

ABSTRACT
Replication-dependent DNA double-strand breaks are harmful lesions preferentially repaired by homologous recombination, a process that requires processing of DNA ends to allow RAD51-mediated strand invasion. End-resection and subsequent repair are two intertwined processes, but the mechanism underlying their execution is still poorly appreciated. The WRN helicase is one of the crucial factors for the end-resection and is instrumental to select the proper repair pathway. Here, we reveal that ordered phosphorylation of WRN by the CDK1, ATM and ATR kinases define a complex regulatory layer that is essential for correct long-range end-resection connecting it to repair by homologous recombination. We establish that long-range end-resection requires an ATM-dependent phosphorylation of WRN at Ser1058 and that phosphorylation at Ser1141, together with dephosphorylation at the CDK1 site Ser1133, is needed to conclude long-range end-resection and support RAD51-dependent repair. Collectively, our findings suggest that regulation of WRN by multiple kinases functions as molecular switch to allow a timely execution of end-resection and repair at replication-dependent DNA double-strand breaks.
INTRODUCTION
DNA double-strand breaks (DSBs) represent a major threat to the integrity of the genome. DSBs can be produced by treatment with physical and chemical agents, such as DNA topoisomerases poisons camptothecin and etoposide (Nitiss, 2002), but it can be induced also at stalled replication forks after their collapse by specialized endonucleases (Dehé & Gaillard, 2017). In eukaryotes, DSBs formed during S-phase are repaired by homologous recombination (HR), however, they must be processed to yield an intermediate suitable for RAD51-mediated strand invasion (San Filippo et al, 2008; Ceccaldi et al, 2015). Processing of DSBs involves different proteins that ultimately are needed to carry out kb-long resection at the DNA ends (Symington & Gautier, 2011; Mimitou & Symington, 2011; Ranjha et al, 2018). This extensive end-resection, on one end, inhibits activation of proteins involved in the non-homologous end-joining (NHEJ) or microhomology-mediated end-joining (MMEJ) pathway of DSBs repair and, on the other hand, allows loading of HR factors (Aparicio et al, 2014; Chapman et al, 2012; Chiruvella et al, 2013; Ceccaldi et al, 2015). In addition, extensive long-range end-resection triggers activation of the ATR-mediated signalling (Flynn & Zou, 2011; Symington, 2016; Saldivar et al, 2017). Interestingly, as the end-processing at DSBs is regulated by ATM, and mostly CDK1, while repair by HR is regulated also by ATR, a switch between the two major regulatory circuits must occur (Symington, 2016; Saldivar et al, 2017). How exactly this switch takes place and which proteins are implicated in the process is partially understood.
Among proteins working at the interface between these two phases of DSBs repair by HR, one particularly interesting is the WRN protein (WRN). WRN is a factor involved in many processes linked to genome maintenance and its loss leads to hypersensitivity to agents inducing DSBs, in particular, those acting during S-phase (Rossi et al, 2010; Pichierri et al, 2000; Franchitto et al, 2000; Pichierri et al, 2011; Poot et al, 1999). Recently, the helicase activity of WRN has been implicated in the long-range end-resection taking place at DSBs, as it stimulates DNA2, another key enzyme in this process (Sturzenegger et al, 2014). In addition, WRN is regulated by the CDK1 kinase during long-range end-resection of replication-dependent DSBs, and its phosphorylation is necessary for proper repair by HR (Palermo et al, 2016). However, WRN is also targeted by the ATM and ATR kinases (Ammazzalorso et al, 2010; Matsuoka et al, 2007) and is a partner of BRCA1 (Cheng et al, 2006). Notably, BRCA1 is required for proper execution of end-resection and repair by HR (Symington, 2016), and it is also targeted by the CDK1, ATM and ATR kinases (Gatei et al, 2001; Johnson et al, 2009; Cortez et al, 1999; Tibbetts et al, 2000; Li et al, 2000; Chen, 2000). WRN might similarly contribute to bridge together the two phases of DSB repair by HR. Unfortunately, loss of WRN leads to complex phenotypes in response to DSBs, since its absence is offset by BLM during end-resection and HR is enhanced in response to CPT (Sturzenegger et al, 2014; Nimonkar et al, 2011; Pichierri et al, 2001).
We took advantage from our WRN mutants to investigate on the cross-talk between CDK1- and ATM-dependent WRN regulation in response to DSB formation, and to assess if WRN regulation was involved in coordinating end-resection with initiation of HR repair. Our results demonstrate that regulation by the CDK1 and ATM kinases is an ordered process involved in establishing long-range end-resection. We show that phosphorylation by CDK1 at Ser1133 stimulates modification at Ser1058 by ATM, which is the regulatory event for correct resection by WRN. Notably, these two “pro-resection” sites must be turned off at the end of long-range end-resection to promote ATM/ATR-dependent modification of Ser1141, which depends on long-resection itself and allow correct repair by HR. Deregulated phosphorylation by ATM leads to end-resection defects alone or in combination with inability to fix DSBs, because of altered recruitment of RAD51 in nuclear foci and persisting BRCA1 foci.
Altogether, our data contribute to unveil that WRN, with its ability to be modified by multiple DSB repair regulators, is able to function as a molecular switch from resection-to-repair mode in response to replication-dependent DSBs formation.
RESULTS
ATM- and CDK1-dependent phosphorylation of WRN are interrelated upon replication fork collapse
We previously demonstrated that WRN is phosphorylated by CDK1 in response to CPT-induced replication fork collapse and ATM following replication stress (Palermo et al, 2016; Ammazzalorso et al, 2010). To understand the existence of a cross-talk between ATM- and CDK1-dependent phosphorylation of WRN in response to replication-dependent DSB formation, we first assessed whether ATM could modify WRN upon CPT-induced DSBs. To this aim, HEK293TshWRN cells were transiently transfected with wild-type Flag-WRN, treated with CPT and the indicated kinase inhibitors. Then, phosphorylation at S/TQ sites was evaluated by anti-pS/TQ Western blotting in the anti-Flag immunoprecipitates. Our results showed that CPT treatment increased the S/TQ phosphorylation of WRN of > 2-fold (Figure 1A). Interestingly, treatment with ATMi (KU-55933) was more efficient than that with ATRi (VE-821) in reducing phosphorylation at S/TQ sites, suggesting that the majority of phosphorylation events depends on ATM after CPT. Notably, phosphorylation of WRN at S/TQ motifs was substantially reduced also by the CDK inhibitor Roscovitine upon treatment, but not under unperturbed conditions (Figure 1A).

