PT3168 him-8(e1489) myIs1[PKD-2::GFP + cc::GFP] IV
PT277 unc-119(ed3)III; him-5(e1490) V
PT1213 myIs4 him-5(e1490) V; mec-5(my2) X
PT1852 pha-1(e2123) III; him-5(e1490) V; Ex [LOV-1::GFP1]
PT2434 dyf-1 (m335); myIs1; him-5
PT2679 him-5(e1490)V;myIs23[Pcil-7::gCIL-7::GFP_3’UTR+ccRFP]
PT2962 him-8(e1489) myIs1; mec-1(e1066) V
PT2963 him-8(e1489) myIs1; mec-1(e1336) V
PT2964 him-8(e1489) myIs1; mec-1(e1292) V
PT2965 him-8(e1489) myIs1; mec-9(e1494) V
PT2966 him-8(e1489) myIs1; mec-9(ok2853) V
PT2967 him-5(e1490) myIs4[PKD-2::GFP + cc::GFP] V; mec-5(e1790) X
PT2968 him-5(e1490) myIs4; mec-5(e1503) X
PT2969 uIs31[MEC-17:: GFP] III; him-5(e1490) myIs4[PKD-2:: GFP + cc::GFP] V; mec-5(u444) X
PT3038 unc-31(e169)IV; him-5(e1490), myIs4 V
PT3203 mec-9(ok2853); pha-1; him-5(e1490); syEx301[pBx+LOV-1::GFP1]
PT3213 mec-9(ok2853)V, him-5(e1490)V;myIs23[pcil-7::gCIL-7:: GFP:: 3’UTR+ccRFP]
PT443 myIs1 pkd-2(sy606) IV; him-5(e1490) V
PT621 myIs4 him-5(e1490) V
RB2140 mec-9(ok2853) V
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.
RESULTS
mec-1, mec-5, and mec-9 regulate PKD-2::GFP localization
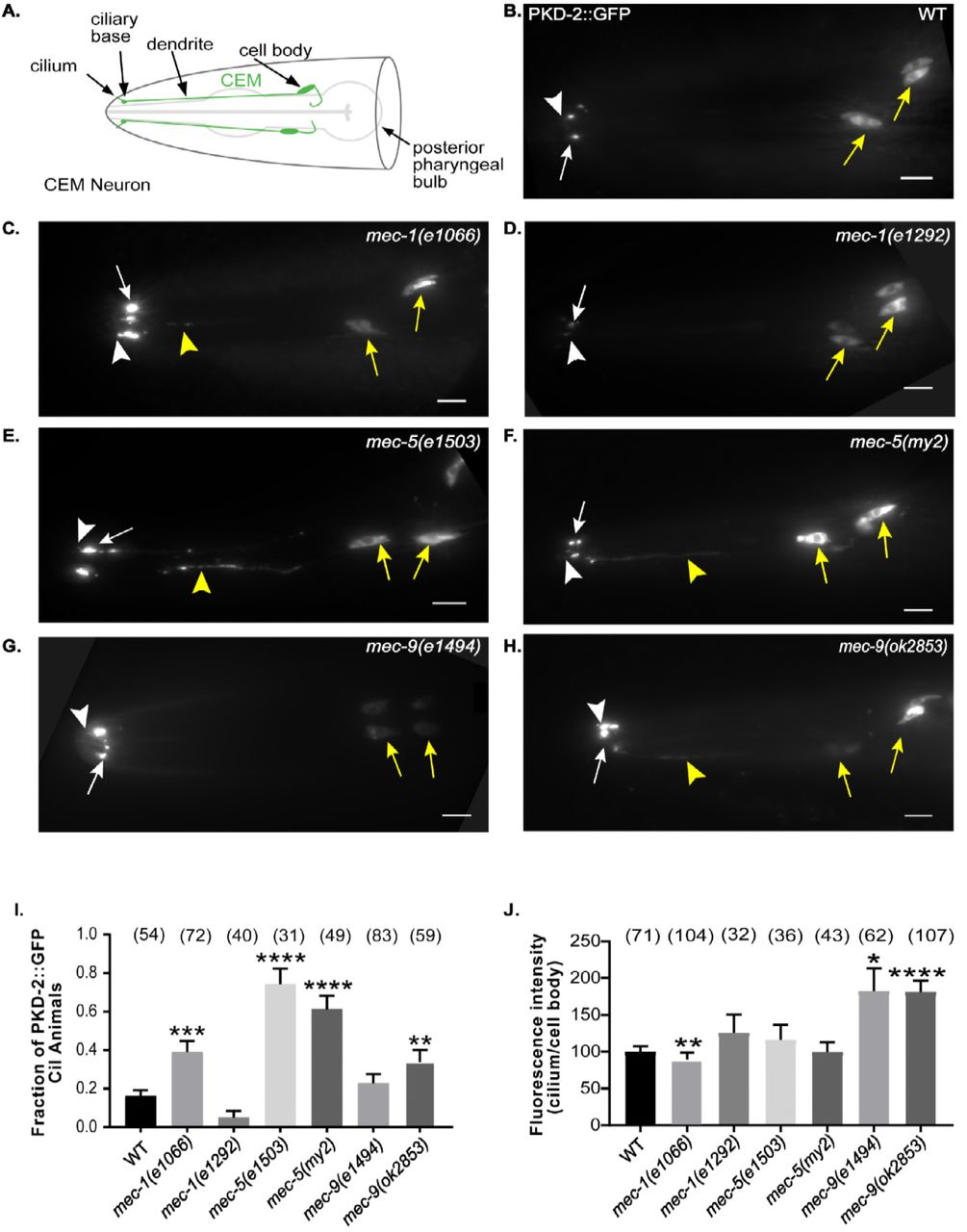
The C. elegans polycystins LOV-1 and PKD-2 localize to cilia and cell bodies of cephalic male-specific (CEM), left and right B-type ray neurons in the tail (RnB; n=neuron 1-9, but not 6), and Hook B (HOB) neurons. (Figure 1 A-B, Supplemental Figure 1 A-B) The polycystins and these male specific neurons are necessary for male mating behaviors (Barret al. 2001; Barr and Sternberg 1999; Liu and Sternberg 1995; Srinivasanet al. 2008). We previously performed a forward genetic screen for genes necessary in PKD-2::GFP localization and identified mutants defective in PKD-2 ciliary receptor localization (the Cil phenotype) (Baeet al. 2008). The cil-2(my2) mutant displays abnormally high PKD-2::GFP levels in the CEM cilia and ciliary base and also has abnormal extracellular, extradendritic PKD-2::GFP accumulation along CEM dendrites (Baeet al. 2008). cil-2(my2) hermaphrodites also displayed temperature sensitive sterility phenotype, that is linked to the Cil phenotype. The extradendritic PKD-2::GFP accumulation and sterility phenotypes are unique to the cil-2(my2) mutant and not the other ten Cil mutants. We used genetic mapping, whole genome sequencing, complementation testing, and rescue experiments of the Cil phenotype to map the my2 lesion to mec-5.

