

ABSTRACT
Objectives TNFAIP3 encodes A20 that negatively regulates nuclear factor kappa light chain enhancer of activated B cells (NF-κB), the major transcription factor coordinating inflammatory gene expression. TNFAIP3 polymorphisms have been linked with a spectrum of inflammatory and autoimmune diseases and recently, loss-of-function mutations in A20 were found to cause a novel inflammatory disease ‘haploinsufficiency of A20’ (HA20). Here we describe a family with HA20 caused by a novel TNFAIP3 loss-of-function mutation and elucidate the upstream molecular mechanisms linking HA20 to dysregulation of NF-κB and the related inflammasome pathway.
Methods NF-κB activation was studied in a mutation-expressing cell line using luciferase reporter assay. Physical and close-proximity protein-protein interactions of wild-type and TNFAIP3 p.(Lys91*) mutant A20 were analyzed using mass spectrometry. NF-κB –dependent transcription, cytokine secretion, and inflammasome activation were compared in immune cells of the HA20 patients and control subjects.
Results The protein-protein interactome of p.(Lys91*) mutant A20 was severely impaired, including inter-actions with proteins regulating NF-κB activation, DNA repair responses, and the NLR family pyrin domain containing 3 (NLRP3) inflammasome. The p.(Lys91*) mutant A20 failed to suppress NF-κB signaling, which led to increased NF-κB –dependent proinflammatory cytokine transcription. Functional experiments in the HA20 patients’ immune cells uncovered a novel caspase-8-dependent mechanism of NLRP3 inflammasome hyperresponsiveness that mediated the excessive secretion of interleukin-1β and -18.
Conclusions The current findings significantly deepen our understanding of the molecular mechanisms underlying HA20 and other diseases associated with reduced A20 expression or function, paving the way for future therapeutic targeting of the pathway.
INTRODUCTION
Tumor necrosis factor α induced protein 3 (TNAP3, also known as A20), encoded by the TNFAIP3 gene, is a key negative regulator of NF-κB activation downstream a large number of immune receptors, including the TNF and Toll/interleukin(IL)-1 receptor families and the T and B cell antigen receptors1-5. A20 achieves this via its dual function as a ubiquitin-editing enzyme: an N-terminal ovarian tumor (OTU) domain removes activating K63-linked polyubiquitin from key intermediate NF-κB signaling molecules to destabilize protein-protein interactions, whereas a C-terminal zinc finger domain catalyzes the attachment of K48-linked polyubiquitin to induce proteasomal degradation3,6,7. In addition, A20 inhibits NF-κB activation via a noncatalytic mechanism involving its binding to linear ubiquitin on NF-κB essential modulator (NEMO)8,9. The mRNA expression of A20 is NF-κB –inducible10, thus generating a negative feedback loop to attenuate NF-κB responses.
Recently Zhou et al. reported six families with heterozygous loss-of-function mutations in the TNFAIP3 gene that led to haploinsufficiency of A20 (HA20) and caused an early-onset Behçet-like disease11. Studies in mouse models have previously demonstrated a crucial role for A20 in auto-inflammation and autoimmunity, as mice with a germ-line deletion of Tnfaip3 spontaneously develop severe multi-organ inflammation and tissue damage resulting in perinatal death12, and cell-specific ablation of Tnfaip3 results in diverse symptoms of immune dysregulation13,14. Similar to Tnfaip3 deficiency in mice12-15, human HA20 led to exaggerated NF-κB responses and increased secretion of NLR family pyrin domain containing 3 (NLRP3) inflammasome target cytokines11. Additional reports from these16 and further identified patients17-21 have expanded the clinical spectrum of HA20 to comprise diseases such as autoimmune lymphoproliferative syndrome, systemic juvenile arthritis, psoriatic arthritis, Crohn’s disease, and Hashimoto’s thyroiditis. The increasing number of diseases associated with A20 have emphasized its role in inflammatory diseases and in autoimmunity and raised the possibility that mutations in TNFAIP3 may be an unexpectedly common under-lying factor in the pathogenesis of rheumatic and other autoimmune diseases.