A) WRN was immunoprecipitated from cells transiently transfected with Flag-WRN wild-type (WRNWT) were treated with 20μM Roscovitine, 10μM ATMi (KU-55933), 10μM ATRi (VE-821), alone or in combination, and then with 5μM CPT for 4 hours. Nine-tenth of IPs was analysed by Western Blotting (WB) with the anti-pS/TQ antibody, while 1/10 was analysed by anti-WRN antibody. Input represent 1/50 of the lysate. Anti-Flag WB was used to verify transfection. An anti-β-Tubulin antibody was used as loading control. Quantification of the representative blots is reported below each lane. B) Cells treated and subjected to anti-Flag-WRN IPs as in “A”. Nine-tenth of IPs was subjected to WB with an anti-pS1141WRN antibody, while 1/10 was detected by anti-Flag antibody, as indicated. Input represent 1/50 of the lysate. Anti-LaminB1 WB was used as loading control. C) Cells transiently transfected as in “A” were treated with 20 μM Roscovitine or 10μM ATMi (KU-55933), either alone or in combination, and then treated with 5μM CPT for 4 hours followed by IP/WB. Nine-tenth of IPs were analysed by WB with the anti-pS1133WRN antibody, while 1/10 was analysed by anti-Flag antibody, as indicated. One-fiftieth of the lysate was used as input.
As WRN contains six S/TQ sites and two of them, S1141 and S1058, are critical for ATM-dependent regulation (Ammazzalorso et al, 2010; Matsuoka et al, 2007), we narrowed our analysis to S1141 phosphorylation using the commercially-available anti-pS1141-WRN antibody. As shown in Figure 1B, treatment with CPT strongly enhanced S1141-WRN phosphorylation. Of note, phosphorylation of S1141 was similarly reduced by either ATMi or ATRi and also decreased substantially by Roscovitine (Figure 1B), recapitulating what observed with the pan-specific pS/TQ antibody (Figure 1A). These results suggest that CDK-dependent phosphorylation is required for the ATM/ATR-dependent regulation of WRN occurring in response to CPT.
We next tested whether also the CDK1-dependent phosphorylation site of WRN, S1133, (Palermo et al, 2016) was dependent on ATM activity. To do this, cells were treated with CPT and the indicated inhibitors, then the phosphorylation of S1133 was assessed by IP/WB. As expected, S1133-WRN phosphorylation was greatly increased after treatment with CPT and reduced almost completely by Roscovitine (Figure 1C). Notably, phosphorylation of S1133 was also substantially reduced by inhibition of ATM (Figure 1C). This result indicates that ATM activity is required for CDK1-dependent phosphorylation of WRN.
Interestingly, in response to CPT treatment, ATM/ATR- or CDK1-dependent phosphorylation of WRN was reduced also by Mirin (Figure EV1), further supporting that both events occur downstream initial end-resection.
Altogether, these findings demonstrate that, in response to replication-dependent DSB formation, ATM/ATR- or CDK1-dependent phosphorylation of WRN is influenced by each other and both requires MRE11 activity.
ATM-dependent phosphorylation of WRN requires prior modification by CDK1
To determine whether reduction of ATM-dependent phosphorylation of WRN depends on loss of S1133 phosphorylation by CDK1 instead of lack of initiation of end-resection because of CDK inhibition by Roscovitine, we examined WRN phosphorylation at S/TQ sites in cells expressing the wild-type form of WRN or its CDK1-unphosphorylable S1133A mutant by IP/WB experiments. We found that ATM-dependent phosphorylation was largely suppressed in the S1133A-WRN mutant in response to CPT but not under unperturbed conditions (Figure 2A). Interestingly, the S1133A-WRN mutant also showed a strong reduction of phosphorylation at the ATM site S1141 under all conditions (Figure 2B).

A) Cells were transiently transfected with the indicated Flag-tagged WRN and treated with 10μM ATMi prior to 5μM CPT for 4 hours. Anti-Flag IPs were analysed by WB using anti-pS/TQ (9/10 of IPs) or anti-WRN antibody (1/10 of IPs). One-fiftieth of the lysate (input) was blotted with an anti-Flag antibody to verify transfection. An anti-LaminB1 antibody was used as loading control. B) Cells were treated as in “A”. Nine-tenth of IPs was analysed by WB using the anti-pS1141WRN antibody while 1/10 was detected by anti-WRN antibody, as indicated. The input was subjected to WB as indicated. C) In vitro ATM kinase assay. For kinase assay, 2 μg of immunopurified GST-tagged WRN wild-type fragment (C-WRNWT) or WRN phosphomimetic mutant fragment (C-WRNS1133D) were phosphorylated in vitro using Flag-tagged ATM kinase, immunoprecipitated with specific anti-Flag-conjugated beads, and detected with anti-pS/TQ antibody. Treatment with 10μM ATM inhibitor (KU-55933) was used as a control, as indicated. Coomassie (CBB) showed GST-C-WRN fragments in the gel, as a control. D) Cells transiently transfected as indicated were treated or not with 5μM CPT for 4 hours. Cells were lysates and WRN protein was immunoprecipitated as in “A”. Nine-tenth of IPs was subjected to SDS–PAGE and detected by WB with the anti-pS1133WRN antibody, while 1/10 was detected by anti-WRN antibody, as indicated. Input is 1/50 of the lysate. The graph shows quantification of WRN phosphorylation at S1133 in each experimental condition from the representative blot.
