

Abstract
Restriction-modification (RM) systems hinder the use of genetic approaches in the vast majority of bacteria. Here, we describe a systematic approach to adapt genetic tools for use in bacteria that are genetically intractable or poorly tractable owing to active RM defenses. In this process, we determine the genome and methylome of a bacterial strain and use this information to define the bacterium’s RM target motifs. We then synonymously eliminate RM targets from the nucleotide sequence of a genetic tool in silico, synthesize an RM-silent ‘SyngenicDNA’ tool and propagate the tool as novel minicircle plasmids, termed SyMPL tools, before transformation. Using SyngenicDNA and SyMPL tools, we achieved a profound, >100,000- fold, improvement in the transformation of a clinically relevant USA300 strain of Staphylococcus aureus demonstrating the efficacy of these approaches for evading RM systems. The SyngenicDNA and SyMPL approaches are effective, flexible, and should be broadly applicable in microbial genetics. We expect these will facilitate a new era of microbial genetics free of the restraints of restriction-modification barriers.
Restriction-modification (RM) systems are bacterial defense mechanisms that have hampered microbial genetic engineering since the inception of recombinant genetics 40 years ago1. Found in ~90% of sequenced bacterial genomes, RM systems enable bacteria to distinguish self from nonself DNA via two enzymatic activities: a restriction endonuclease (REase) and a modification methyltransferase (MTase). The REase recognizes the methylation status of DNA at a highly specific DNA target sequence and degrades unmethylated or inappropriately methylated (i.e., nonself) targets. Its cognate MTase protects the same target sequence across the host’s genome via addition of a methyl group, marking each site as self. RM targets vary greatly in sequence and length, typically ranging from 4-18 bp. To date, >450 different target sequences and >4,000 RM-associated enzymes have been identified2. Additionally, the number of these systems present in a bacterial cell and the targets recognized are hypervariable and highly species specific, often even strain, specific3.
Numerous approaches to overcome RM systems have been developed4, almost all involve a mimicry-by-methylation approach to replicate the specific methylation pattern of the desired bacterial host on human-made DNA by using heterologously expressed methyltransferase (MTase) enzymes5, 6 or, less successfully, crude cell lysates from the strain of interest. Although sometimes effective, mimicry-by-methylation approaches are time, resource, and cost intensive, and they suffer from limited applicability across different strains (Supplementary Note 1).
Here we present SyngenicDNA (Supplementary Note 2), a rapid, systematic, and relatively inexpensive approach to circumvent RM barriers during microbial genetic engineering. In contrast to current mimicry-by-methylation approaches, SyngenicDNA involves a stealth-by-engineering approach (Supplementary Fig. 1). It is inspired by a simple hypothesis: if a synthetic piece of DNA lacks the highly specific target recognition sequences for a host’s RM systems, then it is invisible to these systems and will not be degraded during artificial transformation. Therefore, in the SyngenicDNA approach, we identify the precise targets of the RM systems within an intractable (or poorly tractable) bacterial strain, eliminate these targets from the DNA sequence template of a genetic tool in silico—via single nucleotide polymorphisms (SNPs) or synonymous nucleotide modifications, and synthesize a tailor-made version of the tool that is RM-silent with respect to that specific host (Fig. 1). This stealth-based approach allows for simple reworking of currently available genetic tools, and DNA parts, to permit them to efficiently operate in bacteria with active RM defenses. Additionally, for effective propagation of the genetic tool, we have repurposed minicircle technology to eliminate components required in Escherichia coli but superfluous in the target host (Supplementary Fig. 2). Though not essential, use of minicircle technology reduces the complexity and increases the flexibility of SyngenicDNA.

(A) Identification of RM system target motifs by SMRTseq. Methylome analysis of polymerase kinetics during sequencing permits detection of methylated sites at single-nucleotide resolution across the genome, revealing the exact motifs targeted by innate RM systems (indicated by colored nucleotides, where N is any nucleotide) (Kinetic trace image adapted from www.pacb.com). (B) Assembly in silico of a genetic tool with a desired functionality, followed by screening for the presence of RM target sequences and sequence adaptation, using SNPs or synonymous codon substitutions in coding regions, to create an RM-silent template which is synthetized de novo to assemble a SyngenicDNA tool. (C) Artificial transformation of the bacterium of interest target bacterium. Inappropriately methylated target motifs of the original genetic tool are recognized as nonself-DNA and degraded by RM systems. In contrast, the SyngenicDNA variant retains the form and functionality of the genetic tool but is uniquely designed at the nucleotide level to evade the RM systems and can operate as desired within the target bacterial host.
