

Abstract
The bacterium Mycobacterium tuberculosis (Mtb) causes tuberculosis (TB) and is responsible for more human mortality than any other single pathogen1. Although ~1.7 billion people are infected with Mtb2, most infections are asymptomatic. Progression to active disease occurs in ~10% of infected individuals and is predicted by an elevated type I interferon (IFN) response3–8. Type I IFNs are vital for antiviral immunity, but whether or how they mediate susceptibility to Mtb has been difficult to study, in part because the standard C57BL/6 (B6) mouse model does not recapitulate the IFN-driven disease that appears to occur in humans3–5,8. Here we examined B6. Sst1S congenic mice that carry the C3H “sensitive” allele of the Sst1 locus that renders them highly susceptible to Mtb infections9,10. We found that B6.Sst1S mice exhibit markedly increased type I IFN signaling, and that type I IFNs were required for the enhanced susceptibility of B6. Sst1S mice to Mtb. Type I IFNs affect the expression of hundreds of genes, several of which have previously been implicated in susceptibility to bacterial infections11,12. Nevertheless, we found that heterozygous deficiency in just a single IFN target gene, IL-1 receptor antagonist (IL-1Ra), is sufficient to reverse IFN-driven susceptibility to Mtb. As even a partial reduction in IL-1Ra levels led to significant protection, we hypothesized that IL-1Ra may be a plausible target for host-directed anti-TB therapy. Indeed, antibody-mediated neutralization of IL-1Ra provided therapeutic benefit to Mtb-infected B6. Sst1S mice. Our results illustrate how the diversity of inbred mouse strains can be exploited to better model human TB, and demonstrate that IL-1Ra is an important mediator of type I IFN-driven susceptibility to Mtb infections in vivo.
Introduction
Mycobacterium tuberculosis (Mtb) infects approximately one quarter of all humans worldwide2, with highly diverse outcomes ranging from asymptomatic lung granulomas to lethal disseminated disease. Active TB disease is characterized by the uncontrolled replication of bacteria and pathological inflammation in the lungs and other organs, and arises in approximately 10% of infected HIV-negative individuals. There is no vaccine that reliably protects against pulmonary TB, and although antibiotics can be curative, the long (≥6-month) course of treatment and increasing prevalence of multi-drug resistant Mtb infections has spurred a search for alternative therapeutic approaches13,14. Since only individuals with active TB readily transmit infection, and as humans are the only natural reservoir of Mtb, a favored strategy to contain the TB epidemic is to identify and treat latently infected individuals likely to progress to active disease1,3. Identification of such individuals is challenging, but recent studies have demonstrated that an enhanced type I interferon (IFN) signature correlates with active TB5,6,8 and can predict progression to active TB up to 18 months prior to diagnosis3,4. A partial loss-of-function polymorphism in the type I IFN receptor (IFNAR1) is associated with resistance to TB in humans, suggesting that elevated levels of type I IFNs not only predict but may even be causally linked to TB progression15. In addition, numerous animal studies have demonstrated causal roles for type I IFNs in susceptibility to Mtb6,7,16,17 and other bacterial infections11,12.
Given strong evidence that type I IFNs confer susceptibility to human TB, two major remaining challenges are: (1) to determine the mechanisms by which type I IFNs mediate susceptibility to active TB, and (2) to exploit this knowledge to develop interventions that can reverse the susceptibility. Mechanistic studies and initial trials of possible therapeutic interventions require a robust animal model. However, the most commonly used animal model, the C57BL/6 (B6) mouse, does not robustly recapitulate the interferon-driven TB susceptibility seen in humans. B6 Ifnar−/− mice show mild resistance to Mtb in the spleen and variable but modest effects in the lungs18−20. To better model IFN-driven susceptibility, some investigators treat Mtb-infected B6 mice with poly-IC, a potent inducer of type I IFNs21. Such treatment dramatically increases susceptibility to Mtb in an Ifnar-dependent manner21,22, but because the IFN is induced artificially and not by Mtb itself, it is unclear if poly-IC mimics the course of IFN-driven disease in humans.
As an alternative approach, we sought to exploit the natural diversity of available inbred mouse strains. The 129 mouse strain shows clear IFN-driven susceptibility to Mtb23, but there are limited tools on this genetic background. We therefore turned to a previously described congenic mouse strain, B6.Sst1S, that carries the 10.7Mb ‘super susceptibility to tuberculosis 1’ region of mouse chromosome 1 from C3H on an otherwise B6 genetic background9,10. The B6. Sst1S mice exhibit marked susceptibility to aerosol TB infection9,10, however the mechanism by which the Sst1S locus confers susceptibility remains incompletely understood. Recent work has established that macrophages from B6.Sst1S mice exhibit an enhanced type I IFN response24,25. Here we show that B6.Sst1S mice exhibit an exacerbated type I IFN response in vivo that causes susceptibility to Mtb infection. We exploit this model of type I IFN-driven TB disease to identify the interleukin-1 receptor antagonist (IL-1Ra) as a major driver of type I IFN-induced susceptibility to Mtb. Genetic or antibody-mediated reduction in IL-1Ra levels largely eliminates the IFN-induced susceptibility of B6.Sst1S mice. Our results suggest new therapeutic strategies for tuberculosis.