(B-H) Images were compiled from a 630x maximum intensity Z-series obtained by epifluorescence microscopy. Scale bar is 10 μm. White arrow, ciliary base; white arrow head, cilium; yellow arrow, cell body; yellow arrowhead, dendrite. (A) Schematic of WT cephalic male neurons in the head of a male (green). (B) A lateral view of WT PKD-2::GFP (translational reporter). PKD-2::GFP localized to CEM cell bodies and cilia. (C-D) Lateral view of PKD-2::GFP in mec-1 mutant males. (C) Increased PKD-2::GFP at mec-1(e1066) CEM cilia and ciliary base. (D) mec-1(e1292) was indistinguishable from WT. (E-F) mec-5 PKD-2::GFP ciliary localization. Extra-dendritic and increased ciliary PKD-2::GFP observed in mec-5(e1503) and mec-5(my2) male CEMs. (G-H.) PKD-2 localization in mec-9 mutant males. (G) Increased PKD-2::GFP at mec-9(e1494) CEM ciliary base. (H) Extra-dendritic and increased PKD-2::GFP at mec-9(ok2853) CEM ciliary base. (I) Graph of the fraction of PKD-2::GFP mislocalization in CEM neurons of wild type and mutant males via blind examination. An animal was scored as “Cil” (ciliary localization defective) if CEM cilia, ciliary base, and/or dendrites in male worms had abnormally increased or mislocalized PKD-2::GFP. Values were reported as fraction of animals that are Cil. Significance was determined by Kruskal Wallace test with Dunn’s multiple comparisons test performed to compare groups. **p<0.01, ***p<0.001, ****p<0.0001. In WT, we observed occasional Cil animals (~16% of males). mec-1, mec-5 and mec-9 all had mutant alleles that mislocalized PKD-2::GFP. (J) Ratio (cilium/cell body) of maximum intensity showed that PKD-2::GFP abundance in CEM cilia is increased in comparison with the cell bodies only in mec-9 ECM gene mutants; however, mec-1 and mec-5 alleles also affected PKD-2 abundance (Supplemental Table 1). Background measurements were subtracted from cilium and cell body values for standardization of images and we expressed the measurements in ratio of cilia to cell body FI. Significance was measured by Kruskal-Wallace test, comparisons made using Dunn’s multiple comparisons. *p<0.05; **p<0.01; ***p<0.001; **** p<0.0001. WT values were normalized to 100. The mec-9 mutants had a brighter maximum FI (~1.75x) than WT (Figure 1J).
Expression of the wild-type (WT) mec-5 genomic region using either the mec-5 promoter or muscle-specific myo-3 promoter rescued cil-2(my2) defects (data not shown), confirming that mec-5 was mutated in the cil-2(my2) mutant and that mec-5 acted non-cell autonomously to regulate PKD-2::GFP ciliary localization in male-specific neurons. mec-5 encodes a collagen protein that is produced and secreted by hypodermal cells to anchor the degenerin complex in touch receptor neurons to the extracellular matrix (ECM) (Duet al. 1996). In these non-ciliated neurons, mec-5 and the ECM encoding genes mec-1 and mec-9 act in concert (Duet al. 1996). We therefore determined whether MEC-1, MEC-5, and MEC-9 also regulated PKD-2::GFP ciliary localization.
We characterized PKD-2::GFP ciliary localization in two mutant alleles each of mec-1 (e1066 and e1292), mec-5 (e1503 and my2), and mec-9 (e1494 and ok2853) (Figure 1C-H). First, we visualized ciliary localization in CEM neurons of WT and mutant males via blind examination. A male was scored as Cil if CEM cilia, ciliary base, and dendrites in male worms had abnormally increased or mislocalized PKD-2::GFP. Values were reported as percent of animals that are Cil (Figure 1I). In WT, we saw occasional Cil animals (~16% of males). In contrast, mec-5 males exhibited the most penetrant Cil phenotype: over 60% of mec-5(my2) animals and over 70% of mec-5(e1503) were Cil (p values < 0.0001). Also, mec-9(ok2853) animals had a significantly increased number of Cil males (p=0.008), but mec-9(e1494) males were statistically indistinguishable from WT. mec-1(e1066) males were also Cil (p= 0.0003), but mec-1(e1292) males were similar to WT.
In mec-1(e1066), mec-5(e1503), mec-5(my2), and mec-9(ok2853) males, we observed abnormally increased PKD-2 localization in CEM cilia, ciliary bases, and occasionally dendrites. (Figures 1C, E, F, and H) The extradendritic accumulation in mec-1(e1066), mec-5(e1503) and mec-9(ok2853) was reminiscent of that observed in mec-5(my2). The Cil defect was not observed in mec-1(e1292) and was only occasionally seen in mec-9(e1494). (Figures 1D and G)
mec-1 encodes multiple ECM protein isoforms with multiple disulfide-linked EGF and Kunitz-type protease inhibitor domains (Emtageet al. 2004). The mec-1 alleles cause gene truncations: e1292 results in truncation after Kunitz Domain 3 whereas e1066 truncates after Kunitz Domain 6 (Emtageet al. 2004), both of which remove Kunitz domains but might leave EGF domains intact. mec-9 encodes two protein isoforms of an ECM protein containing multiple EGF domains, multiple Kunitz domains, and a glutamic acid-rich region (Duet al. 1996). mec-9 mutations perturb EGF domains: e1494 is a point mutation in the first set of EGF domains and affects only the mec-9 long (mec-9L) isoform. ok2853 perturbs the second set of EGF domains via deletions and affects both short and long isoforms (Duet al. 1996). Using whole genome sequencing, we found that both mec-5 mutants had lesions in the third intron (data not shown). We conclude that some but not all alleles of mec-1, mec-5 and mec-9 perturbed PKD-2::GFP localization.
We observed that cilia and ciliary bases appeared brighter in some but not all ECM mutants. We measured highest/maximum fluorescence intensity (FI) of PKD-2::GFP in CEM neuron cell bodies and cilia (including both cilium and ciliary base) to quantify PKD-2::GFP abundance. FI is a computed measurement of the pixels illuminated in a selected region of interest. mec-9 mutants had a brighter maximum FI (~1.75x) than WT (Figure 2B): mec-9(e1494) p=0.011 and mec-9(ok2853) p<0.0001 (Figure 1J). mec-1(e1066) mutants were dimmer than WT: p=0.0088. We observed variability in CEM FI of ECM gene mutant cilia, dendrites, and cell bodies (Supplemental Table 1), with mec-9(ok2853) exhibiting the brightest overall FI. Similar to CEM neurons, we also observed variable ray neuron FI in ECM mutant tails (Supplemental Figure 1 and Supplemental Table 1), again with mec-9(ok2853) exhibiting the brightest overall FI. Our data shows that although mec-1, mec-5, and mec-9 were all necessary for PKD-2 ciliary localization, mec-9 also regulated PKD-2::GFP ciliary abundance.