There are four basic steps (Fig. 1) to produce SyngenicDNA-based genetic tools: 1) target identification, 2) in silico tool assembly, 3) in silico sequence adaptation, and 4) DNA synthesis and assembly. Below, we detail each step and illustrate the power of the SyngenicDNA method by applying it to a poorly tractable strain of the human pathogen Staphylococcus aureus (Fig. 2).

{kind=link}
{kind=link}
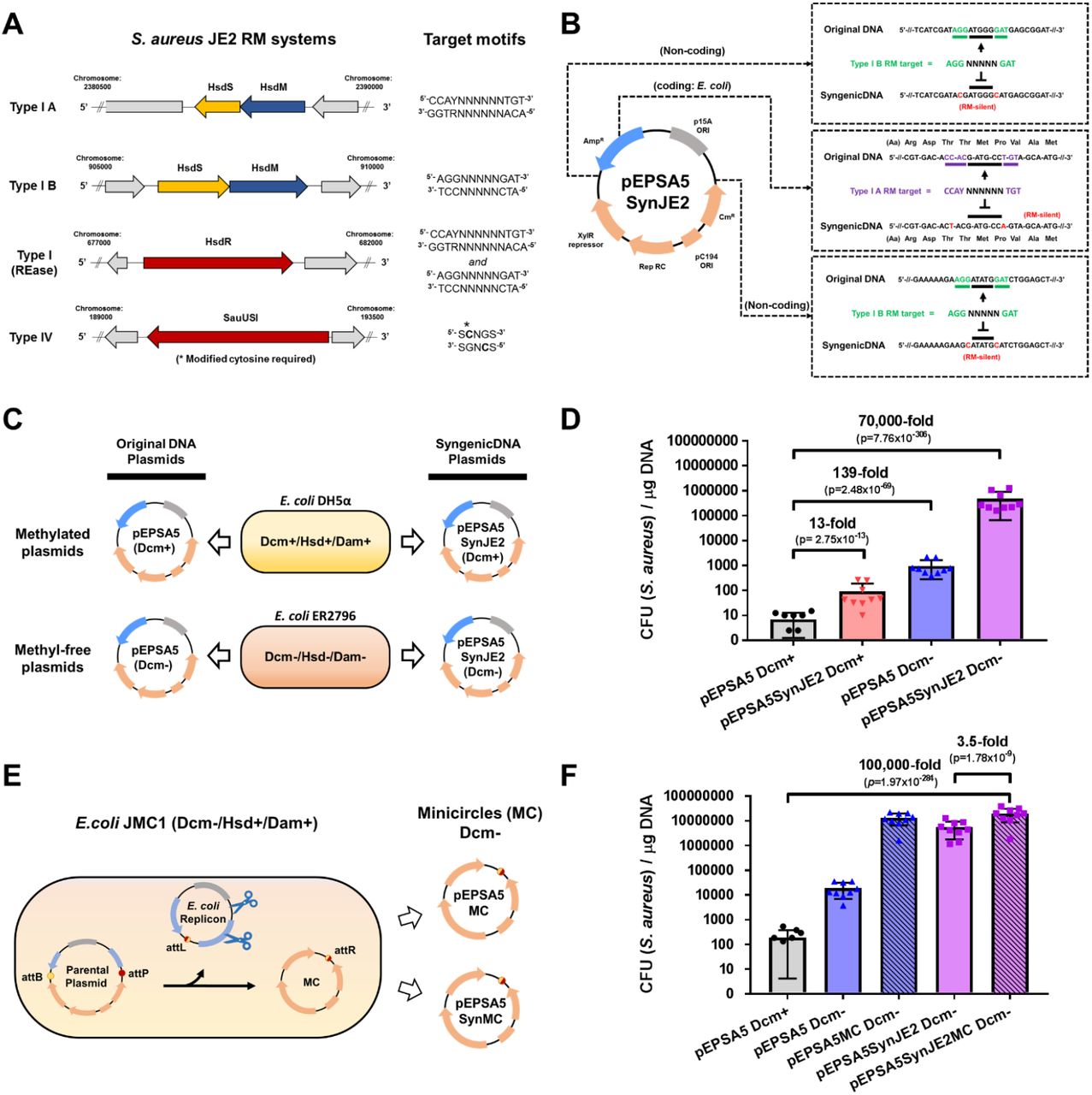
(A) JE2 maintains two Type I RM systems and a Type IV restriction system. Restriction endonuclease (HsdR and SauUSI) and methyltransferase (HsdM) genes are shown in red and blue, respectively, and specificity subunit (HsdS) genes are shown in yellow. RM system operons and their corresponding target motifs (where N is any base) were identified by SMRTseq and REBASE analysis. (B) Construction of pEPSA5SynJE2, which is an RM-silent variant of the pEPSA5 plasmid tailored to JE2. Six nucleotide substitutions (two synonymous codon substitutions and four SNPs) eliminated all Type I RM system targets from pEPSA5 sequence. (C) Plasmid propagation scheme. E. coli host strains produce DNA susceptible (DH5α; Dcm+) or resistant (E. coli ER2796; Dcm-) to the S. aureus JE2 Type IV restriction system. (D) Comparison of plasmid transformation efficiency (CFU/μg DNA) with pEPSA5 and the SyngenicDNA-variant pEPSA5SynJE2. (E) Propagation of minicircles (pEPSA5MC and pEPSA5SynJE2MC) lacking Dcm-methylated sites within SyMPL producer strain E. coli JMC1. (F) Comparison of SyngenicDNA and pEPSA5-based SyMPL plasmid transformation efficiency (CFU/μg DNA) with JE2. Data are means + SEM from nine independent experiments (three biological replicates with three technical replicates each).