Results
In agreement with previous work24,25, we found that bone marrow-derived macrophages (BMMs) from B6.Sst1S mice expressed higher levels of interferon-beta (Ifnb) and interferon-stimulated genes (ISGs) upon stimulation with TNF (Fig. S1). To determine if B6.Sst1S mice also exhibit an enhanced type I IFN signature in vivo, we measured Ifnb transcripts in the lungs of M. tuberculosis-infected mice. Indeed, B6.Sst1S mice exhibited higher levels of Ifnb transcripts as compared to B6 mice (Fig. 1a). To investigate whether this enhanced type I IFN signaling causes the susceptibility of B6.Sst1S mice to Mtb, we infected mice with Mtb and then treated with an IFNAR1-blocking antibody26 to inhibit type I IFN signaling. B6.Sst1S mice treated with the IFNAR1-blocking antibody showed significantly decreased bacterial burdens compared to those that only received an isotype control antibody (Fig. 1b). To provide genetic confirmation of this result, we crossed B6.Sst1S mice to B6.Ifnar−/− mice. Ifnar deficiency largely reversed the enhanced susceptibility of B6.Sst1S mice to Mtb infection. At 25 days post-infection, the bacterial burdens in the lungs of the B6. Sst1SIfnar−/− mice were significantly lower than in the lungs of B6.Sst1S mice, and were similar to B6 mice (Fig. 1c). Infected B6.Sst1SIfnar−/− mice also survived significantly longer than B6.Sst1S mice (Fig. 1d), though there are also clearly Ifnar-independent effects of the Sst1S locus that act at later time points. By contrast, and consistent with prior reports18–20, Ifnar deficiency had little or no effect on Mtb disease in wild-type B6 (Sst1R) mice. Recent data have suggested that the host protein STING is required for interferon induction to Mtb27–31. However, crossing B6.Sst1S mice to STING-deficient Stinggt/gt mice did not significantly reduce bacterial burdens at day 25 compared to B6.Sst1S mice (Fig. S2a). However, the B6. Sst1SStinggt/gt mice did show a slight improvement in survival not seen in the B6 genetic background30 (Fig. S2b). Overall our data demonstrate that the Sst1S locus acts to increase type I IFN signaling in vivo and thereby exacerbate Mtb infection, particularly during the early phases of infection.

a, Expression of Ifnb measured by qRTPCR relative to Rps17 in Mtb-infected lungs at day 25. b, Lung bacterial burdens at day 25 from Mtb-infected mice treated with anti-IFNAR1 or isotype control antibody. c, Lung bacterial burdens at day 25, or d, survival, of Mtb-infected mice. For a-c, genotypes indicated on the x-axis. c-d, Combined results from three independent infections. All except B6 mice were bred in-house (b-d). Error bars are SEM. Analyzed with two-ended Mann-Whitney test (b, c) or Log-rank (Mantel-Cox) Test (c). Asterisk, p ≤ 0.05; two asterisks, p ≤ 0.01; three asterisks, p ≤ 0.001.