(A-C) mec-1(e1066) males were response defective, mec-5(e1503) males were leaving assay defective, mec-9(ok2853) males were defective in all three mating behaviors. (A) Leaving assay measured the probability of males leaving food to search for a mate. WT values normalized to 1. mec-5 (e1503) males were leaving assay defective when compared to WT. mec-9(ok2853) males left food more readily than WT. (B) mec-1(e1066) and mec-9(ok2853) males were defective in responding to hermaphrodite contact. (C) mec-9(ok2853) males were location of vulva defective. Significance determined by Kruskal-Wallis test (Mann-Whitney test for WT vs. mec-5(e1503) only); p values compared by Dunn’s multiple comparisons test. ** p<.01; **** p<0.0001.
To test for specificity of ECM components, we examined PKD-2::GFP localization in a hemicentin mutant. Hemicentin is an ECM component required for adhesion between tissues including touch neuron attachment to the epidermis and gonad (Vogel and Hedgecock 2001). Analysis of the hemicentin mutant him-4(e1267) revealed no differences in PKD-2::GFP ciliary localization or FI. We conclude that the mec-1, mec-5, and mec-9 ECM genes; but not the him-4 ECM gene, were necessary for polycystin localization and abundance.
We generated a monoclonal anti-PKD-2 antibody to visualize and measure endogenous PKD-2 localization in WT and ECM gene mutant males and observed similar Cil defects (Supplemental Figures 2 and 3). In WT, endogenous PKD-2 was limited to the cell bodies and cilia of CEM head neurons and ray RnB and hook HOB tail neurons (Supplemental Figure 3A and 3B). In mec-9(ok2853) males, endogenous PKD-2 mislocalized to dendrites and had increased abundance in cilia and cell bodies as shown in FI measurements (Supplemental Figure 3C and 3D). Endogenous PKD-2 localization was also abnormal in the ray cilia in mec-1(e1066) and mec-5(e1503) (data not shown) male tail but not as severely as mec-9(ok2853). We conclude that all three ECM genes regulate PKD-2 localization with mec-9 mutants displaying the most severe Cil defects.
The partner of Polycystin-2 (PKD-2) is Polycystin-1 (LOV-1). We therefore, examined LOV-1 localization in ECM gene mutants. In WT, LOV-1::GFP localizes to the CEM, RnB, and HOB cell bodies and cilia. In mec-9(ok853) mutants, we observed distal dendritic LOV-1::GFP mislocalization and increased ciliary fluorescence (Supp. Figure 4). We also observed significantly increased LOV-1::GFP FI in CEMs and RnBs of mec-5(e1503) males (data not shown). We conclude that ECM encoding genes mec-1, mec-5, and mec-9 regulate polycystin localization in male-specific ciliated sensory neurons.
mec-1, mec-5, and mec-9 regulate male mating behaviors
pkd-2, lov-1, and the male-specific polycystin expressing neurons are required for the male mating behaviors of mate searching, response to hermaphrodite contact, and location of hermaphrodite vulva (O’Haganet al. 2014). We therefore determined whether the three ECM genes were required for these male sensory behaviors.
C. elegans males leave a food source in search of a mate if no hermaphrodite is present (Liptonet al. 2004). lov-1 and pkd-2 mutant males do not leave food to search for a mate (Barrioset al. 2008). Similarly, mec-5(e1503) males were leaving defective (Figure 2A). In contrast, mec-9(ok2853) mutants left food more readily than WT animals (Figure 2A). Hyper-leaving behavior is associated with defects in male-specific and the shared inner labial type 2 IL2 ciliated neurons (Maguireet al. 2015), suggesting that the mec-9 mutation may affect other neurons in addition to the polycystin-expressing cells.
When male ray neurons detect contact with a hermaphrodite, males initiate the response behavior by stopping forward locomotion, and initiating backing (Barr and Garcia 2006; Barret al. 2018). lov-1 and pkd-2 mutant males are response defective. mec-9(ok2853) and mec-1(e1066) mutants were also defective in response to hermaphrodite contact, while mec-5(my2) mutant males displayed normal response behavior (Figure 2B) (Baeet al. 2006). After response, the male scans the hermaphrodite’s body for her vulva. lov-1 and pkd-2 mutants are location of vulva defective (Lov). Only mec-9(ok2853) males displayed the Lov phenotype (Figure 2C).
We previously showed that mate searching, response, and location of vulva do not require MEC-4 and MEC-10, the touch neuron specific degenerin epithelial sodium channel (DEG/ENaC) receptors (Barr and Sternberg 1999; Barrioset al. 2008). However, mec-1, mec-5, and mec-9 ECM genes are required for these male-specific sensory behaviors. We conclude that mec-1, mec-5 and mec-9 ECMs genes act beyond DEG/ENaC localization and function in touch receptor neurons and are required more broadly for the function of other sensory neurons. Moreover, only mec-9 was required for all examined male-specific behaviors suggesting a distinct role for mec-9 in polycystin-expressing male specific neurons.
mec-9 long and short isoforms have distinct expression patterns: MEC-9S is expressed in ciliated sensory neurons
mec-9 encodes two predicated proteins (Duet al. 1996). The mec-9 long isoform encodes an 839-amino acid protein with five Kunitz protease inhibitor domains, seven EGF domains, and a glutamic acid rich/coiled-coiled region (Figure 3A) (Duet al. 1996). The mec-9 short isoform encodes a 502-amino acid protein with two Kunitz domains, three EGF domains, and a glutamic acid rich/coiled-coiled region (Duet al. 1996) (Figure 3B). Both long and short isoforms encode an N-terminal signal sequence consistent with secreted ECM proteins. mec-9(ok2853) is a 408-base pair in-frame deletion that is predicted to remove two of the three EGF like domains in both isoforms (Consortium 2012) (Figure 3A).
Chalfie and colleagues showed mec-9 long isoform expression in touch receptor neurons and mec-9 short isoform expression in other neurons in the nerve ring and ventral cord in hermaphrodites (Duet al. 1996). We examined long and short isoform expression patterns in males and hermaphrodites using transcriptional reporters (Figure 3A). We find that mec-9Lp::GFP (long isoform transcriptional reporter) was expressed only in the shared touch receptor neurons (found in both males and hermaphrodites) and not male-specific neurons. However, mec-9Sp::GFP (short isoform transcriptional reporter) was expressed in polycystin-expressing CEM, RnB, and HOB male-specific as well as shared ciliated sensory neurons found in both males and hermaphrodites (Figures 3D and 3E).