Next, we performed an in vitro kinase assay using the C-terminal fragment of S1133D WRN mutant as substrate, to investigate if phosphorylation of S1133 improved the ability of ATM to modify its targets on WRN. Since fragments are expressed and purified from bacteria, the wild-type fragment contains unphosphorylated S1133 (Palermo et al, 2016). As shown in Figure 2C, ATM phosphorylation was greatly increased by the S1133D phosphomimetic mutation. These results suggest that CDK1 primes ATM-dependent modification of WRN at its target sites, thus including S1141.
Hence, we verified whether CDK1-dependent phosphorylation of WRN might be influenced by abrogating ATM-dependent phosphorylation or mimicking constitutive modification of WRN. To this end, we analysed phosphorylation of S1133 by WB in immunoprecipitates from cells expressing WRN wild-type, its ATM-unphosphorylable (3AATM) or phosphomimetic (3DATM) mutant. Notably, phosphorylation of S1133 was greatly increased under unperturbed conditions by both mutations, but it was only minimally affected by CPT treatment, especially in the 3DATM mutant (Figure 2D). This result suggests that loss or constitutive phosphorylation of WRN by ATM does not affect CDK1-dependent phosphorylation.
Altogether, these findings indicate that phosphorylation of WRN by ATM/ATR requires prior modification by CDK1 and suggest also that phosphorylation of WRN by CDK1 and ATM is ordered.
Regulated and site-specific phosphorylation of WRN by ATM is crucial for end-resection at DSBs
Our results indicate that CPT stimulates ATM-dependent phosphorylation of WRN, which is downstream that at S1133 by CDK1. Since proper execution of end-resection of CPT-induced DSBs requires WRN S1133 phosphorylation (Palermo et al, 2016), we investigated whether ATM is in the process. To this aim, we evaluated end-resection by the IdU/ssDNA assay (Figure 3A) after treatment with CPT in WS-derived cells complemented with the wild-type WRN (WRNWT), its ATM-unphosphorylable (WRN3AATM) or phosphomimetic (WRN3DATM) mutant or, as a control, S1133A-WRN mutant (WRNS1133A). Since WRN participates to long-range end-resection, we analysed ssDNA formation at 90min of treatment that corresponds to the WRN-DNA2-dependent processing (Palermo et al, 2016). As expected, loss of CDK1-dependent phosphorylation of WRN greatly reduced formation of ssDNA upon CPT exposure (Figure 3B). Of note, formation of ssDNA was also significantly suppressed by mutations abrogating ATM-dependent phosphorylation of WRN and, surprisingly, by mutations mimicking constitutive WRN phosphorylation (Figure 3B). Such defective end-resection phenotype of the ATM phosphomutants of WRN was also confirmed by the analysis of detergent-resistant RPA32 foci in response to CPT (Figure EV2A-B). However, not all the effects of ATM inhibition on end-resection was dependent on loss of WRN phosphorylation. Indeed, ATMi completely abrogated formation of ssDNA in WRNWT cells, but also in WRN3AATM and WRN3DATM cells (Figure EV2C).


A) Cartoon depicting the ssDNA assay by native anti-IdU immunofluorescence. The scheme shows how ssDNA can be visualized at collapsed replication forks. CPT treatment results in one-ended DSBs at replication forks leading to 5′-3′ resection of template DNA by nucleases (pacman) thus exposing nascent ssDNA, which is detected by our native IdU/ssDNA assay. Nascent DNA was pre-labelled for 15 min with IdU before treatment and labelling remained during treatment with CPT. B) Western blotting shows WRN expression levels in each WS-derived cell line complemented with the indicated form of WRN. The graph shows the mean intensity of IdU/ssDNA staining for single nuclei measured from three independent experiments (n=300, each biological replicate), data are presented as mean±SE. Representative images of IdU/ssDNA-stained cells are shown. C-D). WS fibroblasts were transiently transfected with the indicated WRN-expressing plasmid. WB shows WRN expression levels 48hrs after transfection using anti-WRN antibody. The level of ssDNA was analysed at different time points, as indicated. The dot plot shows the mean intensity of IdU/ssDNA staining for single nuclei (n=300, two biological replicates). Data are presented as mean±SE. Representative images of IdU/ssDNA-stained from CPT-treated cells are shown. E) WS-derived SV40-trasformed fibroblasts were transfected with Flag-tagged WRN wild-type or its phosphomutants as indicated. Western blotting shows WRN expression 48 hrs after transfection. Cells were labelled, treated and IdU/ssDNA assay was performed as in “B”. The dot plot shows the mean intensity of ssDNA staining for single nuclei measured from two independent experiments (n=300, each biological replicate), data are presented as mean±SE. Representative images of IdU/ssDNA-stained from CPT-treated cells are shown. Statistical analysis was performed by the ANOVA test (**** = p<0.0001; ** = P<0.01; * = P>0.01; ns = not significant).
These observations prompted us to explore the functional role of each single ATM phosphorylation site for the end-resection. To this end, we expressed the WRN wild-type, (WRNWT) or mutants in which each of the ATM sites was changed into Alanine (unphosphorylable; WRNS1058A) or Aspartate (phosphomimetic; WRNS1058D) in WS cells. Then, we tested the ability of the cells to perform end-resection by the ssDNA/IdU assay at 60 and 90mins of CPT treatment (Figure 3C). As expected, in cells expressing wild-type WRN, CPT induced a time-dependent increase in the formation of ssDNA (Figure 3C). In contrast, expression of the S1058A-WRN mutant significantly impaired end-resection and recapitulated the phenotype of WRN3AATM cells (Figure 3C). Notably, expression of the phosphomimetic S1058D-WRN mutant failed to affect formation of ssDNA (Figure 3C), diverging from the end-resection defect observed in WRN3DATM cells. In sharp contrast, cells expressing the S1141A-WRN mutant showed a wild-type formation of ssDNA after CPT treatment with a slightly higher level of ssDNA at the early time-point while those expressing the phosphomimetic S1141D mutation recapitulated the defective end-resection of WRN3DATM cells (Figure 3D). Of note, the last SQ site targeted by ATM in vitro (S1292; (Ammazzalorso et al, 2010) was phenotypically-neutral for end-resection (Figure EV2D). Therefore, to support end-resection, WRN needs to be phosphorylated at S1133 by CDK1 and at S1058 by ATM, but dephosphorylated at S1141.