Target identification requires the delineation of each methylated site, with single-base resolution, across an entire bacterial genome (i.e., the methylome) and starts with single molecule real-time (SMRT)7 genome and methylome sequencing. Using methylome data, we delineate each of the recognition motifs protected by the MTases of the host’s RM systems and infer the targets recognized and degraded by their cognate REases (Supplementary Note 3). This yields a concise list of a host microbes’ RM targets to be eliminated from the DNA sequence of the selected genetic tool.
In silico tool assembly requires complete annotation of a genetic tool’s sequence with respect to plasmid chassis, replication origins, antibiotic resistance cassettes, promoters, repressors, terminators and functional domains to avoid adverse changes to these structures during subsequent adaptation steps. Ideally, a complete and minimalistic genetic tool with previous demonstrable functionality in a genetically tractable strain is used for initial experiments, allowing for subsequent addition of DNA parts to increase functionality after successful transformation is achieved (Supplementary Note 4).
In silico sequence adaptation of the genetic tool is the most crucial step of the SyngenicDNA approach and exploits an intrinsic evolutionary weakness present in all RM systems: their exquisite specificity in target sequence recognition. REases are toxic in the absence of their cognate MTases and consequently seldom deviate from their recognition specificity8. Accordingly, in this step, we first screen the complete nucleotide sequence of the genetic tool for the presence of RM targets delineated by SMRTseq. We then recode the nucleotides of each RM target in silico to eliminate the target while preserving the functionality of the sequence. In noncoding regions, targets are removed by SNPs. In coding regions, the sequence of the target is removed using synonymous codon substitution. A single nucleotide switch is generally sufficient to remove RM targets but multiple switches can also be used. The preferential codon bias of the desired host is used to avoid introducing rare or unfavorable codons during the synonymous switch (Supplementary Note 5). Upon complete removal of all RM targets in silico, the adapted DNA sequence is RM silent with respect to the host and ready for de novo DNA synthesis.
Synthesis and assembly of RM-silent genetic tools is carried out with commercially available de novo DNA synthesis and standard assembly approaches, ensuring that any laboratory can construct SyngenicDNA tools. During commercial DNA synthesis, polynucleotide sequences are typically cloned onto an E. coli plasmid replicon, which is propagated to yield large amounts of the synthetic DNA. This E. coli replicon is convenient but might include RM targets that could lead to degradation of the overall circular tool after transformation into the host species. We have developed two solutions to this potential issue. One solution is to generate a SyngenicDNA E. coli plasmid backbone for each specific microbial host strain (Fig. 2). However, in routine applications this will increase costs of SyngenicDNA synthesis and, moreover, the E. coli replicon itself becomes redundant after propagation in E. coli, as it is typically nonfunctional in other bacterial species after transformation. Our alternative solution, therefore, is to remove the E. coli replicon entirely using minicircle DNA technology, rather than recode it. This approach also increases flexibility because the same E. coli replicon can be used to generate tools for multiple different host strains.