(A and B) mec-9 long and short transcripts. (A) mec-9 long isoform encodes a secreted ECM protein that has 5 Kunitz domains, 7 EGF repeats, and a glutamic acid-rich/coiled-coil region (Du et al., 1996). e1494 is a point mutation in the first set of EGF domains and ok2853 perturbs the second set of EGF domains via base pair deletions. (B) mec-9 short transcript is 502 aa (Du et al., 1996): the promoter for the mec-9 short transcript located in mec-9 long 9th intron. We used the 2kb upstream sequence with a C terminal GFP tag to construct mec-9Lp::GFP transcriptional reporter. We used the 1.5kb mec-9 long intron 9 sequence with a C terminal GFP tag to construct mec-9Sp::GFP transcriptional reporter. (C) Ciliated sensory neurons expressed mec-9Sp::GFP: CEMs, IL2, amphids, RnB, HOB, phasmids. Male (top), hermaphrodite (bottom). Ventral nerve cord (non-ciliated neurons) expression also observed and shown. (D) mec-9Sp::GFP coexpressed with the kinesin-3 gene, klp-6p::tdTomato transcriptional reporter. klp-6p::tdTomato expressed in male-specific CEM and IL2 neurons in the head and RnB and HOB male-specific neurons in the male tail. Left column is lateral view of male head. mec- 9Sp::GFP expressed in CEMs and IL2 neurons. Right column is lateral view of male tail. mec-9Sp::GFP expressed in HOB and RnB neurons. (E) Some mec-9Sp::GFP expressing ciliated neurons filled with Dil. Dil is a lipophilic dye taken up by amphids and phasmids. Left column is lateral view of hermaphrodite head. mec-9Sp::GFP expresses in amphids in the hermaphrodite and male head. Right column is lateral view of hermaphrodite tail. mec-9Sp::GFP expressed in phasmids.
mec-9Sp::GFP was coexpressed with the kinesin-3 gene klp-6 transcriptional reporter in the shared IL2 neurons and male-specific CEM neurons in the head and RnB and HOB neurons in the tail (Figure 3D). These 27 klp-6 expressing neurons are called extracellular vesicle releasing neurons (EVNs) based on their ability to shed and release ciliary EVs (Wang et al., 2015). mec-9Sp::GFP was also expressed in the amphid neurons in the head and phasmid in the tail that take up the lipophilic fluorescent dye Dil (Figure 3E).
MEC-9 regulates extracellular vesicle biogenesis and release
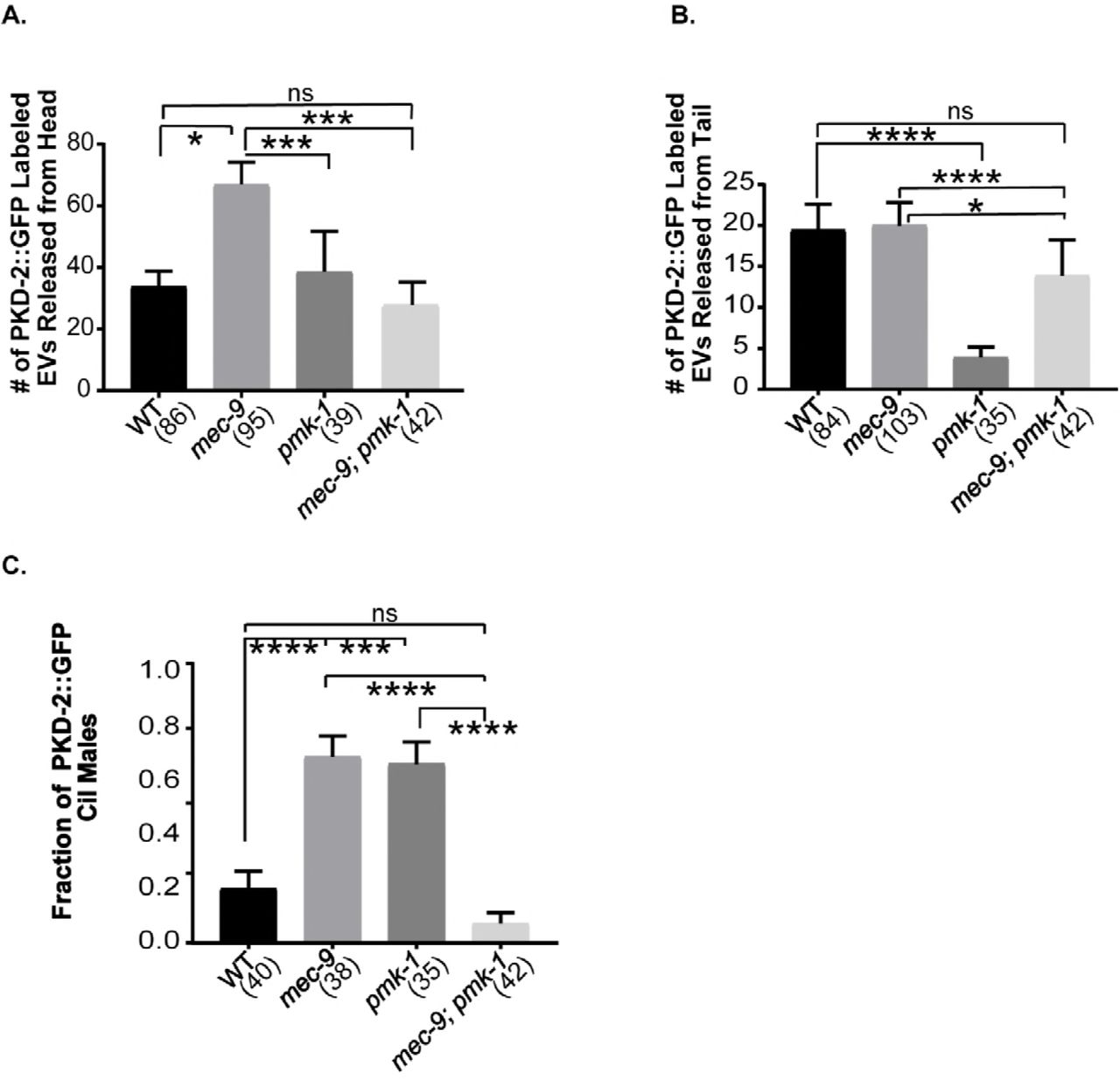
EVs are shed and released from ciliated IL2, CEM, and RnB neurons (Wanget al. 2014), all of which expressed mec-9Sp::GFP. To determine if mec-9 regulates EV biogenesis, we counted PKD-2::GFP containing EVs that were shed and released into the local environment from the male head and tail. WT adult males released an average of 26 PKD-2::GFP labeled EVs from the CEM neurons compared to 78 EVs in mec-9 mutants (p=0.0008): a threefold increase (Figure 4C). The EV hypersecretion phenotype of mec-9 mutants contrasts other previously described EV hyposecretion mutants that are deficient in EV release (Maguireet al. 2015; O’Haganet al. 2017; Silvaet al. 2017; Wanget al. 2014).