To confirm that both S1133 and S1058 of WRN are essential and equally-important during end-resection, we analysed formation of ssDNA upon CPT treatment in WS cells complemented with S1058A+S1133A (WRNAA), S1058D+S1133D (WRNDD), S1058D+S1133A (WRNDA) or S1058A+S1133D (WRNAD) WRN mutant (Figure 3E). Supporting our hypothesis, a small amount of ssDNA was detected in cells expressing WRNAA suggesting a defective end-resection, while cells expressing WRNDD mutant showed a wild-type phenotype (Figure 3E). Of note, formation of ssDNA in response to CPT was defective also in cells expressing WRNAD mutant, while it was comparable to a wild-type in those transfected with WRNDA mutant (Figure 3E). These results suggest that phosphorylation at S1058 by ATM predominates on that performed at S1133 by CDK1.
Collectively, these findings show that proper execution of end-resection at CPT-induced DSBs requires concerted and ordered phosphorylation of WRN by the CDK1 and ATM kinases. Furthermore, they demonstrate that the modification of WRN at S1058 by ATM is the crucial event for end-resection, and that constitutive phosphorylation of WRN at S1141 negatively impacts on end-resection.
S1141 of WRN is phosphorylated during long-range end-resection and requires dephosphorylation at S1133
Having shown that ATM-dependent phosphorylation of WRN at S1141 apparently counteracts resection of DSBs induced by CPT, we tested the possibility that it takes place during or after long-range end-resection. Thus, we reasoned that phosphorylation of WRN at S1141 might be inversely-regulated with that performed at S1133 by CDK1, which correlates with long-range resection (Palermo et al, 2016). To test this, we performed IP/WB to evaluate phosphorylation at S1133 and S1141 in response to CPT at 1h, which corresponds to end-resection ramping, at 4h and after recovery, which corresponds to full engagement of end-resection and repair in a wild-type condition (Figure EV3, Figure 5 and later on). Phosphorylation at S1133 was detectable already at 1h of treatment and remained constant at 4h (Figure 4A). However, and most strikingly, phosphorylation at S1133 of WRN declined after recovery (Figure 4A). In contrast, phosphorylation at S1141 was barely detectable at 1h of treatment, increased strongly at 4h and remained elevated also during recovery from CPT (Figure 4A).


A) Cells were transiently transfected with an empty vector or a vector expressing Flag-tagged WRN wild-type (WRNWT) and were treated with 5μM CPT for different time points as indicated. In addition, cells were treated with CPT for 4h and recovered for 2 hours in drug-free medium. Flag-WRN was immunoprecipitated and 9/10 of IPs was analysed by WB with both the anti-pS1133WRN and the anti-pS1141WRN antibodies, while 1/10 was detected by anti-WRN, as indicated. One-fiftieth of the lysate was blotted with an anti-Flag antibody to verify transfection. An anti-LaminB1 antibody was used as loading control. The graph show quantification of the level of pWRN and data are from the representative experiment. B) Cells were transiently transfected with an empty vector or a vector expressing the indicated Flag-tagged WRN plasmids and were treated with 5μM CPT for different time points. Flag-WRN was immunoprecipitated and 9/10 of IPs was analysed by WB with the anti-pS1141WRN antibody, while 1/10 was detected by anti-WRN, as indicated. One-fiftieth of the lysate was blotted with an anti-Flag antibody to verify transfection. An anti-LaminB1 antibody was used as loading control. The quantification of the level of pWRN is reported below each lane and data are from the representative experiment. C) Cells were transiently transfected with an empty vector or a vector expressing the WRN wild-type (Flag-WRNWT) or helicase-dead WRN mutant (Flag-WRNK577M), and then treated with 5μM CPT for 4 hours. Flag-WRN was immunoprecipitated and 9/10 of IPs was analysed by WB with the anti-pS1141WRN antibody, while 1/10 was detected by anti-WRN, as indicated. One-fiftieth of the lysate was blotted with an anti-Flag antibody to verify transfection. An anti-LaminB1 antibody was used as loading control. The quantification of the level of pWRN is reported below each lane and data are from the representative experiment. D) WS-derived SV40-trasformed fibroblasts were transfected with the indicated WRN plasmid and treated with 5μM CPT at different time point, as indicated. The end-resection was analysed by the non-denaturing IdU/ssDNA assay. The dot plot shows the mean intensity of ssDNA staining for single nuclei measured from two independent experiments (n=300, each biological replicate), data are presented as mean±SE. Statistical analysis was performed by the ANOVA test (**** = P<0.0001; *** = P<0,001; ns = not significant).