Minicircles (MCs) are minimalistic circular expression cassettes devoid of a plasmid backbone9. These are primarily used in gene therapy applications to drive stable expression of transgenes in eukaryotic hosts10. MCs are produced by attaching a parental plasmid (PP) to a transgene cassette; cultivating this construct in an E. coli host grown to high-cell density; inducing construct recombination to form an isolated transgene on a MC and a separate, automatically degraded, PP containing the E. coli replicon; and, finally, purifying isolated MCs by using standard plasmid methods9 (Supplementary Fig. 2). Because any DNA sequence can take the place of the transgene, we first hypothesized that MC technology could be repurposed to carry entire microbial plasmids and facilitate the removal of superfluous E. coli replicons from shuttle vectors. We demonstrated that the incorporation of SyngenicDNA sequences into a PP allowed us to create Syngenic DNA minicircle plasmid (SyMPL) tools. SyMPL tools include replication, selection, and functional domains for operation in a specific non-E. coli host, but lack an E. coli replicon despite being isolated at high concentrations from the MC-producing E. coli strain. In our SyMPL strategy, we attach a synthesized (and assembled) SyngenicDNA tool to the nonSyngenicDNA E. coli PP and propagate this construct in a MC-producing E. coli strain. The induction of MCs via recombination, with concurrent induction of a specific endonuclease that eliminates the PP, allows for easy isolation of a minimalistic SyngenicDNA-based genetic tool ready to transform into the desired host strain (Supplementary Fig. 2).
Notably, tool propagation in E. coli leads to the addition of methylation signatures on plasmid DNA by innate E. coli MTase enzymes. We demonstrated that this is also true of the MC-producing E. coli host (Supplementary Fig. 3). These methylation signatures can activate Type IV restriction systems, which target and degrade methylated DNA motifs, if such systems are present in a desired host strain. Accordingly, to ensure the functionality of our SyMPL approach in a broad range of microbial species, we applied iterative CRISPR-Cas genome engineering11 (Supplementary Fig. 4–6) to generate a suite of E. coli hosts each capable of producing MCs with different methylation signatures, including a methylcytosine-deficient MC producer strain (JMC1: dcm-, hsdM+, dam+) (Supplementary Note 6).
RM systems are a known critical barrier to genetic engineering in most strains of Staphylococcus aureus12. Based on its public health importance, we selected S. aureus JE2, a derivative of the epidemic USA300 community-associated methicillin-resistant S. aureus (MRSA) LAC strain13 to demonstrate proof of principle for our stealth-by-engineering approaches. We first applied our SyngenicDNA approach to the E. coli-S. aureus shuttle vector pEPSA5 (Fig. 2A-D, Supplementary Fig. 7) and generated pEPSA5SynJE2, a variant that differed by only six nucleotides (99.91% identical at nucleotide level), eliminating three RM target motifs present in the original sequence. We demonstrate a ~70,000-fold (p=7.76×10−306) increase in transformation efficiency (CFU/μg DNA) using the completely RM-silent pEPSA5SynJE2 (propagated in dcm-E. coli) compared to the original pEPSA5 plasmid (propagated in dcm+ E. coli) (Supplementary Note 7).
Subsequently, we sought to determine if a further increase in transformation efficiency could be achieved using the SyMPL minicircle approach. We generated a SyngenicDNA pEPSA5 minicircle for JE2 (pEPSA5SynJE2MC); 38% smaller than pEPSA5 and free of the original E. coli replicon (Supplementary Fig. 8). This pEPSA5SynJE2MC variant achieved ~2 x 107 transformants/μg DNA, a further 3.5-fold increase (p=1.78×10−9) in transformation efficiency over pEPSA5SynJE2 and >100,000-fold increase (p=1.97×10−284) compared to the original unmodified pEPSA5 plasmid (propagated in dcm+ E. coli). (Fig. 2E-F, Supplementary Table 3 and Supplementary Note 8). These results demonstrate the profound increase in transformation efficiency that can be achieved by systematic evasion of RM system barriers through stealth-by-engineering approaches.
SyngenicDNA and SyMPL approaches are effective, flexible, and broadly applicable methods to circumvent the RM barriers that heretofore have stymied the advancement of research and development in basic-, synthetic-, and translational-microbiology14–16. These methods may also be useful for evasion of other microbial defense mechanisms that rely on distinct target recognition sequences to discriminate self from nonself DNA. The stealth-by-engineering approaches we have developed offer the promise of an era in which microbial genetic system design is unrestrained by a microbe’s innate defense mechanisms.