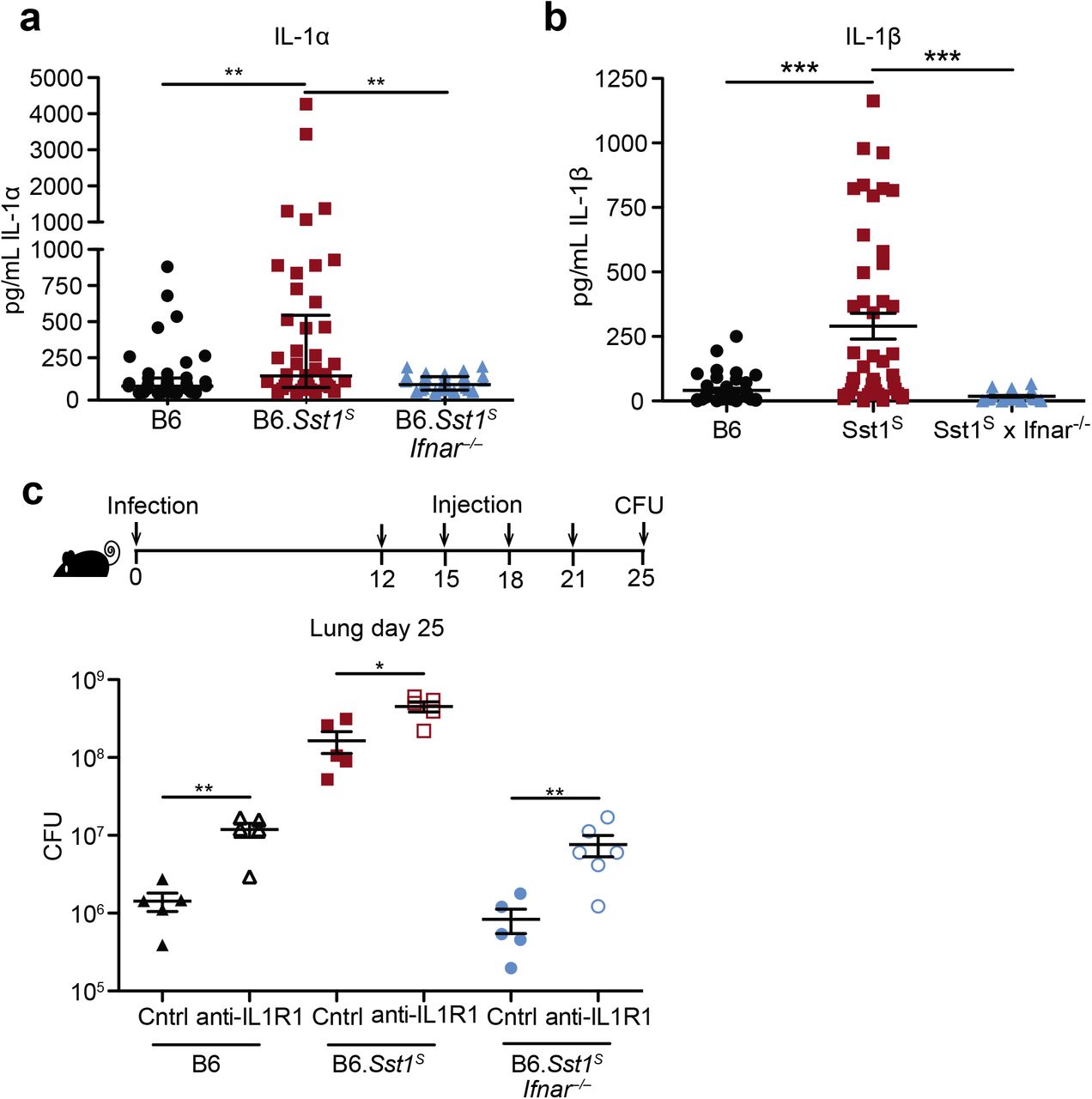
Type I IFN negatively regulates anti-bacterial immune responses via multiple mechanisms6,7, including through increased IL-10 levels32, decreased IFNγ signaling33, induction of cholesterol 25-hydroxylase (Ch25h)34, and/or decreased IL-1 levels35,36. We did not observe significant differences in IL-10 or IFNγ levels in the lung during in vivo Mtb infection (Fig. S3a,b). In addition, crossing B6.Sst1S mice to B6.Ch25h−/− mice did not alter day 25 lung bacterial burdens (Fig. S3c). Moreover, despite clear evidence that type I IFN and IL-1 counter-regulate each other37, the Sst1S locus did not appear to act to decrease the levels of IL-1 in vivo; in fact, we unexpectedly observed higher levels of both IL-1α and IL-1β in the lungs of B6.Sst1S mice at 25 days post-infection as compared to B6 mice (Fig. 2a,b). Other inflammatory cytokines, including TNF and CXCL1 were similarly elevated in the B6.Sst1S mice (Fig. S3d,e) as was the frequency of CD11b+Ly6G+ cells (neutrophils) in the lungs (Fig. S3f). The elevated inflammation in B6.Sst1S mice was a consequence of elevated type I IFNs, as inflammatory cytokines and neutrophils were reduced in B6.Sst1SIfnar−/− mice, but the underlying mechanism was not apparent.

a-b, Protein levels of IL-1α (a) and IL-1β (b) were measured in the lungs of Mtb-infected mice by ELISA at day 25. Combined results of four independent experiments. c, Lung bacterial burdens at day 25 from Mtb-infected mice treated with anti-IL1R1 or isotype control antibody. All animals except B6 were bred in-house (a-c). Error bars are SEM. Analyzed with two-ended Mann-Whitney test (a-c). Asterisk, p ≤ 0.05; two asterisks, p ≤ 0.01; three asterisks, p ≤ 0.001.
We reasoned that the high levels of IL-1α/β in B6.Sst1S mice may be a consequence of the higher bacterial burdens in these mice, or alternatively, may be causing increased bacterial replication via induction of a pro-bacterial inflammatory milieu, as previously proposed38,39. To distinguish these possibilities, we inhibited IL-1 signaling in vivo using an anti-IL-1R1 blocking antibody40, beginning 12 days post-infection (Fig. 2c, S4a). Both B6 and B6.Sst1S mice treated with IL-1R1 blocking antibody exhibited increased bacterial burdens compared to mice treated with an isotype control antibody. These results confirm prior evidence that IL-1 plays a protective role in B6 mice41–46, and extend this observation to B6.Sst1S mice as well. Thus, elevated IL-1 levels do not explain the exacerbated infections of B6.Sst1S mice.