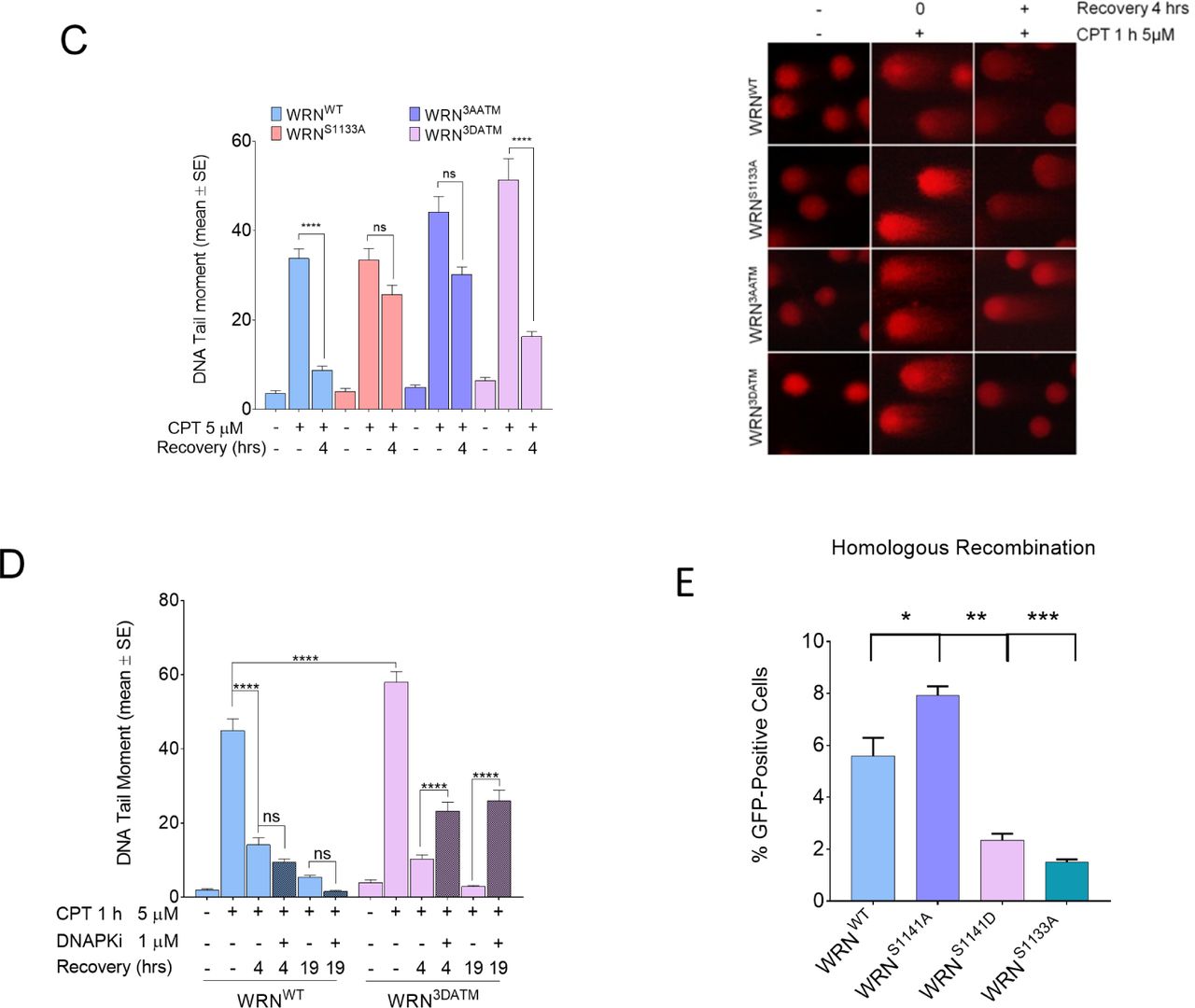
A) Analysis of DSB repair efficiency. WS-derived SV40-trasformed fibroblasts were transfected with the indicated WRN plasmids and then were treated with 5 μM CPT for 1 h prior to the indicated recovery (0h recovery = 1h CPT; - = untr). DSB repair was evaluated by the neutral Comet assay. In the graph, data are presented as mean tail moment±SE from three independent experiments. Representative images from the neutral Comet assay are shown. B) WS-derived SV40-trasformed fibroblasts were transfected with the indicated WRN plasmids, treated with 5 μM CPT for 1 h and allowed to recover in the presence or not of the different inhibitors as indicated. The presence of DSBs was evaluated by the neutral Comet assay. In the graph, data are presented as percent of mean tail moment±SE from three independent experiments normalised against the value of the 0h recovery. Statistical analysis was performed by Anova test (* = P<0.05; ****P<0.0001; ns= not significant; n=300, each biological replicate). C) WS-derived SV40-trasformed fibroblasts were transfected with the indicated WRN plasmids and then were treated with 5 μM CPT for 1 h prior to the indicated recovery (0h recovery = 1h CPT; - = untr). DSB repair was evaluated by the neutral Comet assay. In the graph, data are presented as mean tail moment±SE from three independent experiments. Representative images from the neutral Comet assay are shown. D) WS-derived SV40-trasformed fibroblasts were transfected with the indicated WRN plasmids and then were treated with 5 μM CPT for 1 h prior to the indicated recovery (0h recovery = 1h CPT; - = untr). DSB repair was evaluated by the neutral Comet assay. In the graph, data are presented as mean tail moment±SE from three independent experiments. E) Efficiency of HR-mediated repair by reporter assay. HEK293TshWRN cells were co-transfected with the indicated WRN forms, the I-SceI expression vector pCBASce and the pDRGFP HR reporter plasmid, as described in Methods. The graph shows the percentage of GFP positive cells measured by flow cytometry. Data are presented as mean±SE from three independent experiments (ns = not significant; * = P<0.05; ** = P<0.01; *** = P<0.001; ANOVA test; n=3 × 105 events each biological repeat).
As we previously reported that expression of the phosphomimic S1133D-WRN mutant enhanced end-resection (Palermo et al, 2016), we reasoned that if S1141 targeting occurred at the end of resection, then it might be delayed or reduced in the S1133D-WRN mutant. To test this hypothesis, we analysed phosphorylation at S1141 after 2 or 4h of CPT treatment by WB in WRN immunoprecipitated from cells transfected with wild-type WRN or S1133D-WRN mutant. As shown in Figure 4B, CPT-induced phosphorylation at S1141 increased with time in WRNWT cells. Notably, at 2h, the presence of the phosphomimetic S1133D-WRN mutation did not affect S1141 phosphorylation, while it substantially reduced its level at 4h.