METHODS
Materials
Escherichia coli NEBalpha competent cells were purchased from New England Biolabs (NEB) and used as intermediate cloning hosts. E. coli ER2796 was provided by the laboratory of Rich Roberts (NEB) and used to produce methylation-free plasmid DNA. E. coli MC (ZYCY10P3S2T; original minicircle-producing strain) was purchased from System Biosciences (SBI). Antibiotics and chemicals were purchased from Millipore-Sigma (St. Louis, MO) (Kanamycin, ampicillin, chloramphenicol, spectinomycin, isopropyl-D thiogalactopyranoside; IPTG) or Cayman Chemicals (Anhydrotetracycline). Growth media were purchased from Millipore-Sigma (Luria–Bertani, Brain Heart Infusion) or Oxoid (Vegetable Peptone). DNA isolation kits were purchased from Lucigen (Masterpure Gram Positive kit) and Qiagen (QIAprep Spin Miniprep Kit). Cloning reagents and DNA enzymes were purchased from NEB (Phusion High-Fidelity DNA Polymerase, HiFi DNA Assembly Master Mix, Q5 Site-Directed Mutagenesis Kit, EpiMark Bisulfite Conversion Kit) or Takara (EpiTaq HS for bisulfite-treated DNA). Plasmids were purchased from System Biosciences (SBI) (Parental plasmid; pMC vector), Elitra Pharmaceuticals (pEPSA5), Addgene (pCas; plasmid #42876, pTargetF; #62226) or obtained from the laboratory of George Church, Harvard University (pCKTRBS1) or Rich Roberts, NEB (pRRS). Oligonucleotides were purchased from IDT Technologies (Coralville, IA). Electroporation cuvettes (1 mm-gap) were purchased from BioRad and transformations performed on a BioRad Gene Pulser instrument. De novo DNA synthesis services and polynucleotide fragments were purchased from Synbio Technologies (Monmouth Junction, NJ). Plasmid DNA sequencing services were purchased from Macrogen (Cambridge, USA) or the DNA core at the Center for Computational and Integrative Biology, Massachusetts General Hospital (Cambridge, MA).
SMRTseq and RM system identification
The principle of single molecule, real-time sequencing (SMRTseq) and related base modification detection has been detailed previously2. SMRTseq was carried out on a PacBioRSII (Pacific Biosciences; Menlo Park, CA, USA) with P6/C4 chemistry at the Johns Hopkins Deep Sequencing & Microarray Core Facility, following standard SMRTbell template preparation protocols for base modification detection (www.pacb.com). Genomic DNA samples were sheared to an average size of 20 kbp via G-tube (Covaris; Woburn, MA, USA), end repaired and ligated to hairpin adapters prior to sequencing. Sequencing reads were processed and mapped to respective reference sequences using the BLASR mapper (https://github.com/PacificBiosciences/blasr) and the Pacific Biosciences’ SMRTAnalysis pipeline (https://www.pacb.com/documentation/smrt-pipe-reference-guide/) using the standard mapping protocol. Interpulse durations were measured and processed for all pulses aligned to each position in the reference sequence. To identify modified positions, we used Pacific Biosciences’ SMRTanalysis v2.3.0 patch 5, which uses an in silico kinetic reference and a t-test-based kinetic score detection of modified base positions. Using SMRTseq data, RM system identification was performed essentially as previously described3, using the SEQWARE computer resource, a BLAST-based software module in combination with the curated restriction enzyme database (REBASE)4. Prediction was supported by sequence similarity, presence, and order of predictive functional motifs, in addition to the known genomic context and characteristics of empirically characterized R-M system genes within REBASE and enabled the reliable assignment of candidate methyltransferase genes to each specificity based on their RM types.
Bioinformatics and SyngenicDNA adaptation in silico
DNA sequence analysis and manipulation was performed using the Seqbuilder and Seqman programs of the DNASTAR software package (DNASTAR, Madison, WI). Codon usage analyses and synonymous substitutions were determined using a combination of CodonW and the Codon Usage Database (www.kazusa.or.jp/codon/), and introduced within Seqbuilder to maintain the amino acid integrity of coding regions within E. coli. Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/) was used to align DNA and amino acid sequences from original ORFs and SyngenicDNA variants. Plasmid DNA (dsDNA) conversions from weight (μg) to molarity (pmol) was performed with Promega BioMath Calculators (https://www.promega.com/a/apps/biomath/).