Type I IFNs induce the expression of hundreds of target genes. However, given that IL-1 signaling is essential for resistance to Mtb41,42, we were particularly interested in Il1rn, which encodes the secreted IL-1 receptor antagonist (IL-1Ra)47. IL-1Ra binds to IL-1R1 without generating a signal and blocks binding of both IL-1α and IL-1β48 (Fig. 3a). Il1rn is known to be induced by type I IFN signaling49,50, and as expected, B6.Sst1S BMMs strongly upregulated Il1rn in an Ifnar-dependent manner when stimulated with TNF (Fig. 3b). Similarly, B6.Sst1S mice infected with M. tuberculosis had higher levels of IL-1Ra protein in their lungs, as compared to infected B6 or B6.Sst1SIfnar−/− mice (Fig. 3c, S4b). These results raised the possibility that the high IL-1 protein levels in B6.Sst1S mice are inadequate to protect against infection because of a block in IL-1 signaling. To test this possibility, we examined the amount of functional IL-1 signaling in lung homogenates from infected mice using HEK-Blue-IL-1RTM reporter cells (Invivogen). Despite higher levels of IL-1 proteins, lung homogenates from infected B6.Sst1S mice had less functional IL-1 signaling capacity as compared with B6 mice (Fig. 3d, S3c). The reporter appeared to be a reliable indicator of functional IL-1 as responses were blocked by anti-IL-1R1 antibody (Fig. S4c). The lower levels of IL-1 signaling seen in B6.Sst1S mice was reversed in B6.Sst1SIfnar−/− mice. These data underline the importance of distinguishing IL-1 protein levels from signaling capacity, and suggest that B6.Sst1S mice may be susceptible to Mtb because of reduced functional IL-1 signaling, despite increased IL-1 protein levels. Consistent with these observations, some data suggest IL-1Ra may also exacerbate TB in humans51–54.

a, Schematic of IL-1R signaling. b, Il1rn expression in BMMs treated with 10 ng/ml TNFα for 24 hours. c, IL-1Ra protein levels and d, IL-1 reporter assay measuring functional IL-1 activity in lungs of Mtb-infected mice at day 25. Combined results of three independent infections (d) or representative of at least two independent experiments (b, c). All animals except B6 were bred in-house (a-c). Error bars are SEM. Analyzed with two-ended Mann-Whitney test (c, d). Asterisk, p ≤ 0.05; two asterisks, p ≤ 0.01; three asterisks, p ≤ 0.001.
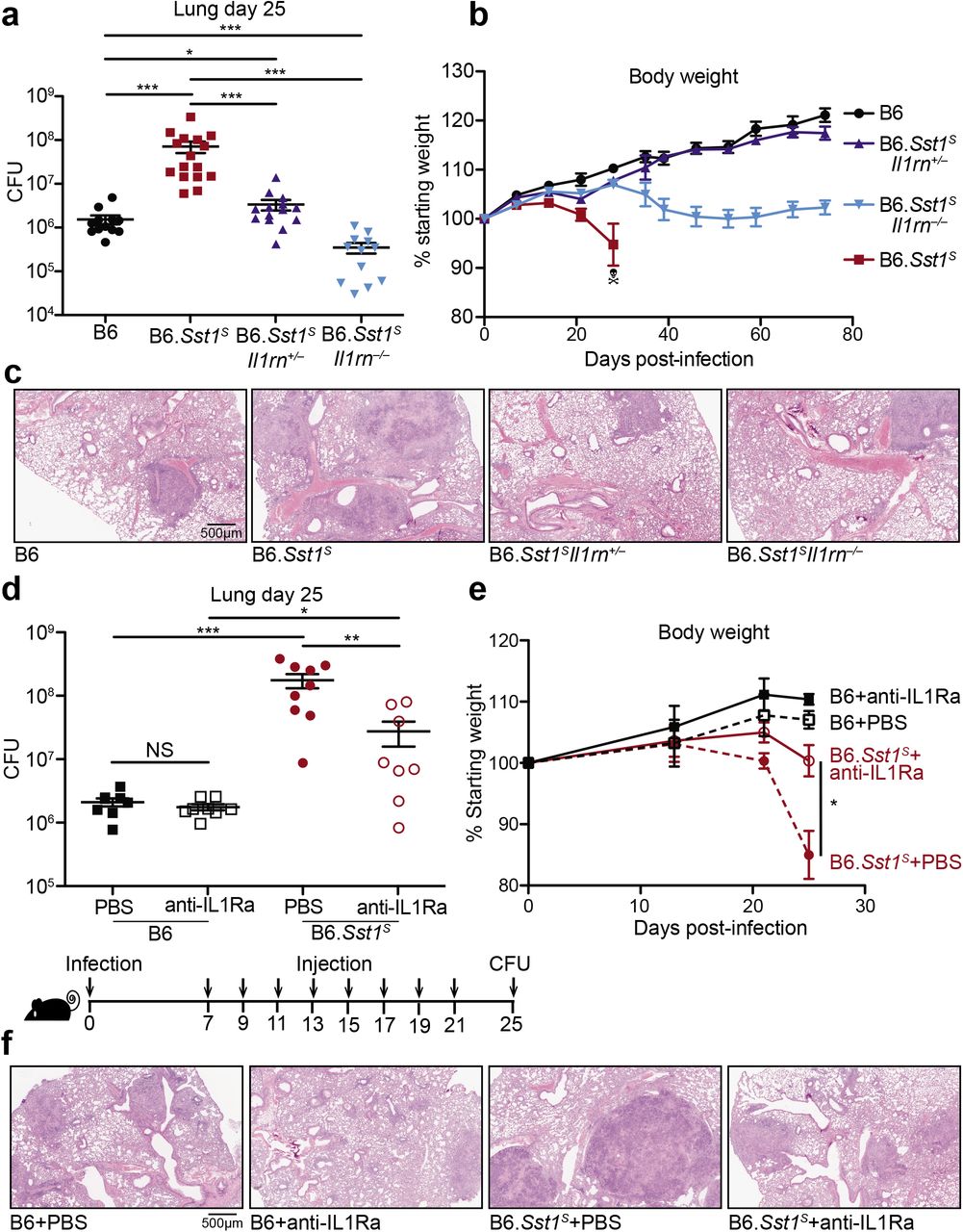