We describe here a family with polyautoimmunity and autoinflammatory symptoms caused by a novel heterozygous TNFAIP3 p.(Lys91*) loss-of-function mutation resulting in haploinsufficiency. Our aim was to further elucidate the upstream molecular mechanisms linking HA20 to dysregulated NF-κB activation and NLRP3 inflammasome responses. Thus, we analyzed protein-protein interactions of wild-type (wt) and p.(Lys91*) A20 in HEK cells and performed functional experiments in patients’ immune cells to study NLRP3 inflammasome responses. As A20 is rapidly emerging as a key regulator of immune activation and autoimmunity in humans, the detailed characterization of A20 effector mechanisms paves the way for future therapeutic targeting of the pathway.
MATERIALS AND METHODS
Ethics statement
Blood samples from patients and controls were collected under written consent in accordance with the Declaration of Helsinki; also the parents of the pediatric subjects signed a written informed consent. The study protocol was approved by the Ethical Committee for Clinical Science of Oulu University Hospital and by the Coordinating Ethics Committee of The Hospital District of Helsinki and Uusimaa.
Genetic analyses
DNA samples were extracted from total peripheral blood using standard methods. Whole-exome sequencing and data analysis were performed as previously described.22,23 All the common (frequencies above 0.01 in the general population) and non-coding variants were discarded. We searched for rare heterozygous variants, ac-cording to the inferred autosomal dominant in-heritance pattern (Fig. 1A). The variant identified in the TNFAIP3 gene (Ensembl ENSG00000118503:ENST00000433680: exon2:c.A271T: p.(Lys91*) was confirmed using PCR and capillary electrophoresis.

A) A pedigree showing the TNFAIP3 p.(Lys91*) mutation carriers (-/+, solid symbols) in three generations (I-III). Autoimmune thyreoiditis was diagnosed in all carriers (I-2, II-1, II-4, III-1) at an early age. In addition, the index patient (II-1) suffered from psoriasis, articular symptoms, atrophic gastritis, severe autoinflammatory lung reaction, anemia, genital papillomatosis, and repeated genital HSV infections. Daughter of the index (III-1) was diagnosed with polyarticular juvenile idiopathic arthritis at the age of 4 years, and mother of the index (I-2) died of liver failure at the age of 46 years. B) Schematic illustration of A20 structure showing the protein domains (upper panel) and the folding (lower panel). Position of the novel p.(Lys91*) mutation is indicated in blue. C) 3D structure models of wild-type and mutant p.(Lys91*) A20 (based on PDB accession 5LRX). OTU domain is highlighted with pink.
Creating A20 expressing Flp-In 293 T-REx cell lines
A20 mutant plasmids were created using Quick-Change Site-Directed Mutagenesis kit (Agilent Technologies) to wild-type TNFAIP3 obtained from the human Orfeome collection (Horizon Discovery). TNFAIP3 constructs were further subcloned into N-terminal MAC-tag Gateway® destination vector24. All generated constructs were confirmed with direct sequencing. The MAC-tagged expression constructs were transfected into Flp-In 293 T-REx cells (Invitrogen, Life Technologies) with FuGENE® HD (Promega). The cells were grown according to manufacturer’s instructions under selection with Hygromycin B (Thermo Fisher Scientific) to create stable cell lines. Expression of stable transgenes was confirmed by Western blotting using anti-hemagglutin (HA) primary (Biolegend, MMS-101R) and horseradish peroxidase-conjugated secondary antibody.
Luciferase assay
For reporter assays HEK293 cells were transfected with in total 50 ng A20 mutant or wild-type MAC-tagged construct and GFP or empty MAC-tag-vector24, 40 ng of NF-κB luciferase reporter plasmid (Cignal 45), and 1.4 ng Renilla luciferase reporter plasmid (pRL-SV40). Transfected cells were stimulated with indicated amounts of TNF-α (R&D Systems) for 16 hours, lysed with 1x passive lysis buffer (Promega), and luciferase assays performed with Dual-Glo Luciferase Assay System according to manufacturer’s instructions (Promega).