DNA synthesis and assembly of SyngenicDNA plasmids
A SyngenicDNA-variant of the pEPSA5 plasmid (pEPSA5Syn) was assembled by replacing a 3.05 kb fragment of the original plasmid, encompassing three JE2 RM target sites, with a de novo synthesized DNA fragment that was RM-silent with respect to S. aureus JE2 (Fig. 2, Supplementary Fig.7). Primers used are listed in Supplementary Table 1. The original pEPSA5 plasmid was used as the amplification template for the unmodified backbone, while the plasmid pKan-Frag (Synbio Technologies) was used to amplify the modified RM-silent fragment. PCR amplicons were treated with DpnI to digest non-amplified template DNA and the pEPSA5SynJE2 plasmid was assembled using Gibson cloning. Plasmid nucleotide integrity was confirmed by resequencing. The pEPSA5 and pEPSA5SynJE2 plasmids were propagated within E. coli NEBalpha (dam+, dcm+, hsdM+) to produce methylated plasmid DNA or E. coli ER2796 (dam-, dcm-, hsdM-) to produce methylation-free plasmid DNA for evasion of Type IV RM systems. Methylation status of plasmid DNA was confirmed by DpnI treatment and agarose gel electrophoresis whereby only methylated plasmids were subject to digestion.
Constructing an anhydrotetracycline inducible CRISPR-Cas9/λ-Red gene editing system
We utilized a CRISPR-Cas9/λ-Red multigene editing strategy to introduce scarless MTase gene deletions in the E. coli MC strain (ZYCY10P3S2T). This strategy, initially described5 by Jiang et al., uses a two-plasmid system, pCas and pTarget (Supplementary Fig. 4A). In the original system, the pCas plasmid maintains a constitutively expressed cas9 gene and an arabinose-inducible regulatory promoter/repressor module (araC-Pbad) controlling the λ-Red system (Gam, Beta, Exo), both present on a temperature sensitive replicon (repA101Ts). The compatible pTarget plasmid has a sgRNA scaffold for the desired Cas9-target under control of the constitutive promoter (J23119) and a pMB1 origin of replication.
However, since MC formation within the E. coli MC strain is also regulated by chromosomally integrated araC-Pbad modules, arabinose induction of λ-Red recombination using the original system would cause unintentional induction of MC-assembly enzymes (the ΦC31 integrase and I-SceI homing endonuclease) during gene editing. To avoid this, we replaced the arabinose-inducible module of the λ-Red system with an alternative tetracycline-inducible module. Primers utilized are listed in Supplementary Table 1. A 1318-bp region of pCas, upstream of the λ-Red gam gene, containing the araC-Pbad module was replaced with 818-bp tetracycline-inducible regulatory promoter/repressor unit (TetR/PtetO) (Supplementary Fig. 4B). The plasmid pCKTRBS served as template DNA for amplification of the TetR/PtetO module, which was spliced to an 11.3 -kb amplicon of pCas (lacking the arabinose module) using Gibson assembly to form pCasTet-λ. The modified pCasTet-λ plasmid, in combination with the original pTarget, allowed for CRISPR-Cas9/λ-Red recombineering using anhydrotetracycline, a derivative of tetracycline that exhibits no antibiotic activity, instead of arabinose as an inducer molecule.
Genome editing of E. coli MC strain
The E. coli MC strain contains three active MTases (Dcm+, Hsd+, Dam+) encoded by the dcm, hsdMS, and dam genes respectively. To create a suite of E. coli MC strains, each capable of producing MCs with different methylation signatures, we sequentially deleted (in three-rounds) these MTase genes from the E. coli MC genome using our modified anhydrotetracycline-inducible CRISPR-Cas9/λ-Red recombineering strategy (Supplementary Fig. 4–6). In this strategy, λ-Red mediated recombination with a DNA editing template eliminates the MTase gene from the chromosome, followed by CRISPR-Cas9 mediated targeting of the MTase gene in unedited cells. Double-stranded DNA breaks introduced by CRISPR/Cas9 are toxic in bacteria, so only cells for which the target sequences have been edited can survive, allowing for positive selection of recombination events. MTase deletion template plasmids were constructed by assembling PCR amplicons of regions 5’ and 3’ of each MTase (reflecting the desired deletion event) onto a pRRS plasmid backbone (Supplementary Fig. 4C). These pRRS-based template plasmids were then used to PCR amplify linear editing templates for λ-Red recombineering. To remove template plasmid-carryover during electrotransformation, editing template amplicons were DpnI treated and PCR purified prior to use.