To test whether excessive type I IFN signaling neutralizes IL-1 signaling via IL-1Ra, we sought to reduce IL-1Ra levels in B6.Sst1S mice during Mtb infection. To do this, we first crossed B6.Sst1S to Il1rn−/− mice55. Since uninfected homozygous Il1rn / mice exhibit signs of inflammatory disease due to dysregulated IL-1 signaling55,56, and because heterozygous Il1rn+/− mice have a partial decrease in IL-1Ra levels55, we generated both heterozygous B6.Sst1SIl1rn−/− and homozygous B6.Sst1SIl1rn−/− mice. Both heterozygous and homozygous Il1rn deficiency protected B6.Sst1S mice from Mtb. In fact, bacterial burdens in B6.Sst1SIl1rn−/− mice were even lower than those found in ‘resistant’ B6 mice (Fig. 4a). Notably, a partial reduction in IL-1Ra levels due to heterozygous deficiency of Il1rn was sufficient to almost entirely reverse the enhanced IFN-driven susceptibility of Sst1S mice (Fig. 4b). Histological samples of infected lungs showed significant reduction in lesion sizes in both B6.Sst1SIl1rn−/− and B6.Sst1SIl1rn−/− mice as compared to B6.Sst1S (Fig. 4c). Despite concerns that enhanced IL-1 signaling may cause immunopathology, both B6.Sst1SIl1rn−/− and B6.Sst1SIl1rn−/− mice retained more body weight and exhibited increased survival as compared to infected B6.Sst1S mice (Fig. 4b, Fig.S5a).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
a-c, Lungs of Mtb-infected mice at day 25 were measured for (a) bacterial burdens in the lungs, (b) body weight, or (c) lung sections were stained with hematoxylin and eosin (H&E) for histology. d-f, Mtb-infected mice were treated with anti-IL1Ra antibody or PBS and (d) lung bacterial burdens were measured at day 25, or (f) day 25 lung sections were stained with H&E, or (e) body weights were recorded over time. All were bred in-house, and all except B6 and B6.Sst1S were littermates (a-c); B6.Sst1S were bred in-house (d-f). Error bars are SEM. Analyzed with two-ended Mann-Whitney test (a, b, d, e). Asterisk, p ≤ 0.05; two asterisks, p ≤ 0.01; three asterisks, p ≤ 0.001.
The dramatic protective effects of even partial reductions in IL-1Ra suggested that IL-1Ra might be a suitable target for host-directed therapy during Mtb infection. To test this idea, we treated infected B6.Sst1S mice with an anti-IL-1Ra antibody57 to block IL-1Ra and restore IL-1 signaling (Fig. 4d). B6.Sst1S mice that received the antibody for two weeks had significantly lower bacterial burdens in their lungs as compared to those receiving a control injection of PBS. In addition, mice treated with anti-IL-1Ra antibody retained significantly more body weight than their control counterparts (Fig. 4e), and exhibited reduced lung lesions (Fig. 4f), suggesting that the treatment did not cause detrimental inflammation. Overall these data indicate that genetic or antibody-mediated reduction of IL-1Ra rescues the type I IFN-driven susceptibility in B6.Sst1S mice without overt detrimental immunopathology.