Affinity Purification and mass spectrometry
For each pull-down approximately 5×107 cells (5 × 15 cm dishes) were induced with 2μg/ml tetracycline (Thermo Fisher Scientific) for 24 hr (for BioID additional 50 μM biotin was added). After induction, cells were washed, harvested, pelleted by centrifugation, snap-frozen, and stored at -80 °C until analysis. Affinity purification of AP-MS and BioID was performed as described in Turunen et al. 25 and Heikkinen et al. 26, respectively. Sample preparation for mass spectrometry and LC-MS/MS analysis on Orbitrap Elite ETD mass spectrometer was performed as in Heikkinen et al. 26. After peptide reconstitution, 4 μl were analyzed from both Strep-Tag and BioID-samples.
Culture and stimulation of peripheral blood mononuclear cells (PBMCs)
PBMCs were isolated by density gradient centrifugation in Ficoll-Paque PLUS (GE Healthcare) and seeded at 1.5 × 106 cells/ml in Macrophage-SFM medium supplemented with penicillin-streptomycin or in RPMI 1640 supplemented with GlutaMAX, penicillin-streptomycin, and 10 % FBS (all from Gibco). PBMCs were allowed to rest for a minimum of 3 h before starting the stimulations with 1 μg/ml lipopolysaccharides (LPS) from E.coli O111:B4 (Sigma), 1 μg/ml Pam3Cys-SKKKK (Pam-3Cys; EMC microcollections), 10 μg/ml polyinosinic-polycytidylic acid (poly(I:C); InvivoGen), and 5 mM adenosine 5’-triphosphate (ATP; Sigma) for the indicated times. Inhibitors Z-YVAD-FMK (20 μM; R&D Systems), Z-IETD-FMK (5 μM; BD Bioscience), and necrostatin-1 (30 μM; Enzo Life Sciences) were added to the cells simultaneously with LPS in the 16 h stimulations, or 30 min before LPS in the LPS 6 h +ATP 45 min stimulations.
Measurement of cytokine secretion
TNF-α and the mature, cleaved forms of IL-1β and IL-18 were detected from PBMC culture media supernatants using Human TNF-α DuoSet ELISA, Human IL-1β/IL-1F2 DuoSet ELISA, and Human Total IL-18 DuoSet ELISA (all from R&D Systems).
Quantitative real-time PCR
RNA was isolated from PBMCs using RNeasy Plus Mini Kit (Qiagen) and cDNA synthesized with iScript kit (Bio-Rad). Quantitative real-time PCR was performed from 10 ng of cDNA per reaction using LightCycler480 SYBR Green I master (Roche) and LightCycler96 instrument (Roche); primer sequences are listed in Supplementary table 1. Relative gene expression was calculated using the 2(-δδCt)method, normalizing to the geometric mean of expression of two housekeeping genes, ribosomal protein lateral stalk subunit P0 and beta-2 microglobulin.
Monocyte caspase-1 activity measurement
Aliquots of EDTA-anticoagulated blood were incubated for 6 h at +37 °C under gentle rocking with or without 10 ng/ml LPS (Sigma). ATP at 5 mM (Sigma) was added for 20 min, followed by 2h incubation at +37 °C with a cell-permeable, irreversibly binding FAM-FLICA fluorescent caspase-1 substrate (ImmunoChemistry Technologies). After red blood cell lysis and staining with the monocyte marker anti-human CD14-APC (Miltenyi Biotec) for 15 min at +4 °C, the cells were analyzed using BD Accuri C6 flow cytometer (BD Biosciences; instrument maintained by the Bio-medicum Flow Cytometry Unit).
RESULTS
Identification of the mutation and the clinical manifestations of the patients
Exome sequencing of the index patient (II-1) identified a novel nonsense mutation in the exon 2 of the TNFAIP3 gene: c.A271T: p.(Lys91*) (ENSG0 0000118503:ENST00000433680)(Fig. 1A-B). The mutation is not listed in Genome Aggregation Database (gnomAD, Cambridge, MA, USA), and was predicted to be pathogenic according to the ACMG Standards and Guidelines27. The TNFAIP3 p.(Lys91*) mutation was confirmed in the patient’s genomic DNA and screened in the other members of the family. Two other affected family members (II-4, III-1) were observed to also harbor the mutation, (I-2 is an obligate carrier). The clinical manifestations of the carriers included autoimmune thyroiditis, which was diagnosed in all carriers (I-2, II-1, II-4, III-1) at an early age. The index patient suffered from psoriasis, articular symptoms, atrophic gastritis, severe inflammatory lung reaction, anemia and repeated genital HSV infections. Daughter of the index was diagnosed with polyarticular juvenile idiopathic arthritis at the age of 4. See also the legend of Fig. 1A.