(A-B) Head images of WT and mec-9(ok2853) mutant males show EVs were released. Image brightness was increased and inset was magnified to visualize EVs. Scale bars 10μm. White arrow, ciliary base; white arrow head, cilium; yellow arrow, cell body; yellow arrowhead, dendrite. (B) mec-9(ok2853) males produced significantly more PKD-2::GFP tagged EVs than WT (A). (C) mec-9(ok2853) released significantly more PKD-2::GFP-tagged EVs. Significance determined by Mann-Whitney test; ***p<0.001 (D) Schematics of WT and mec-9 mutant cephalic sensilla. In WT, EVs are released from cilium and are stored in lumen created by the sheath glial cell. Dashed lines (E-G) denote cross section level observed in images 4E, F, and G. mec-9(ok2853) mutants store and release excess EVs and contain dark and light matrix vesicles that contain EVs. (E-F) Cross section of the cephalic sensillum at the level of the transition zone in WT and mec-9(ok2853) males. CEM neurons shaded green, CEP neurons shaded blue. Scale bars 500 nm (E) Two EVs (arrows) observed in WT; one in the lumen and one in CEM cilium. (F) mec-9(ok2853) had a distended lumen with a significant increase of EVs and a lightly shaded matrix filled vesicle itself containing EVs. (G) Dark electron-dense matrix filled vesicles contained EVs. These vesicles were located at the level of the distal dendrite (See dotted line marked G in Figure 4D).
Next, we used transmission electron microscopy (TEM) to more closely examine the ultrastructure of the male cephalic organs containing EVs in ECM and lumenal spaces (Wanget al. 2014). In WT males, EVs are shed from the ciliary base of the CEM neuron and occupy a lumenal space formed by the glial support cells (Figure 4E). In mec-9 mutant males, we observed three striking defects. First, in the mec-9 cephalic lumen we observed a dramatic accumulation of EVs that ranged in diameter from 45nm to 226nm (Figure 4F). Second, we saw an increase in the volume of the glial lumen occupied by the EVs. The 2D area of the mec-9 mutant lumen cross section was massively distended, perhaps due to EV hypersecretion and increased EV storage (Figure 4F). Eight WT and 12 mec-9 cephalic sensilla were compared and revealed increased occurrences and larger diameters of lumenal spaces surrounding cilia and distal dendrites. The enlarged EV containing lumen is a phenotype observed in mutants that shed but do not environmentally release EV. Third, mec-9 cephalic sensilla were filled with remarkable light and dark matrix filled vesicles, themselves also containing EVs. For example, the lightly shaded vesicle shown in Figure 4E was 506 nm in diameter and contained smaller vesicles ranging in diameter from 40-90 nm. Dark matrix vesicles found at the level of the distal CEM dendrite were 898 and 1056 nm in diameter and contained vesicles that ranged from 51-171 nm (Figure 4G). For both abnormal EV and matrix-filled vesicle phenotypes in mec-9, we observed larger complex vesicles in the glial cytoplasm as well as in the lumenal spaces surrounding the cilia, ciliary transition zone (TZ), and distal dendrite (data not shown). The presence of these larger vesicles with complex contents is a phenomenon not previously described in any of our other EV biogenesis mutants (Maguireet al. 2015; O’Haganet al. 2017; Silvaet al. 2017; Wanget al. 2014).. We propose that the mec-9 and likely the mec-5 extradendritic PKD-2::GFP ciliary localization (Cil) phenotype may correlate to extradendritic accumulation of the abnormal EVs in the ECM surrounding the CEM dendrite.
mec-9 and pmk-1 act antagonistically in EV biogenesis and release
We wondered if the EV hypersecretion and excessive EV storage phenotypes seen in mec-9 mutants were due to abnormal EV biogenesis or to defects in neuronal integrity. To test the former possibility, we examined genetic interactions with a positive regulator of EV shedding and release – the p38 MAPK pmk-1. We previously showed that pmk-1 mutants do not accumulate EVs in the cephalic lumen and release fewer PKD-2::GFP labeled EV from ray neurons into the environment (Wanget al. 2015). In contrast, mec-9 mutants exhibit the opposite phenotypes: hypersecretion and excessive release of cephalic EVs. WT and the mec-9(ok2853); pmk-1(km25) double mutant release comparable numbers of PKD-2::GFP labeled EVs (Figure 5A). In a mec-9(ok2853); pmk-1(km25) double mutant we see that EV release from ray neurons is restored to WT levels (Figure 5B), suggesting that mec-9 and pmk-1 may act antagonistically, with mec-9 as a negative regulator, pmk-1 as a positive regulator. Further, PKD-2::GFP fluorescence intensity (FI) and blind study of ciliary localization was analyzed and although the mec-9 and pmk-1 single mutants exhibit an increase of PKD-2::GFP at the ciliary base, the ciliary localization phenotype and increased ciliary FI is ameliorated in the double mutant (Figure 5C). We conclude that MEC-9 is a negative regulator of EV biogenesis, with the mutant shedding and releasing excessive amounts of EVs.

(A) p38 MAPK pmk-1(km25) mutants are defective in RnB EV release (Wang et al., 2015). pmk-1 mutation suppressed the mec-9(ok2853) abnormal EV hypersecretion in the head. (B) mec-9 suppressed the pmk-1 RnB EV release defect. (C) pmk-1; mec-9 double mutant suppressed PKD-2::GFP Cil defect observed in both mec-9 and pmk-1 single mutant males. Significance was determined by Kruskal-Wallace test and Dunn’s multiple comparisons test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
mec-9 maintains CEM and IL2 neuronal morphology
Given that mec-9S was expressed in many ciliated sensory neurons, we used transmission electron microscopy (TEM) to examine ultrastructure of sensory organs in the head of fixed age-matched young adult WT and mec-9 males (Figure 6A). The head contains four cephalic sensilla, six inner labial sensilla, and two amphid sensilla, each of which performs distinct sensory functions in the worm (Inglis 2007).