Discussion
There is increasing interest in developing host-directed therapeutics for Mtb14. Although such therapeutics have not yet proven to be curative, and may thus be unlikely to replace antibiotics, host-directed therapies may offer some advantages in specific scenarios. For example, host-directed therapy could serve as an adjunct to antibiotic regimens in multidrug-resistant or extensively-drug resistant tuberculosis, where mortality is at 20%58,59. It may also be more difficult for Mtb strains to evolve resistance to a host-directed therapy, an important consideration given the increasing prevalence of multi-drug resistant Mtb strains. In addition, our results, along with those of others3,4, suggest a strategy in which latently infected individuals could be screened for the type I IFN signature known to be a strong predictor of progression to active TB. If the type I IFN signature in these individuals could then be reset or reversed by a host-directed therapy targeting IL-1Ra, or other host factors22, then it might be possible to prevent progression to active TB — the transmissible form of the disease — without the prolonged (6-month) antibiotic treatments that are associated with poor compliance and emergence of drug-resistant strains. This therapeutic strategy, though only presented here as a proof-of-concept in animals, may help focus treatments on those who are likely to transmit among the billions of latently infected persons. The success of such a strategy would not necessarily require microbiological cure, but would depend on identification of highly potent master-regulators of interferon-driven TB disease that can be readily targeted clinically in TB endemic areas. Identification of such regulators and development of therapies appropriate for resource-limited settings will require better animal models of interferon-driven TB disease in vivo. Our results show that the B6.Sst1S mouse may represent a useful model of IFN-driven TB disease in humans and suggest that therapeutic targeting of IL-1Ra may be a potent strategy for reversing type I IFN-driven susceptibility in vivo.
Methods
Mice
All mice were specific pathogen-free, maintained under a 12-hr light-dark cycle (7AM to 7PM), and given a standard chow diet (Harlan irradiated laboratory animal diet) ad libitum. All mice were sex and age-matched at 6-8 weeks old at the beginning of infections. C57BL/6J (B6), B6.129S-Il1rntm1Dih/J (Il1rn−/−), B6(Cg)-Ifnar1tm1.2Ees/J (Ifnar−/−) and B6.129S6-Ch25htm1Rus/J (Ch25h−/−) were originally purchased from Jackson Laboratories and subsequently bred at UC Berkeley. B6J.C3-Sst1C3HeBFeJKrmn mice (referred to as B6.Sst1S throughout) were from the colony of I. Kramnik at Boston University and then transferred to UC Berkeley. Stinggt/gt mice were previously described1. All animals used in experiments were bred in-house unless otherwise noted in the figure legends. All animal experiments complied with the regulatory standards of, and were approved by, the University of California Berkeley Institutional Animal Care and Use Committee.
Mycobacterium tuberculosis infections
Mtb strain Erdman (gift of S.A. Stanley) was used for all infections. Frozen stocks of this wild-type strain were made from a single culture and used for all experiments. Cultures for infection were grown in Middlebrook 7H9 liquid medium supplemented with 10% albumin-dextrose-saline, 0.4% glycerol and 0.05% Tween-80 for five days at 37°C. Mice were aerosol infected using an inhalation exposure system (Glas-Col, Terre Haute, IN). A total of 9ml of culture was loaded into the nebulizer calibrated to deliver ~20 to 50 bacteria per mouse as measured by colony forming units (CFUs) in the lungs 1 day following infection (data not shown). Mice were sacrificed at 14 days or 25 days post-infection to measure CFUs and/or cytokines. All but 1 lung lobe was homogenized in PBS plus 0.05% Tween-80 or processed for cytokines (see below), and serial dilutions were plated on 7H11 plates supplemented with 10% oleic acid, albumin, dextrose, catalase (OADC) and 0.5% glycerol. CFUs were counted 21 days after plating. The remaining lobe was used for histology or for RNA extraction. For histology the sample was fixed in 10% formalin for at least 48 hours then stored in 70% ethanol. Samples were sent to Histowiz Inc for embedding in wax, sectioning and staining.
Cytokine measurements
Cell-free lung homogenates were generated as previously described2. Briefly, lungs were crushed through 100μm Falcon cell strainers in sterile PBS with 1% FBS and Pierce Protease Inhibitor (Thermo Fisher). An aliquot was removed for measuring CFU by plating as described above. Cells and debris were then removed by low-speed centrifugation (300×g) and the resulting cell-free homogenate was filtered twice with 0.2μm filters to remove all Mtb for work outside of BSL3. All homogenates were aliquoted, flash-frozen in liquid nitrogen and stored at –80°C. Each aliquot was thawed a maximum of twice to avoid potential artifacts due to repeated freeze-thaw cycles. All cytokines except IL-10 and IL-1Ra was measured using Cytometric Bead Assay (BD Biosciences) according to manufacturer protocols. Results were collected using BD LSRFortessa (BD Biosciences) and analyzed using FCAP Array v3.0. IL-10 levels were measured using Mouse IL-10 ELISA Ready-SET-Go! (2nd Generation, eBioscience). IL-1Ra levels were measured by ELISA using Mouse IL-1ra/IL-1F3 Quantikine ELISA Kit (R&D Systems) according to manufacturer protocols.