Effect of the TNFAIP3 p.(Lys91*) mutation on protein structure and expression
The 3D protein structures of wild-type (wt) and p.(Lys91*) A20 were visualized by processing the PDB (www.rcsb.org28) accession number 5LRX (chain A29) with Chimera30, which yielded a severely truncated protein lacking the complete OTU domain (Fig. 1C). To further analyze the expression and possible physical and functional changes caused by the TNFAIP3 p.(Lys91*) mutation, stable Flp-In 293 T-REx cell lines expressing wt and p.(Lys91*) A20 were created. The wt HA-tagged A20 was expressed in Western blot, but the p.(Lys91*) mutant was detected only with an N-terminal tag (Fig. 2A), suggesting that only an N-terminal fragment is produced and no alternative start codons are used. Peptide identification using mass spectrometry confirmed that only the N-terminal region was detectable in the p.(Lys91*) mutant (Fig. 2B). The expression levels of the N-terminal peptides/protein of the wt and p.(Lys91*) mutant A20 were highly comparable (Fig. 2B).
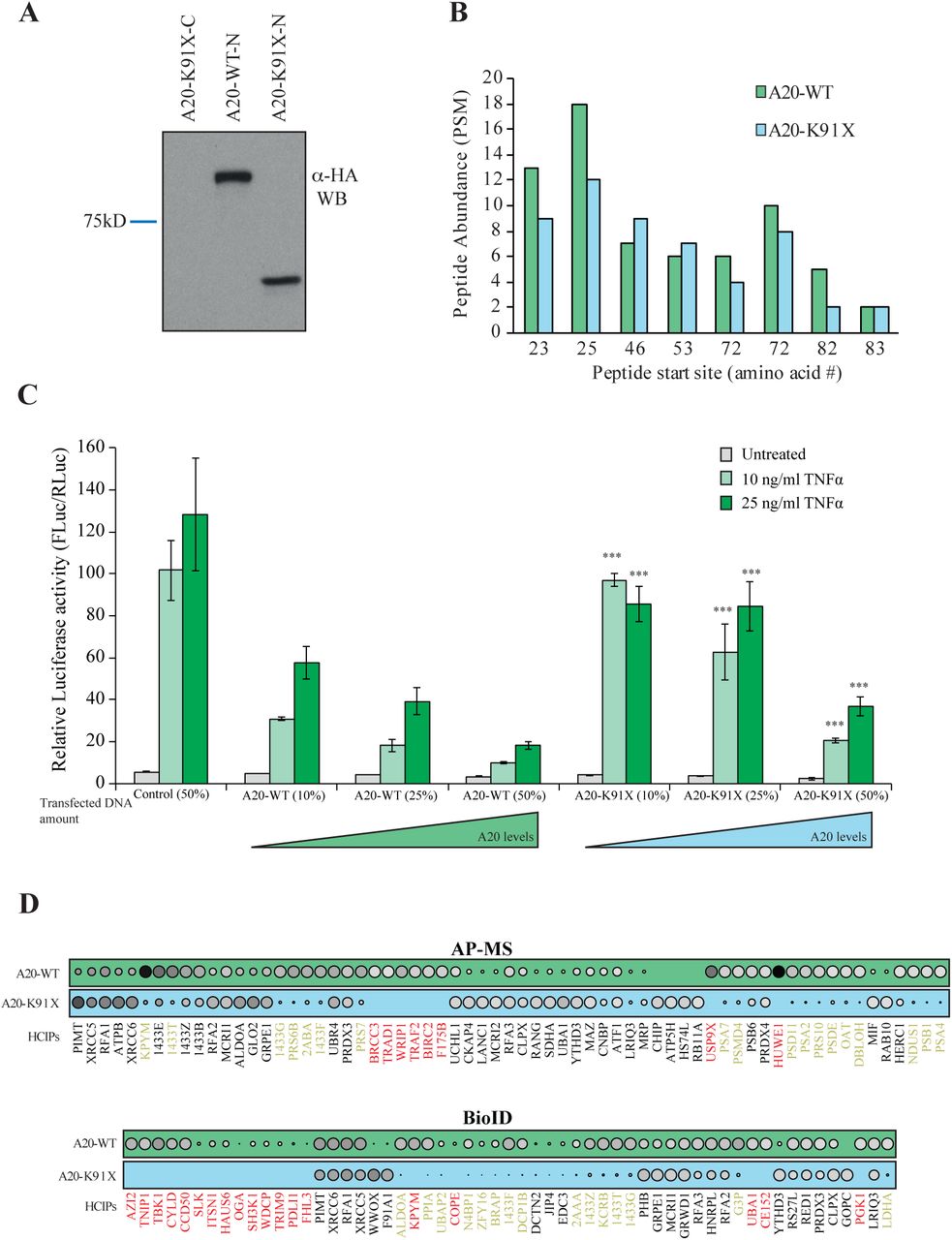
A) Expression profile analysis of wt A20 and the p.(Lys91*) mutant in HEK293 cells. The wt protein is detected with both as N- and C-terminally –tagged constructs, whereas the p.(Lys91*) mutant is only detected as N-terminally –tagged illustrating that no alternative start site is used and only short N-terminal fragment is produced. B) Quantitative mass spectrometry-based proteomics analysis shows that the N-terminal part of the wt A20 and p.(Lys91*) mutant are expressed in similar levels as shown by the quantitative abundance of the peptides derived from the N-termini. C) wt A20 dose-dependently inhibits the NF-κB signaling induced by TNF-α (Key:light green=10ng/ml and dark green=25ng/ml), whereas the p.(Lys91*) mutant show clearly diminished inhibitory activity suggesting haploinsufficiency. The asterisks represent the significance values; * <0.05, ** <0.01, *** <0.001. D) AP-MS (upper panel) and BioID (lower panel) analysis of wt and p.(Lys91*) mutant A20 shows clear differences in their stable protein-protein interactions and BioID analysis displays differential molecular context of the p.