mec-9 regulated CEM, IL2, and amphid channel ciliary lengths and maintained CEM neuronal integrity. (A) Amphid, double ciliated (ADL, ADF), amphid single ciliated (ASE, ASG, ASI, ASJ, ASK), cephalic (CEP) and male-specific CEM (ventral and dorsal), and inner labial (IL2) neurons were measured using serial sectioned electron micrographs (each layer~70 nm). Lengths were determined by counting number of 70 nm layers from transition zone to ciliary tip of each cilium. mec-9(ok2853) amphid cilia were shorter than WT and mec-9(ok2853) CEM, CEP and IL2 cilia were longer than WT cilia. Significance measured by Kruskal-Wallace test and Dunn comparisons test. *p<0.05, **p<0.01 (B-D.) mec-9 maintained dendritic integrity. Images are maximum intensity Z-stacks of WT and mec-9(ok2853) males that expressed a pkd-2p::GFP transcriptional reporter. Images were brightened to observe dendritic morphology. Scale bars are 10 μm. Cell bodies denoted by yellow arrow. (B) Varicosities were observed in WT male CEMs, examples denoted by arrows. (C) mec-9 mutant males have more varicosities. Large, “gross” varicosities denoted by arrows. (D) A line plot of WT (black) and mec-9(ok2853) (red) dendritic fluorescent intensity disclosed increased number of varicosities (E), increased gross varicosities (F), and that varicosities had a greater fluorescence. FI measured here using transcriptional reporter which allows for observation of neuron morphology only, not protein abundance measurements (G). Total varicosities and gross varicosities measured by blind study. Significance measured by Mann-Whitney test. Varicosity Fluorescence (G) normalized to 100. *p<0.05, ***p<0.001, ****p<0.0001.
In the male, the cephalic sensillum includes the cephalic male CEM neuron and the sex-shared CEP neuron. We viewed the CEM in cross section and measured from the ciliary transition zone to the ciliary tip and found WT CEM cilia to be ~2.1 μm in length (Figure 6A). In mec-9 mutants, the CEM neurons were 1.2-1.5 times longer than WT. CEP cilia length was not significantly different between WT and mec-9 mutant animals.
The inner labial sensilla house the IL1 and IL2 neurons. WT dorsal and ventral IL2 cilia averaged 1.155 μm and the lateral IL2s averaged 1.085 μm in length (Figure 6A). In the mec-9(ok2853) mutant, dorsal and ventral IL2 cilia were about twice as long as WT and the mec-9(ok2853) mutant lateral IL2s were ~1.3 times longer than WT (Figure 6A). IL1 ciliary lengths were similar in WT and mec-9, consistent with mec-9S expression in IL2 but not IL1 neurons.
The ECM encoding genes mec-1, mec-5, and mec-9 regulate touch receptor neuron attachment and integrity (Panet al. 2011). In mec-5 and mec-9 CEM neurons, PKD-2::GFP accumulated in extradendritic spaces (Figure 1 E,F, H). We therefore examined CEM neuronal morphology using a soluble GFP reporter. In WT and mec-9 CEMs, pkd-2p::GFP was expressed and localized throughout the neuron in the cell body, dendrite, and cilium (Figure 6B). We did not observe extradendritic accumulation of soluble GFP in mec-9 mutant, therefore the neurons are not fragile or leaky. However, mec-9 mutant dendrites appeared brighter and more irregular than WT (Figure 6C).
To determine if there were differences in the dendritic morphology of WT and mec-9 mutants, we compared FI peaks along the dendrite from the CEM cilium to cell body. These peaks represent dendritic varicosities and the example shown is indicative of the increased number and size of the varicosities in the mec-9 mutant compared to WT (Figure 6D). WT animals averaged 30.6 varicosities per animal, whereas mec-9 mutant animals averaged 36 varicosities (p= 0.0211) (Figure 6E).
We defined gross varicosities as those that are noticeably larger than normally observed and measure this phenomenon by blind investigation and FI. WT averaged 3.7 gross varicosities per animal and mec-9 mutants averaged almost double (7 gross varicosities per animal) (p=0.0007; Figure 6F) The FI of the peaks/varicosities in WT were measured and normalized to 100. In comparison, mec-9 mutant animals’ varicosities averaged 159% of WT intensity (p<0.0001) (Figure 6G). We conclude that the ECM gene mec-9 was important for CEM dendritic morphology.
To determine if defects in dendritic morphology impacted protein transport in CEMs, we used time-lapse microscopy to measure the number and velocity of PKD::GFP particles moving along the dendrite. mec-9(ok2853) mutants had significantly fewer moving puncta (Table 1). In mec-9(ok2853), PKD-2::GFP anterograde and retrograde velocities were significantly slower than WT (Table 1). Combined, these results indicate that the MEC-9 ECM component also regulated the CEM neuronal cytoskeleton and transport.
The number and velocity of PKD::GFP particles was measured using time lapse fluorescent videos. TOTAL PARTICLES/μM: The average number of anterograde and retrograde PKD-2:GFP particles moving along the dendrite are reported here in particles/μm. mec-9(ok2853) mutants have significantly fewer particles than WT. ANTEROGRADE AND RETROGRADE VELOCITY: Anterograde and retrograde PKD-2::GFP particles velocities were measured in microns per second along the entire CEM dendrite. In mec-9(ok2853) males, PKD-2::GFP overall dendritic anterograde and retrograde particle velocity was slower than WT. Time lapse exposure per frame: 300 ms. Significance measured using Mann-Whitney test for particle number and Kruskal-Wallace test with Dunn’s comparison test for velocities.
mec-9 regulates amphid ciliary length, positioning, and fasciculation
mec-9S was also expressed in the dye-filling amphid sensory neurons that are housed in bilateral amphid sensilla (Figure 2H-I). Dye filling assays assess ciliary integrity: WT animals dye fill whereas ciliary mutants are dye filling defective (Dyf). mec-1(e1066) animals have a slight Dyf phenotype (Perkinset al. 1986). We performed dye filling assays to determine whether mec-5 and mec-9 have similar ciliary defects as seen in mec-1. mec-1(e1066), mec-5(e1503), and mec-9(ok2853) had subtle amphid Dyf defects with 1-4 cells of 12 not filling (Figure 7A). mec-1(e1066) and mec-5(e1503) phasmids are also Dyf, with 1-2 out of four cells not filling (Supplemental Figure 5). Therefore, the amphid and phasmid ciliated sensory neurons also require proteins encoded by mec-1, mec-5, and mec-9 ECM genes.

(A) Number of amphid neurons out of 12 that filled with dye. (B-C) Schematics of WT and mec-9(ok853) amphid sensory organ (not all neurons shown). Proximal and medial sections of WT amphid cilia were in the sheath. Distal cilia were surrounded by socket and inverted cuticle with cilia tips exposed to the environment. Dashed lines denote levels of cross sections shown in (D-I). (C) mec-9(ok2853) amphid cilia were short and displaced with few ciliary tips exposed to the environment. (D) Left amphid channel pore and its schematic showing ten cilia at level of singlet microtubules. WT double ciliated amphids were found dorsally and ventrally in the pore with the ADF (medial) and ADL (lateral). The WT single ciliated amphids are found in the center two rows of amphids. The top row of single ciliated amphid channel neurons from medial to lateral are ASE, ASH, and ASG. The bottom row: ASI, ASJ, and ASK. WT left and right (not shown) amphids are mirror images of each other. Neurons are labeled by final amphid letter. (E) WT middle segment contained 10 cilia with microtubules arranged concentrically. (F) We observed the transition zones of most WT amphid cilia at this level and they were embedded within the amphid sheath. (G) Only one cilium out of ten was visible in mec-9 mutant channel pore. Socket and sheath had abnormal morphology as compared to WT. (H) mec-9 mutant middle segment: six out of ten cilia were visible in the lumen and there was increased matrix. mec-9 mutant amphids were abnormally arranged in three rows of two cilia. The top row: ASG and ASH, the second row: ASK and ASE, and the bottom row: ASI and ASJ, all of which are single ciliated amphids. Increased numbers of matrix filled vesicles were observed. (I) The double ciliated amphids (ADFs and ADLs) were in adjacent, aberrant electron dense spaces (increased matrix). Scale bar 200 nm.