IL-1 bioactivity reporter assay
Mice were infected with ~25-35 bacteria per mouse and sacrificed at 25 days-post infection to prepare cell-free lung homogenates. Assays were performed using HEK-Blue™ IL-1R cells (InvivoGen) with minor modifications. Cells were cultured under antibiotic-selection according to manufacturer protocols. Cells were plated at 5×104 cells/well in 50μl in 96-well plates, and allowed to adhere overnight in DMEM supplemented with 10% FBS, 2mM glutamine, 100 U/ml streptomycin and 100 μg/ml penicillin in a humidified incubator (37°C, 5% CO2). 50μl cell-free lung homogenates or standard curve made from recombinant mouse IL-1β (R&D systems 401-ML-005) were added to the cells and incubated overnight in a humidified incubator. Assays were developed using QUANTI-Blue (InvivoGen) according to manufacturer protocols. Experiments in which the initial CFU was either too high or too low produced inconsistent results in this assay.
Flow cytometry
Lungs were perfused with 10 ml of cold PBS and strained through 40μm cell strainers. Aliquots were removed for quantifying CFU. Cells were washed and stained with fixable viability dye (Thermo Fisher 65-0865-14). An aliquot of cells from each sample were removed and mixed with counting beads (Thermo Fisher C36950) for later enumeration. The rest of the cells were incubated with anti-mouse CD16/CD32 monoclonal antibody to block Fc receptors (Thermo Fisher 14-0161-81), then with antibodies for surface staining. The following antigens were stained for: CD45 (30-F11, Biolegend 103107), CD11b (M1/70, Thermo Fisher 48-0112-82), CD11c (N418, Biolegend 117335), Ly6G (1A8, BD Biosciences 740554), Ly6C (HK1.4, Thermo Fisher 17-5932-80), CD24 (M1/69, BD Bioscience 564664), MHC II (M5/114.15.2, Biolegend 107625), SiglecF (E50-2440, BD Biosciences 562680). Cells were fixed with fixation buffer (BD Biosciences 554714) for at least 1 hour at room temperature and stored in PBS with 1% FBS and 2mM EDTA overnight at 4°C in the dark. Data were acquired on a BD Fortessa X-20 flow cytometer and analyzed with FlowJo v10.
Bone marrow-derived macrophages (BMMs) and TNF-treatment
Bone marrow was harvested from mouse femurs and tibias, and cells were differentiated by culture on non-tissue culture-treated plates in RPMI supplemented with supernatant from 3T3-MCSF cells (gift of B. Beutler), 10% fetal bovine serum (FBS), 2mM glutamine, 100 U/ml streptomycin and 100 μg/ml penicillin in a humidified incubator (37°C, 5%CO2). BMMs were harvested six days after plating and frozen in 95% FBS and 5% DMSO. For in vitro experiments, BMMs were thawed into media as described above for 4 hours in a humidified 37°C incubator. Adherent cells were washed with PBS, counted and replated at 1.2 × 106 ~ 1.5 × 106 cells/well in a TC-treated 6-well plate. Cells were treated with 10 ng/ml recombinant mouse TNFα (410-TRNC-010, R&D systems) diluted in the media as described above.
Quantitative RT-PCR
Total RNA from BMMs was extract using RNeasy total RNA kit (Qiagen) according to manufacturer specifications. Total RNA from infected tissues was extracted by homogenizing in TRIzol reagent (Life technologies) then mixing thoroughly with chloroform, both done under BSL3 conditions. Samples were then removed from the BSL3 facility and transferred to fresh tubes under BSL2 conditions. Aqueous phase was separated by centrifugation and RNA was further purified using an RNeasy total RNA kit (Qiagen). Equal amounts of RNA from each sample were treated with DNase (RQ1, Promega) and cDNA was made using Superscript III (Invitrogen). Complementary cDNA reactions were primed with poly(dT) for the measurement of mature transcripts. For experiments with multiple time points, samples were frozen in the RLT buffer (Qiagen) or RNAlater™ solution (Invitrogen). Quantitative PCR was performed using QuantiStudio 5 Real-Time PCR System (Applied Biosystems) with Power Sybr Green PCR Master Mix (Thermo Fisher Scientific) according manufacturer specifications.