(Lys91*) mutant in comparison with the wt A20. (Key: the size and the color gradient show the relative abundance of the interacting prey protein detected in association with A20, red typeface designates the complete loss of the corresponding interactions and yellow >50% decrease compared to the wt A20).
A20 p.(Lys91*) mutant fails to suppress NF-κB activation and shows severely impaired protein-protein interactome
To analyze the possible functional consequences of the p.(Lys91*) mutation and the loss of OTU domain on the A20 functions, we monitored the NF-κB pathway activity using a luciferase-based reporter assay. The wt A20 dose-dependently inhibited TNF-α induced NF-κB pathway activation, whereas the p.(Lys91*) mutant failed to do so, clearly suggesting a haploinsufficiency (Fig. 2C).
The physical effects of the p.(Lys91*) mutation were further analyzed by proteome-wide affinity purification mass spectrometry (AP-MS) (see Supplementary Fig. 1 for details). These analyses revealed that the p.(Lys91*) mutant completely loses physical interactions with BRCC3, TRAD1, WRIP1, TRAF2, BIRC2, F175B, USP9X and HUWE1, with several additional weakened interactions (Fig. 2D). To obtain more insight into the molecular mechanisms of A20 and especially on the p.(Lys91*) mutation, we further quantitatively analyzed the functional and close-proximity inter-actions using BioID MS24(see Supplementary Fig. 1 for details). This analysis revealed complete loss of interactions with AZI2, TNIP1, TBK1, CYLD, CCD50, SLK, ITSN1, HAUS6, OGA, SH3K1, WDCP, TRIM9, PDLI1, FHL3, KPYM, COPE, UBA1, CE152 and PGK1. Many more interactions were severely diminished (Fig. 2D). Majority of the detected interactions of wt A20 were novel (Fig. 3). Functional annotation revealed extensive interactions with ubiquitinylation-, proteasome- and NF-κB -related proteins, as well as with proteins involved e.g. in cell fate decisions (14-3-3 proteins), DNA repair (53BP1 complex), and glycolysis (Fig. 3). Of these, the interactions with ubiquitinylation- and NF-κB -related proteins were most severely affected in the A20 p.(Lys91*) mutant.