E. coli MC competent cells (System Biosciences) were first transformed with pCasTet-λ to form E. coli JMC, which constitutively expressed the Cas9 protein but lacked a gRNA target (Supplementary Fig. 5). JMC electrocompetent cells (harboring pCasTet-λ) were generated as previously described6. For λ-Red induction of JMC cells, anhydrotetracycline (200 ng/ml; ~0.5 μM) was added to the growing (30°C) culture 30 min prior to making cells competent, as described for the arabinose-based system6.
In the first round of genome editing, electrocompetent JMC cells were transformed with the dcm-deletion editing template and pT-Dcm (pTarget with a single gRNA targeting the dcm gene, under control of the J23119 constitutive promoter). For electroporation, 50 μl of cells were mixed with a 5 μl combination of 100 ng pT-Dcm plasmid and 200ng dcm-deletion editing template DNA; electroporation was performed in a 2-mm Gene Pulser cuvette (Bio-Rad) at 2.5 kV. Cells were recovered at 30°C for 1 h before selective plating at 30°C on LB agar containing kanamycin (50 μg/ml) and spectinomycin (50 μg/ml). Transformants were identified by colony PCR and DNA sequencing. Primers are listed in Supplementary Table 1. After confirmation of dcm deletion, the edited colony harboring both pCasTet-λ and pT-Dcm was cured of the latter plasmid by IPTG induction (0.5 mM), essentially as described previously5. Briefly, IPTG induces the production of gRNA, which targets the origin of replication of pT-Dcm after interaction with the constitutively expressed Cas9 protein. This gRNA is encoded on the pCasTet-λ plasmid under transcriptional control of the lacO/LacI (IPTG-inducible) system. The resulting E. coli strain, (dcmΔ/pCasTet-λ+) was made competent once again for the next round of editing, or cured of the pCasTet-λ plasmid by incubation at 37°C for four continuous inoculums, to form a plasmid-free minicircle producing strain E. coli JMC1 (dcm-, hsdM+, dam+).
In the second round of genome editing, the entire process was repeated targeting the Hsd MTase system. E. coli dcmΔ/pCasTet-λ+ was transformed with the Hsd-deletion editing template and the pT-Hsd plasmid (pTarget with a single gRNA targeting the hsdM gene). The resulting E. coli strain, (dcmΔ,hsdMΔ, pCasTet-λ+) was cured of the pCasTet-λ plasmid to form the E. coli JMC2 strain (dcm-, hsdM-, dam+). In the third round, the entire process was repeated targeting the Dam MTase system. E. coli dcm-, hsdM-, pCasTet-λ+ was transformed with the dam-deletion editing template and the pT-Dam plasmid (pTarget with a single gRNA targeting the dam gene). The resulting E. coli strain (dcm-, hsdM-, dam-) was cured of both plasmids to form the completely methyl-free E. coli JMC3 strain (dcm-, hsdM-, dam-).
After each round of genome editing, the phenotypic effect of dcm, hsdM, and dam gene deletions were confirmed using bisulfite sequencing, SMRTseq, and methyl-dependent restriction enzyme analysis, respectively (Supplementary Fig. 6). Site directed bisulfite sequencing and DpnI methyl-dependent restriction analysis of gDNA were performed essentially as we described previously7.
Production of SyMPL tools
The 4.3 kbp S. aureus replicon of both pEPSA5 plasmids (pEPSA5 and the pEPSA5SynJE2) were PCR amplified and spliced to the MC parental plasmid (pMC; Systems Biosciences) to form pEPSA5P and pEPSA5SynJE2P (P denotes parental). Primers listed in Supplementary Table 1. To evade the Type IV restriction system of S. aureus JE2, which targets Dcm-methylated cytosine residues, we used our dcm-deficient MC-producing E. coli strain JMC1 (dcm-, hsdM+, dam+). Competent plasmid-free E. coli JMC1 cells, prepared as described previously, were transformed with pEPSA5P and pEPSA5SynP. Minicircle induction and isolation was performed per manufacturers recommendations for the original E. coli MC strain (ZYCY10P3S2T). The resulting SyMPL tools pEPSA5MC and pEPSA5SynMC were eluted in high pure H20 and normalized to 250 ng/μl prior to transformation. Plasmid nucleotide integrity was confirmed by resequencing.