Next, we asked whether phosphorylation of WRN at S1141 should be similarly reduced by abrogation of long-range resection. Since the helicase-dead mutant of WRN disables WRN-DNA2-dependent long-range resection (Sturzenegger et al, 2014; Palermo et al, 2016), we examined the level of S1141 phosphorylation in cells expressing the helicase-dead K577M-WRN mutant by IP/WB. As expected, treatment with CPT induced S1141 phosphorylation in cells expressing wild-type WRN (Figure 4C). Loss of WRN helicase activity resulted in elevated basal level of S1141 phosphorylation and decreased after CPT (Figure 4C). This experiment reveals that phosphorylation of WRN at S1141 may regulate the extent of long-range end-resection.
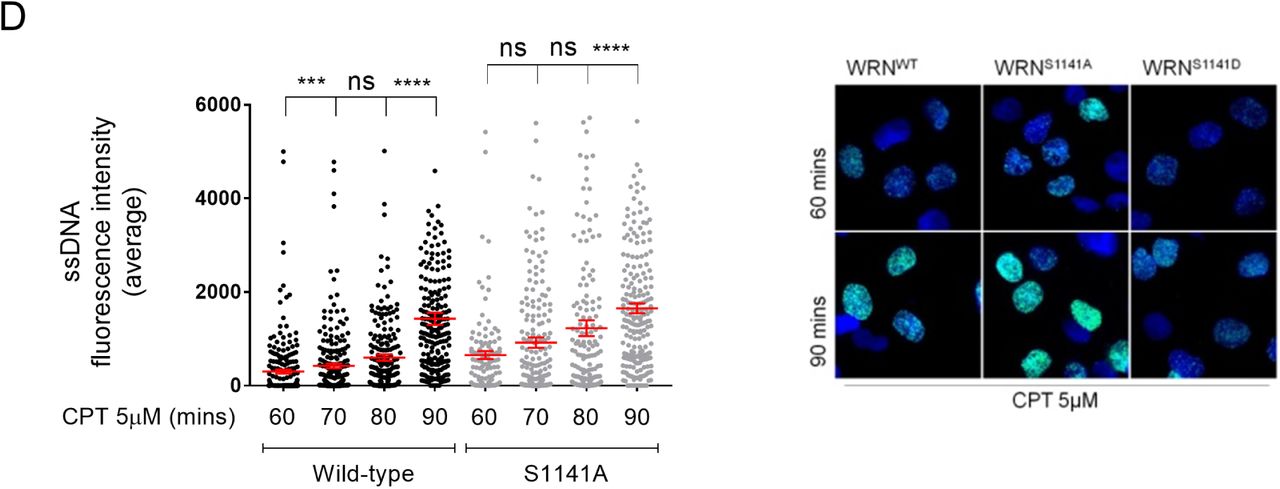
To further investigate the role of phosphorylation of WRN at S1141 in end-resection, we used cells expressing the S1141A-WRN mutant and performed a tight kinetics analysis of ssDNA formation after CPT exposure. In WRNWT cells, formation of ssDNA increased with time from 60 to 90min with highest value at 90min (Figure 4D). In WRNS1141A cells, formation of ssDNA was more elevated than in WRNWT cells at 60min, it did not increase from 60-80min but increased at 90min, when no statistically-significant differences were observed with the WRNWT cells (Figure 4D). Of note, a substantial fraction of cells expressing the S1141A-WRN mutant showed a very high ssDNA labelling, as indicated by the upshifted distribution of values in the dot plots (Figure 4D).
Altogether, our results indicate that phosphorylation at S1133 and at S1141 of WRN are inversely-regulated and suggest that modification at S1141 is linked to proper execution of long-range end-resection.
Deregulated S1141 phosphorylation of WRN abrogates DSBs repair
We demonstrate that loss of ATM-dependent phosphorylation or incorrect regulation of S1141 phosphorylation of WRN similarly impairs end-resection process in response to CPT.
Since end-resection is required for subsequent DSBs repair, we investigated whether expression of S1141A-WRN or S1141D-WRN mutant could affect repair of CPT-induced DSBs and induce a pathway switch. To this aim, we analysed DSBs repair by neutral Comet assay during recovery from a short treatment with CPT. We found that in WRNWT cells, DSBs were repaired already at 4h of recovery and abrogation of S1141 phosphorylation (WRNS1141A) did not have any effect (Figure 5A). In contrast, WRNS1141D cells showed a compromised DNA repair ability (Figure 5A).
To determine which pathway was engaged to repair DSBs, we evaluated their extinction in cells recovered from CPT in the presence of inhibitors of specific DNA repair pathways. We used the RAD51 inhibitor B02 (Huang et al, 2011) to interfere with HR, the DNAPK inhibitor NU7441 (Leahy et al, 2004) to block NHEJ, and the LigaseI/III inhibitor Li67 (Chen et al, 2008) to prevent repair by alt-EJ. As shown in Figure 5B, WRNWT cells repaired the large majority of DSBs by HR and a small fraction by NHEJ. Cells expressing the S1141A-WRN mutant also repaired DSBs by HR (Figure 5B). Notably, cells expressing the S1141D-WRN mutant did not show any significant change (Figure 5B).
In parallel experiments, we analysed DSBs repair in cells expressing the 3AATM-WRN or 3DATM-WRN mutant, which are both end-resection deficient (Figure 3B). Surprisingly, WRN3DATM cells, although defective in end-resection as WRNS1141D cells, repaired DSBs as efficiently as WRNWT cells, while the WRN3AATM cells failed to repair (Figure 5C). Such paradoxical phenotype prompted us to verify whether DSBs repair in the 3DATM-WRN mutant occurred through NHEJ rather than HR. Since NHEJ requires DNAPK activity, we assessed repair by neutral Comet assay after recovery from CPT treatment in the presence or not of the DNAPKi. As shown in Figure 5D, inhibition of NHEJ significantly reduced DSBs repair in WRN3DATM cells, indicating that mutations that mimic constitutive phosphorylation of WRN by ATM induce a repair pathway switch at CPT-induced DSBs.