Affinity purification mass spectrometry and BioID analysis of A20 identified 98 high-confidence protein-protein interactions (known interactions are shown with green lines, novel interaction detected on this study with blue and known prey-prey interactions with a dashed line). The interacting proteins are grouped bases on their molecular functions/complexes. Interactions, which are lost with the p.(Lys91*) mutation, are illustrated with red node fill color.
Blood immune cells of TNFAIP3 p.(Lys91*) carriers display strongly increased secretion of inflammasome-controlled cytokines IL-1β and IL-18
The NF-κB pathway and A20 have been shown to play regulatory roles in the activation of NLRP3 inflammasome, a pathway triggering caspase-1-mediated proteolytic maturation and secretion of proinflammatory cytokines IL-1β and IL-1815,31,32. Excessive NLRP3-dependent cytokine secretion was demonstrated in HA2011, yet the mechanism(s) linking reduced A20 to NLRP3 inflammasome activation in patient cells were not elucidated. We stimulated peripheral blood mononuclear cells (PBMCs) of the HA20 patients with Toll-like receptor (TLR) 4 agonist LPS, with or without a subsequent ATP pulse; both stimuli trigger the NLRP3 inflammasome in monocytes via distinct mechanisms and with different kinetics31. We found elevated secretion of IL-1β and IL-18 in PBMCs of the TNFAIP3 p.(Lys91*) carriers in response to LPS+ATP-induced ‘canonical’ (Fig. 4A-B; left panels) as well as LPS-induced ‘alternative’ pathway of NLRP3 inflammasome activation (Fig. 4A-B; right panels). To control for potential confounding factors in FBS preparations, PBMC cytokine secretion was analyzed both in commercial serum-free monocyte-macrophage medium (Fig. 4) and in RPMI containing 10 % FBS (Supplementary Fig. 2), yielding similar results. Also, TNF-α secretion in response to LPS, Pam3Cys, and poly(I:C) was increased in the patients (Fig. 4C, Supplementary Fig. 2C). We further studied the levels of IL-1β and IL-18 in patients’ plasma by ELISA and found increased levels of circulating IL-18 in TNFAIP3 p.(Lys91*) carriers compared to the age- and sex-matched controls (Supplementary Fig. 3A), whereas IL-1β was not detectable.



PBMCs from TNFAIP3 p.(Lys91*) mutation carriers, patients 1 (II-1, female, 30 years old) and 2 (III-1, female, 8 years old), were compared to sex- and age-matched controls 1 and 2, respectively. (A-C, left panels) The cells were primed with LPS for 6 h, followed by treatment with ATP for 45 min. (A-C, right panels) The cells were treated with TLR agonists for 16 h. Inhibitors necrostatin-1 (nec), Z-YVAD-FMK (yvad), and Z-IETD-FMK (ietd), or solvent control dimethyl sulfoxide (dmso), were added as indicated and cytokines were detected from PBMC culture supernatants by ELISAs.