We proceeded to examine amphid sensilla ultrastructure using TEM. In WT, 10 amphid cilia are enclosed in a pore of the bilateral papilla at the worm nose and surrounded by the cuticle (Figures 7B and 7D). mec-9 worms exhibited a misshapen cuticular pore encapsulating only the ASE cilium (Figure 7C). In the WT amphid channel, ten cilia are present in an invariant and precise arrangement (Figures 7D and 7G). In mec-9 mutants, we observed only cilia from the ASE, ASG, ASH, ASI, ASJ, and ASK (the single ciliated amphids) in the more proximal amphid channel; ADF and ADL (the double ciliated neurons) were displaced in an adjacent, aberrant electron dense space (Figure 7I). Consistent with a function in these neurons, mec-9shp::GFP was expressed in ADL and ADF. We conclude that mec-9 was necessary for cilia alignment and placement in amphid sensilla.
We measured ciliary lengths in these double ciliated amphid neurons from the bifurcation at the transition zone to ciliary tip. In contrast to the longer CEM and IL2, EV releasing cilia in mec-9 mutants, the cilia of the ADL and ADF amphid neurons were significantly shorter than WT by ~1.5x (Figure 6A). We also measured single ciliated amphid axonemes and found the ASI cilia significantly shorter than WT (Figure 6A). These amphid cilia length defects may contribute to dye filling defects observed in mec-9 mutants (Figure 7A). Not only were mec-9 amphid cilia shorter and misplaced, mec-9 mutant amphid transition zones were disorganized and abnormally dispersed along the anterior-posterior axis (compare WT in Figure 7F to mec-9 in Figure 7I), suggesting that the mec-9 ECM component is necessary for regulating dendritic fasciculation and/or transition zone placement.
Perkins et al. describe the presence of dark matrix vesicles in the space between the amphid cilia and sheath in WT animals and highlighted several mutants (osm-1 and osm-3) that abnormally accumulate large matrix vesicles (Perkinset al. 1986). In mec-9(ok2853) mutants, we observed a substantial increase in electron dense extracellular spaces that we identified as large matrix filled vesicles (Figure 7H). mec-9 mutants also had excessive matrix filled spaces in the amphid neuron sheath surrounding the amphid neurons that decouples cilia from glia (Figures 7H). Because of these spaces, there is a dysmorphic amphid socket and sheath in mec-9 mutant animals, which may contribute to the disorganization of the amphid channel cilia (Figures 7G-I). We conclude that mec-9 regulates ECM deposition in amphid and cephalic sensilla (Figure 4H-I).
DISCUSSION
ECM is important for neuronal anatomy and organization of the brain and nervous system (Gardiner 2011). For example, the ECM glycoprotein Reelin is necessary for migration of neocortical radial cells in the mammalian brain (Francoet al. 2011). In C. elegans, ECM components dex-1 and dyf-7 regulate amphid dendritic extension, which then affects cilia placement (Heiman and Shaham 2009). Mutations in the RNA splicing gene mec-8 or ECM component mec-1 mutants cause ciliary fasciculation defects (Perkinset al. 1986). The ECM gene mec-9 is expressed in the C. elegans nervous system where it provides mechanical support to multiple cell types including non-ciliated touch receptor neurons (Duet al. 1996) and, as shown here, ciliated sensory neurons.
mec-9 encodes two isoforms (Duet al. 1996). The mec-9 long isoform is expressed in touch receptor neurons (Duet al. 1996). Here, we show that the mec-9 short isoform is expressed in the shared and male-specific ciliated nervous system. In amphid and phasmid sensory organs, mec-9 mutants are dye filling defective, which reflects abnormalities in stereotypical ciliary positioning and fasciculation. In cephalic sensory organs of males, mec-9 mutants display distended extracellular spaces that contain excessive amounts of ciliary EVs and longer cilia. In CEM neurons, mec-9 is also required for dendritic integrity, with mec-9 mutant dendrites showing varicosities and waves as opposed to more linear trajectories. This abnormal dendritic morphology is typically observed in the nervous system of aged animals, and these age-dependent defects are accelerated by mutations that disrupt neuronal excitability or mechanosensation, including the mec-1, mec-5, and mec-9 ECM genes (Panet al. 2011). Further studies are needed to ascertain if MEC-9 protein physically restrains the dendrite or attaches to the ciliary or dendrite membrane to properly position the neuron. However, it is clear that MEC-9 and the ECM support the development and function of the C. elegans sensory nervous system (Figure 8).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The ECM genes mec-1, mec-5, and mec-9 regulate PKD-2::GFP localization and male mating behaviors (Figure 1 and 2). mec-9 also is necessary for PKD-2::GFP dendritic transport (Table 1), negative regulation of EV biogenesis, storage, and release (Figure 4), and neuron anatomy, such as dendritic integrity (Figure 6), cilia length (Figure 6) and organization (Figure 7), and matrix deposition (Figures 4G and 7H-I).
ECM and cilia share an intimate association. In aortic valves, primary cilia are restricted to ECM zones (Toomeret al. 2017). Chondrocytes have cilia embedded in ECM (Ruhlen and Marberry 2014). In umbilical cord mesenchyme, ECM regulates ciliary orientation (Nandadasaet al. 2015). ECM regulates ciliogenesis and organogenesis of Kupffer’s vesicle, the zebrafish equivalent of the mammalian embryonic node (Compagnonet al. 2014; Hochgreb-Hageleet al. 2013). In Drosophila, the ECM protein artichoke is required for morphogenesis of ciliated organs (Andreset al. 2014). The ECM gene spacemaker/Eyes shut/RP25 is necessary for ciliary pocket morphology and photoreceptor survival (Yuet al. 2016), with mutation causing photoreceptor degeneration in retinitis pigmentosa patients (Abd El-Azizet al. 2008).