Next, to confirm that expression of the phosphomimetic S1141D-WRN mutant affects HR, we used a reporter assays to test the efficiency of endonuclease-induced DSBs repair by HR (Pierce et al, 2001). The pDRGFP HR reporter was transiently transfected in HEK293TshWRN cells together with a plasmid expressing the wild-type, S1141A or S1141D variants of WRN and a plasmid expressing the I-SceI endonuclease. As an internal control, we also analysed HR efficiency in cells expressing the S1133A-WRN mutant, which is HR-defective (Palermo et al, 2016). At 72 h post-transfection, repair was evaluated by flow cytometry analysing the number of GFP-positive cells. As shown in Figure 5E, the efficiency of HR was slightly enhanced in cells expressing the S1141A-WRN, while it was substantially reduced by the presence of the WRN-S1141D phosphomimetic mutation.
Therefore, constitutive phosphorylation of WRN at S1141 prevents DSBs repair after CPT treatment and substantially reduce ability of WRNS1141D cells to perform HR. In addition, our data show that concomitant presence of all constitutive ATM-dependent phosphorylations of WRN channels CPT-induced DBSs to NHEJ.
Phosphorylation of WRN at S1141 influences the interaction with BRCA1 and relocalisation of RAD51
The different end-resection and repair phenotypes observed in WRNS1141D and WRN3DATM cells, prompted us to investigate if dynamic WRN phosphorylation could be involved in the recruitment of HR factors at DSBs. A crucial protein involved in late processing of DSBs and in HR is BRCA1, which is essential for end-resection but also to mediate RAD51-dependent repair. Thus, we analysed formation of BRCA1 foci by immunofluorescence in cells expressing the wild-type form of WRN or each S1141 mutant upon treatment with CPT and during recovery. To quantitatively assess the ability of BRCA to form foci, we evaluated the intensity of fluorescence. In the presence of a wild-type WRN, BRCA1 showed increased localization in nuclear foci in response to treatment, which decreased with time during recovery (Figure 6A). Cells expressing the S1141A-WRN mutant, showed less BRCA1 foci during treatment that were more persistent during recovery respect to WRNWT cells (Figure 6A). In contrast, in WRNS1141D cells, the number of BRCA1 foci during treatment was comparable to that of WRNWT cells, reduced at 2h of recovery and remained stable at 4h (Figure 6A). These results indicate that inability of cells expressing the S1141D-WRN mutant to properly support long-range end-resection and repair of DSBs is unrelated to defective BRCA1 localisation.


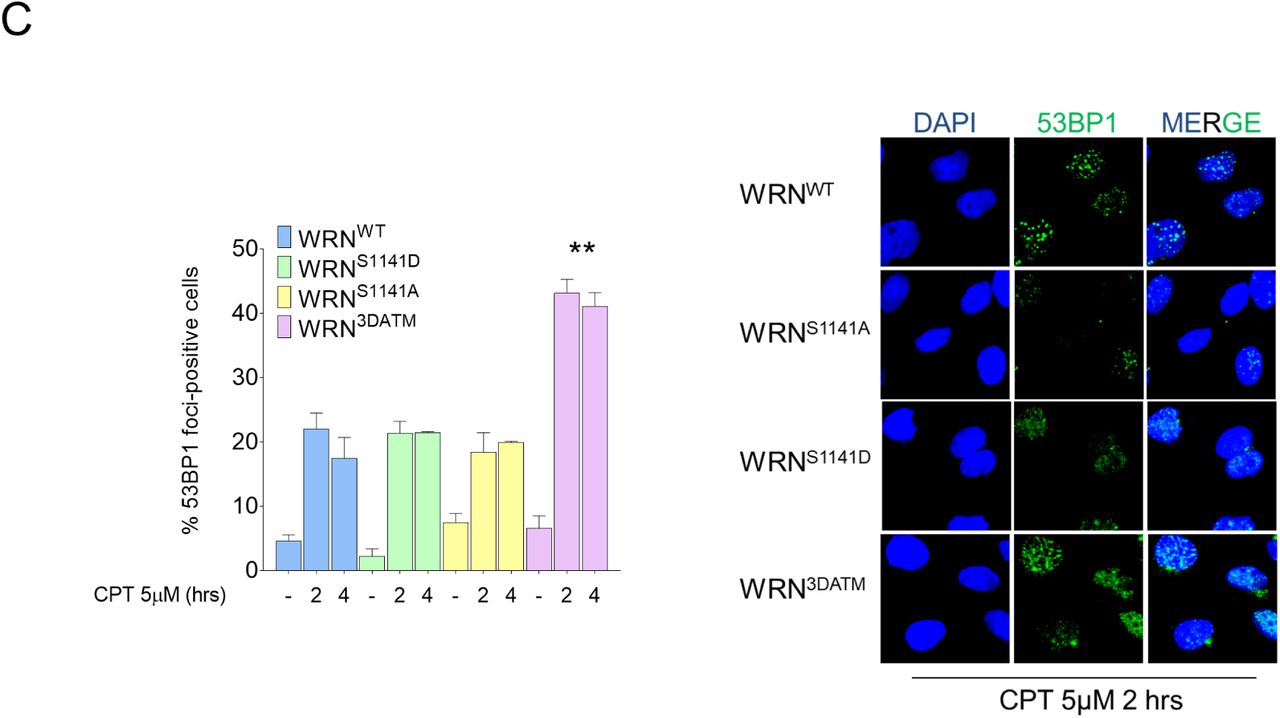
WS-derived SV40-trasformed fibroblasts transiently transfected with different Flag-tagged WRN mutants as indicated were treated with 5 μM CPT for 2 hrs and allowed to recover for different time points as indicated. The BRCA1-foci staining was analysed by IF. The dot plot shows the mean intensity of BRCA1 staining for single nuclei measured from two independent experiments (n=300), data are presented as mean±SE. Representative images of BRCA1 staining in response to treatment are shown. DAPI was used to counterstain nuclei. (ns = not significant; **** = P<0.0001, each biological replicate). B) Cells transiently transfected with different Flag-tagged WRN mutants were treated as in “A”, and then RAD51-foci staining was analysed by IF. The dot plot shows the mean intensity of RAD51 staining for single nuclei measured from two independent experiments (n=300), data are presented as mean±SE. Representative images of RAD51 staining in response to treatment are shown. (ns = not significant; **** = P<0.0001, each biological replicate). C) Cells transiently transfected with different Flag-tagged WRN mutants were treated as in “A”, and then 53BP1-foci staining was analysed by IF. The graph shows the percentage of 53BP1-foci positive cells as obtained from three independent experiments (n=200, each biological replicate), data are presented as mean±SE. Representative images of 53BP1 staining in response to treatment are shown. (ns = not significant; ** = P<0.01 compared with wild-type. ANOVA test).