Staphylococcus aureus transformations
Electrocompetent S. aureus JE2 cells were prepared using a modified version of that used by Löfblom et al.8 Briefly, overnight cultures of S. aureus JE2 (~OD600nm=1.8) in vegetable peptone broth (VPB) were diluted to an OD600nm of 0.25 in fresh prewarmed VPB. In initial experiments to test the efficacy of the SyngenicDNA method, cultures were grown at 37°C with shaking (100 rpm) until they reached an OD600nm between 0.8-0.95 (~3 hours). However, in the interim of SyngenicDNA experiments and SyMPL method experiments, we observed increased JE2 cell competency was achieved when cultures were grown to an OD600nm between 1.5-1.7 (~6 hours). Therefore, we performed all SyMPL experiments with cells harvested at this higher optical density. In both cases, when culture tubes reached the desired OD, culture flasks were chilled on wet ice for 15 min. Cells were harvested by centrifugation at 5000 x g at 4°C for 10 min, washed once in equal volumes of ice-cold sterile water and pelleted at 4°C. The cells were then washed in 1/10 volume ice-cold sterile 10% glycerol, repeated with 1/25 volume ice-cold sterile 10% glycerol, repeated with 1/100 volume ice-cold sterile 10% glycerol, resuspended in 1/160 volume of ice-cold sterile 10% glycerol and then aliquoted (250 μl) into 1.5 ml tubes. Electrocompetent cell aliquots were frozen at −80°C until use.
For electroporation, a single aliquot was utilized for each individual experiment for accurate comparison of transformation efficiency between plasmids. The aliquot was thawed on ice for 5 min, transferred to room temperature for 5 min, centrifuged at 5000 x g for 1 min and resuspended in 250 μl sterile electroporation buffer (10% glycerol, 500 mM sucrose). A 50 μl volume of competent cells was mixed with 1 μg plasmid DNA (250 ng/ul in sterile water) and added to a sterile 1mm-gap electroporation cuvette. The cells were pulsed once using a Bio-Rad Gene Pulser System (settings: 25 μF, 100 Ω, 2.1 kV with a 2.3 millisec time constant) and outgrown in 1 ml of trypic soy broth with 500 mM sucrose for 1 hour at 37°C, diluted for spreading on trypic soy agar plates with 15 μg/ml Cm and incubated overnight at 37°C.
Scientific Rigor and Experimental Design
Transformation efficiencies (presented in Figure 2 D and F) were determined based upon nine independent experiments. We prepared three independent batches of electrocompetent S. aureus cells (Biological Replicate 1,2, and 3; Supplementary Table 3). Three aliquots from each batch of electrocompetent cells were used to perform three independent transformation experiments, typically on consecutive days (Technical Replicates A, B, and C; Supplementary Table 3). A single plasmid preparation (for each pEPSA5 variant) was used for all technical replicates within a batch. A fresh plasmid preparation (for all pEPSA5 variants) was used for each new batch of cells to account for variation associated with plasmid propagation/isolation from E. coli strains and the effect of freeze-thaw on plasmid DNA. In independent experiments, a single 250 μl aliquot of electrocompetent S. aureus was used for all plasmids (50 μl/plasmid) within each of the nine experiments, so that data within technical replicates could be treated as paired, or “clustered” across the four plasmids, and plasmid transformation efficiencies could be compared validly and efficiently. The average of CFU counts from a minimum of three replicate agar plates was used when determining transformation efficiencies for individual plasmids within experiments.
Statistical analysis
Statistical analyses were carried out using Graphpad Prism (version 7.04; GraphPad Software, San Diego, CA) and Stata version 12.1 (StataCorp. 2011. Stata Statistical Software: Release 12. College Station, TX: StataCorp LP). Means with standard error (SEM) are presented in each graph. As appropriate for count data, we compared transformation efficiency across plasmids by fitting negative binomial regression models with two-sided alpha=0.05. We used a generalized estimating equations (GEE) framework and robust standard errors to account for clustering within technical replicates of competent cells. For each experiment designed as a 2×2 factorial design, we fit main effects and multiplicative interaction terms (see Experimental Design). This can be thought of as a difference-in-differences analysis, quantifying how the effect of one condition (e.g. SyngenicDNA plasmid versus unmodified plasmid) differs in the presence or absence of another condition (e.g. propagated in a Dcm+ or a Dcm-E. coli host).
Data availability
Complete genome sequences and associated methylome annotations of Staphylococcus aureus USA300 JE2_Forsyth and Escherichia coli MC_Forsyth have been submitted to REBASE (http://rebase.neb.com/) for public release under organism # 21742 and # 21741, respectively. The nucleotide sequences of each plasmid used in this study are included here as Supplementary files. Raw CFU colony count data for determination of transformation efficiencies, along with data for associated analyses, are presented in Supplementary Tables 3 – 5.
References
Methods References
Supplementary Note References