ECM influences ciliary length in multicellular animals. Here we show that ECM regulates ciliary length in a cell-type specific manner in C. elegans. In amphid channel neurons, mec-9 is a positive regulator, with mec-9 mutants having shorter cilia. Conversely, in EV-releasing IL2 and CEM neurons, mec-9 is a negative regulator of ciliary length, with mec-9 mutants having significantly longer cilia. In mammalian skin, ECM component laminin-511 and its receptor integrin-b1 are required for primary cilia formation (Gaoet al. 2008). In mouse embryonic fibroblast 3T3-L1 cells, type 1 collagen promotes primary ciliary growth by repressing the HDAC6-autophagy pathway (Xuet al. 2018). In zebrafish Kupffers Vesicle, laminin-1 is a positive regulator of ciliary length (Compagnonet al. 2014; Hochgreb-Hageleet al. 2013). The mechanisms by which ECM controls ciliary length are largely unknown. A positive or negative feedback loop may act cell autonomously (between the ciliated cell and ECM secreted by the cell itself) or non-autonomously (between the ciliated cell and ECM secreted by neighboring cells). Our data are consistent with both possibilities. Rescue experiments indicate that mec-5 acts non-autonomously while mec-9S expression in ciliated neurons suggest cell autonomous function.
EVs are components of the ECM and EVs themselves may carry ECM proteins as cargo (Rackovet al. 2018; Rillaet al. 2017). Chlamydomonas ciliary EVs carry ECM proteins and ECM-degrading proteases, including a proteolytic enzyme that degrades ECM necessary for hatching (Longet al. 2016; Woodet al. 2013). Here we show that mec-9 mutants display dramatic increases in EV shedding and release (Figure 4C) and abnormal dark/light matrix vesicles containing EVs (Figure 4G), suggesting that the MEC-9 ECM component negatively regulates both EV shedding and release. We do not understand how ECM regulates EV biogenesis. However, genetic analysis revealed that mec-9 and the p38 MAPK pmk-1 acted antagonistically in EV biogenesis and release. pmk-1 mutants were defective in EV shedding and EV release (Wanget al. 2015). Interestingly, pmk-1 suppressed the mec-9 EV hypersecretion phenotype and mec-9 suppressed pmk-1 EV hyposecretion (Figure 5), suggesting that these genes act in opposing pathways that control EV biogenesis. An intriguing possibility is that MEC-9/ECM and pmk-1 kinase regulate the same target(s) such as a cell surface ECM receptor. In mice, CELSR3 (cadherin EGF LAG seven-pass G-type receptor 3) has MEC-9-like EGF domains in its N-terminal ectodomain and CELSR3 interacts with a kinase that regulates extension and guidance of sensory neurons (Goffinet and Tissir 2017). Mice deficient in CELSR2 and CELSR3 are defective in ependymal cilia development and develop hydrocephalus, a ciliopathy phenotype (Goffinet and Tissir 2017). The ligand(s) that activates CELSR2 and CELSR3 are not known.
The CELSR family is categorized as adhesion GPCRs (Liebscher and Schoneberg 2016). Adhesion GPCRs (aGPCRs) contain a large N-terminal ectodomain that contains a tetherized agonist Stachel sequence (Liebscher and Schoneberg 2016). Removal or structural changes to the N-terminal ectodomain exposes the Stachel sequence, which in turn activates the GPCR. Proposed mechanisms of Stachel release and activation of aGPCR signaling include mechanical stress and binding of ECM proteins to the N-terminus (Liebscheret al. 2014; Luoet al. 2014; Scholzet al. 2016).
The polycystin-1 family, while an 11-transmembrane spanning receptor class, has some features similar to an aGPCR (Cazorla-Vazquez and Engel 2018; Langenhanet al. 2015; Trudelet al. 2016). The function of the polycystins remains an enigma, even thirty years after the cloning of PKD1 and PKD2 (Maet al. 2017). Based on their ciliary localization, the polycystins were thought to be ciliary mechanosensors, but this model was disproven by the Clapham lab {Delling, 2013 #14868}(DeCaenet al. 2013). In mice, an ECM receptor integrin signaling pathway is essential for the development of ADPKD (Leeet al. 2015). An intriguing possibility is that the ECM itself acts in permissive fashion to allow cell-type specific polycystin activation and signaling, a mechanism used by adhesion GPCRs. Several lines of evidence support this idea. - Polycystin-1 binds to many ECM proteins including collagen I, II, and IV, fibronectin, and laminin (Malhaset al. 2002). Inactivation of integrin-b1 or integrin-linked kinase inhibits cystogenesis in Pkd1 mutant mice (Leeet al. 2015; Ramanet al. 2017). In zebrafish, pkd2 deficiency causes increased collagen synthesis via upregulated protein secretion and downregulation of this secretory pathway rescues cystogenesis (Le Correet al. 2014; Mangoset al. 2010). Combined, these studies reflect the close but poorly understood association between ECM, cilia, and the polycystins.
Our data reveal the profound importance of ECM components in nervous system of the worm. Model Figure 8 depicts the activity of ECM genes on ciliated sensory neurons. mec-1, mec-5, and mec-9 regulated PKD-2::GFP localization and male mating behaviors. mec-9 also influenced PKD-2::GFP dendritic transport and negatively regulated EV biogenesis, storage, and release. mec-9 and ECM was also important for neuronal anatomy, dendritic integrity, ciliary length and organization, and matrix deposition. Notably, abnormal ECM is implicated in the pathogenesis of ADPKD (Calvet 1993; Songet al. 2017) and the polycystins interact with ECM and focal adhesion proteins (Drummond 2011; Retailleau and Duprat 2014). Renal fibrosis observed in PKD is characterized by excessive deposition of ECM proteins (Drummond 2011; Songet al. 2017). Here we show that ECM is necessary for the health and well-being of ciliated neurons and neural organs in the nematode. Our findings highlights the promiscuity of ECM components, reveal ECM activity in ciliated neurons of the worm, and broadens the scope of activity of the ECM proteins originally named for their roles in mechanosensory touch receptor neurons. That ECM proteins contribute to ciliary localization and function of the polycystins in C. elegans advances the understanding of ciliopathies like ADPKD.
Acknowledgements
We thank Brian Coblitz and Martin Chalfie (Columbia University) for mec-5(+) rescue constructs; Richard Poole (Hobert lab, Columbia University) for assistance with whole genome sequencing of cil-7/mec-5(my2); Aranzta Barrios for advice on leaving assays and statistical analysis; Geoff Perumal and Leslie Gunther-Cummins for their help with HPF/FS processing; Monica Driscoll, Marion Gordon, Sunita Kramer, Barth Grant, Martha Soto, Christopher Rongo, and the Rutgers Super Worm Group for essential feedback during D.D.’s graduate career; the Barr lab for ongoing discussion and constructive criticisms, and especially Robert O’Hagan for his expertise in mechanotransduction; Gloria Androwski for outstanding laboratory support; WormBase and WormAtlas for online resources. This work was funded by National Institutes of Health grants DK059418 and DK111214 (to M. M. B.), OD 010943 (to D. H. H), and F31DK103550 (to D.D.). Some strains were provided by the National BioResource Project and the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs [P40 OD010440]. Authors declare no competing financial interests or any funding that can compromise the integrity of this work.
References