The increased NLRP3 inflammasome response in TNFAIP3 p.(Lys91*) carriers is dependent on enhanced pro-IL-1β transcription and caspase-8 activity, but not on RIPK1
To elucidate the mechanism of altered NLRP3 inflammasome activation in the HA20 patients’ PBMCs, we analyzed the mRNA expression of inflammasome pathway components and cytokines and the effect of inflammasome-related inhibitors on cytokine secretion. PBMCs of patient III-1 displayed strongly increased baseline expression of pro-IL-1β and NLRP3 receptor, whereas patient II-1 cells showed only moderate changes (Supplementary Fig. 3B). After LPS stimulation, PBMCs from both patients showed slightly elevated levels of pro-IL-1β mRNA, and NLRP3 receptor expression remained moderately elevated only in patient III-1 (Fig. 5A). LPS-induced expression of NF-κB target cytokines IL-6, TNF-α, and IL-8 was elevated in patient III-1, and IL-6 also in patient II-1 (Fig. 5B). Receptor interacting serine/threonine-protein kinase 1 (RIPK1) inhibitor necrostatin-1 did not affect IL-1β or IL-18 secretion in the patients’ PBMCs (Fig. 4A-B), although it normalized the excessive TNF-α response to 16 h LPS treatment (Fig. 4C, right panel). As expected, both canonical and alternative NLRP3 inflammasome responses were efficiently blocked by the caspase-1 inhibitor Z-YVAD-FMK (Fig. 4A-B). How-ever, levels of active caspase-1 in monocytes after whole blood inflammasome stimulation were increased only in patient II-1 (Fig. 5C), supporting a more indirect enhancement of inflammasome complex function. Remarkably, caspase-8 inhibitor Z-IETD-FMK efficiently suppressed IL-1β and IL-18 secretion in PBMCs of the TNFAIP3 p.(Lys91*)- positive patients, particularly after canonical NLRP3 inflammasome activation (Fig. 4A-B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Samples from TNFAIP3 p.(Lys91*) mutation carriers, patients 1 (II-1, female, 30 years old) and 2 (III-1, female, 8 years old), were compared to sex- and age-matched controls 1 and 2, respectively. Relative expression of (A) NLRP3 inflammasome components and target cytokines and (B) inflammasome-independent proinflammatory cytokines was analyzed in PBMCs by quantitative PCR and normalized against housekeeping gene expression (arbitrary units). (C) Whole blood was stimulated with LPS and ATP, followed by the detection of NLRP3 inflammasome-triggered caspase-1 activity in monocytes using a fluorescent FLICA probe; the data are presented as median fluorescence intensity (MFI).
DISCUSSION
HA20 is a recently described familial autoinflammatory disease originally reported to manifest as an early-onset Behçet-like disease11, yet the range of known clinical manifestations is rapidly expanding16-21. Here we studied patients with HA20 caused by a novel heterozygous TNFAIP3 p.(Lys91*) mutation resulting in severe truncation and production of an N-terminal protein fragment with highly limited functional capacity. The patients exhibit an unusual combination of early onset autoimmune diseases, autoinflammatory symptoms and immunodeficiency, reflecting the widespread functions of A20 in controlling inflammation. To gain further insight into the molecular mechanism underlying the development of this complex disease, we focused on scrutinizing two pathways strongly linked with A20 function; the NF-κB pathway and the NLRP3 inflammasome.
Mutations causing HA20 were previously reported to increase TNF-α –induced IKKα/ IKKβ and MAPK phosphorylation and activation of NF-κB via defective removal of K63-linked ubiquitin from RIPK1, TNF receptor-associated factor 6, and NEMO11,18,19. As expected, also the A20 p.(Lys91*) mutant was defective in suppressing TNF-α –induced NF-κB activation. We employed state-of-the-art proteomics tools to comprehensively map global changes in A20 protein-protein interactions caused by the p.(Lys91*) mutation. In agreement with A20 ubiquitin-editing functions, the results revealed loss of physical interactions with ubiquitin ligases (BIRC2, TRAF2, HUWE1), deubiquitinases (BRCC3, USP9X), and other ubiquitinylation modulators (WRIP1, TRAD1, F175B/ ABRX2). BIRC2/3 and TRAF2/3 form a regulatory complex that suppresses apoptotic functions of caspase-8 to enforce canonical (inflammatory) NF-κB signaling, while blocking noncanonical NF-κB activation33,34. A20 modulates this balance by inducing TRAF2 degradation to suppress canonical NF-κB activation, and by binding c-IAP1/2 to support their caspase-8-inhibiting function while blocking their noncanonical NF-κB-inhibiting function35-37. Thus, the loss of A20 p.(Lys91*) interaction with BIRC2 and TRAF2 is compatible with enhancement of caspase-8 activity and canonical NF-κB signaling. In turn, BRCC3, F175B/ABRX2, WRIP1, and HUWE1 have specific functions in DNA damage responses38-41, which could be a significant finding regarding the association of reduced A20 levels with systemic lupus characterized by autoantibodies against nuclear antigens14. Moreover, BRCC3 and F175B/ ABRX2 are also components of the BRISC deubiquitinase complex that cleaves K63-linked ubiquitin and regulates mitotic spindle assembly42, type I interferon signaling43, and NLRP3 inflammasome activation44. Also, functional close-proximity protein-protein interactions of the A20 p.(Lys91*) mutant, particularly those related to ubiquitinylation and NF-κB signaling, were severely impaired, whereas interactions with proteasome, 53BP1-containing DNA repair complex, and 14-3-3 regulatory proteins remained partially functional.