Transcript levels were normalized to housekeeping genes Rps17, Actb and Oaz1 unless otherwise specified. The following primers were used in this study. Rps17 sense: CGCCATTATCCC CAGCAAG; Rps17 antisense: TGTCGGGATCCACCTCAATG; Oaz1 sense: GTG GTG GCC TCT ACA TCG AG; Oaz1 antisense: AGC AGA TGA AAA CGT GGT CAG; Actb sense: CGC AGC CAC TGT CGA GTC; Actb antisense: CCT TCT GAC CCA TTC CCA CC; Ifnb sense: GTCCTCAACTGCTCTCCACT; Ifnb antisense: CCTGCAACCACCACTCATTC; Il1rn sense:
CGCCCTTCTGGGAAAAGACC, Il1rn antisense: CCGTGGATGCCCAAGAACAC; Irf1 sense: TGAGGAAGGGAAGATAGCCG; Irf1 antisense: TGTATGCCTATCCCAATGTCCC; Irgm1 sense: AAAACCAGAGAGCCTCACCA; Irgm1 antisense: ATGTTGGGGAGTAGTGGAGC; Gbp4 sense: TGAGTACCTGGAGAATGCCCT; Gbp4 antisense: TGGCCGAATTGGATGCTTGG; Gbp5 sense: TGTTCTTACTGGCCCCTGCT; Gbp5 antisense: CCAATGAGGCACAAGGGTTC; Ifit3 sense: AGCCCACACCCAGCTTTT; Ifit3 antisense: CAGAGATTCCCGGTTGACCT; Stat1 sense:
CAGAAAAACGCTGGGAACAGA; Stat1 antisense: CAAGCCTGGCTGGCAC; Gbp7 sense: AGCAAGCCCAAGTTCACACT; Gbp7 antisense: TCCGCTCTGTCAGTTCTGTG.
RNA sequencing and analysis
Total RNA was isolated as described above. Illumina-compatible libraries were generated by the University of California, Berkeley, QB3 Vincent J. Coates Genomics Sequencing Laboratory. The libraries were multiplexed and sequenced using one flow cell on HiSeq4000 (Illumina) as 100bp paired-end reads. The data were aligned using Sleuth3 and analyzed using Kallisto4.
Antibody-mediated neutralization
For all antibody treatments, the schedules are indicated in the figures. All treatments were delivered by intraperitoneal injection. Mouse anti-mouse IFNAR1 (MAR1-5A3) and isotype control (GIR208, mouse anti-human IFNGR-α chain) were purchased from Leinco Technologies Inc. Each mouse was given 500μg per injection. Hamster anti-IL1R1 antibody (mIL1R-M147) was obtained from Amgen. Isotype control was Ultra-LEAF Purified Armenian Hamster IgG Isotype Antibody from Biolegend (400940). Each mouse was given 200μg per injection. Armenian hamster anti-IL1Ra antibody was produced in-house using a previously published hybridoma line5. Cells were grown in Wheaton CELLine Bioreactor Flasks (Fisher Scientific) according to manufacturer instructions. Media in the cell compartment used ultra-low IgG FBS (ThermoFisher 16250078) to minimize bovine IgG contamination during purification. Cell-free supernatant from the cell compartment was purified using protein G resin (GenScript). IgG was eluted using 0.1M acetic acid, then 10% of total volume of 1M Tris pH8 and 0.5M NaCl was added to neutralize. Size exclusion and buffer exchange to PBS was performed using Amicon Ultra-4 Centrifugal Filter Units (EMD Millipore). The final product was filter sterilized and stored at –80°C. For injections antibody stocks were diluted in sterile PBS and each mouse received 500μg per injection.
Statistical analysis
All survival data were analyzed with Log-rank (Mantel-Cox) Test. All other data were analyzed with Mann-Whitney test unless otherwise noted. Both tests were run using GraphPad Prism 5. Asterisk, p ≤ 0.05; two asterisks, p ≥ 0.01; three asterisks, p ≤ 0.001. All error bars are s.e.
Acknowledgements
We thank the Stanley and Cox labs for discussions and for support with Mtb and BSL3 experiments. We thank the Barton lab for discussions, L. Flores, P. Dietzen and R. Chavez for technical assistance, H. Nolla and A. Valeros and the Cancer Research Laboratory for flow cytometry. We thank A. Sandstrom, I. Rauch and P. Mitchell for helpful discussions and technical support. We thank J. Price, L. DiPeso and R. Merl for support with the RNAseq. We thank B. Penn for comments on the manuscript. Generation of the anti-IL1RA antibody was supported by the Extramural Collaborative Research Program of Cancer Research Institute, Kanazawa University. K.H.D and R.E.V. were supported by Investigator in the Pathogenesis of Infectious Diseases awards from the Burroughs Wellcome Fund. R.E.V. is an HHMI Investigator and is supported by NIH grants AI075039 and AI066302.
Footnotes
↵* e-mail: rvance{at}berkeley.edu
References