The roles of A20 in regulation of the NLRP3 inflammasome, both NF-κB –dependent and –independent, are just beginning to be uncovered15,32. Mice deficient in myeloid A20 develop erosive polyarthritis specifically due to poor control of NLRP3 inflammasome activation by A2015. Moreover, increased NLRP3-dependent secretion of IL-1β and IL-18 was reported in HA20 patients11,17, yet the upstream mechanism(s) linking this effect to A20 in patient immune cells remain unknown. We found increased IL-1β and IL-18 secretion in PBMCs of TNFAIP3 p.(Lys91*)- positive patients in response to both canonical and alternative NLRP3-activating stimuli. In mouse macrophages, A20 suppresses the NF-κB-induced mRNA expression of NLRP3 receptor and pro-IL- 1β, but also blocks RIPK1/3-dependent pro-IL-1β ubiquitination that supports the inflammasome-mediated processing into mature IL-1β15,32. Our patients’ PBMCs showed elevated LPS-induced transcription of pro-IL-1β, but NLRP3 receptor expression was not consistently increased. The RIPK1 inhibitor necrostatin-1 that reduced aberrant IL-1β secretion in Tnfaip3-deficient mouse macrophages32 had no effect on IL-1β nor IL-18 secretion in the patients’ PBMCs, which was, instead, strongly suppressed by caspase-8 inhibition. This suggests a novel caspase-8-dependent mechanism linking reduced A20 function to enhanced NLRP3 inflammasome activation. The finding is in line with the increased caspase-8 activity reported in Tnfaip3-deficient mouse cells32,45and with the caspase-8-suppressing functions of A2037,45, and further supported by the loss of physical interaction between A20 p.(Lys91*) mutant and BIRC2, which is required for caspase-8 inhibition by A2037. Previous studies suggest that alongside its apoptotic functions, caspase-8 facilitates the priming and activation of NLRP3 inflammasome via multiple mechanisms46. In addition, the loss of interaction between A20 p.(Lys91*) mutant and the BRCC3-containing BRISC complex suggests a further novel NLRP3- regulating function of A20 that is lost in haplo-insufficient cells, as BRCC3 deubiquitinates the NLRP3 receptor directly promoting its activation44.
In summary, we report here a TNFAIP3 p.(Lys91*) loss-of-function mutation leading to HA20 and autoimmune/inflammatory symptoms. We utilize this mutation as a model to uncover novel molecular mechanisms related to the role of A20 in disease development. We defined, for the first time, the global physical and functional interactome of wild-type A20 and found severely diminished interactions in the p.(Lys91*) mutant. Moreover, we discovered a novel caspase-8- dependent mechanism linking HA20 to hyperactivation of the NLRP3 inflammasome in patient immune cells. Collectively, these data greatly deepen our understanding of the molecular mechanisms underlying the clinical disease phenotype of HA20 and other diseases associated with reduced A20 expression or function.
ACKNOWLEDGEMENTS
The study was supported by Finska Läkaresäll-skapet (K.K.E., D.N), The Canadian Institutes of Health Research (THC 135230; K.K.E.), the Stock-mann foundation (K.K.E.), and the Paulo foundation (K.R.). Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Bio-computing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). Exome sequencing of the index patient was conducted in the Institute for Molecular Medicine Finland (FIMM) Technology Centre. Sini Miettinen is thanked for expert mass spectrometry skills, and Xiaonan Liu for help with Chimera.
REFERENCES