Since during HR BRCA1 is needed to support localisation of RAD51 (Bhattacharyya et al, 2000; Feng & Jasin, 2017), we verified whether RAD51 localisation was influenced by altered WRN phosphorylation at S1141. To this aim, we performed immunofluorescence analysis of RAD51 foci formation after CPT and during recovery in wild-type cells or in cells expressing the S1141A or S1141D-WRN mutant. In WRNWT cells, RAD51 foci were easily detectable at 4h of treatment, increased substantially after 2h of recovery and declined at 4h after treatment (Figure 6B and C). The amount of RAD51 foci in CPT was reduced in cells expressing S1141A-WRN, however, their number followed a wild-type kinetics during recovery (Figure 6B). In contrast, expression of the S1141D-WRN mutant substantially increased formation of RAD51 foci in CPT-treated cells and the effect was particularly-relevant before recovery when RAD51-positive cells were low in wild-type cells (Figure 6C). In contrast, BRCA1 and RAD51 foci were low in the 3DATM-WRN mutant as they are in the 3AATM-WRN mutant (Figure EV4A, B).
The take-over of NHEJ at DSBs usually requires increased 53BP1 localization (Panier & Boulton, 2014). Since the 3DATM but not the S1141D phosphomimetic WRN mutant is DNA repair proficient, we analysed 53BP1 foci formation by immunofluorescence. As shown in Figure 6C, very few 53BP1-positive cells were found in WRNWT cells, consistent with proficiency in end-resection and DSBs repair by HR. Notably, more 53BP1-positive cells were found in WRN3DATM cells but not in WRNS1141D cells (Figure 6C), which are unable to repair CPT-induced DSBs. This experiment suggests that end-resection and HR defects are unrelated to 53BP1 take-over at DNA ends in cells expressing WRN-S1141D. Our results indicate that cells expressing the S1141D-WRN mutant recruits BRCA1 and RAD51 faster and stronger than a wild-type cells, although long-range resection is severely reduced. Thus, we tested the possibility that unproductive recruitment of BRCA1 and/or RAD51 may correlate with the DNA repair defect of WRNS1141D cells. To this end, we examined DSBs repair by neutral Comet assay in WRNS1141D cells upon depletion of BRCA1 by RNAi (Figure 7A) or after treatment with the RAD51 inhibitor B02 during recovery from CPT. Depletion of BRCA1 restored the ability to perform DSBs repair in cells expressing the S1141D-WRN mutant, and repair was sensitive to the DNAPK inhibitor, indicating that it mainly relies on NHEJ (Figure 7B, C). Notably, inhibition of RAD51 partially reverted defective DSBs repair in WRNS1141D cells (Figure 7C). However, most of the repair observed after inhibition of RAD51 in WRNS1141D cells was not sensitive to the DNAPK inhibitor (Figure 7C).

A) Western blotting shows depletion of BRCA1 in cells transiently expressing WRN wild type or its S1141D mutant. Whole-cell extracts were prepared at 48 hrs after transfection with BRCA1 siRNA and analysed by WB with the anti-BRCA1 antibody.
b) Analysis of DSB repair efficiency by neutral Comet assay. WS-derived SV40-trasformed fibroblasts were transfected with the indicated WRN plasmid. Control (Ctrl)-depleted or BRCA1-depleted cells were treated with 5 μM CPT for 1 h and allowed to recover for different time points as indicated. In the graph, data are presented as mean tail moment±SE from two independent experiments Statistical analysis was performed by ANOVA test (ns = not significant; ****P<0.0001; n=300, each biological replicate).
c) WS-derived SV40-trasformed fibroblasts were transfected with the indicated WRN plasmids, treated with 5 μM CPT for 1 h and allowed to recover in the presence or not of the different inhibitors as indicated. The presence of DSBs was evaluated by the neutral Comet assay. In the graph, data are presented as percent of mean tail moment±SE from three independent experiments normalised against the value of the 0h recovery (* = P<0.05; ** = P<0.01, ANOVA test).
D) Experimental scheme depicts how ssDNA assay was performed in presence or not of RAD51 inhibitor. Cells were labelled and treated with 5μM CPT as indicated, then the RAD51 inhibitor was added the last hour before sampling. The end-resection was analysed by the non-denaturing IdU/ssDNA assay. The dot plot shows the mean intensity of ssDNA staining for single nuclei measured from two independent experiments (n=300, each biological replicate), data are presented as mean±SE. Statistical analysis was performed by the ANOVA test (** = P<0.01).
Next, we verified whether inhibition of RAD51 could rescue the end-resection defect observed in WRNS1141D cells. Thus, we analysed ssDNA formation in cells expressing the S1141D-WRN mutant at 2h of CPT in the presence of the RAD51 inhibitor B02 in the last hours of treatment. RAD51 inhibitor did not affect end-resection in WRNWT cells, as evaluated by ssDNA assay (Figure 7D). In contrast, inhibition of RAD51 nucleofilaments partially restored end-resection in WRNS1141D cells (Figure 7D).
Collectively, our data indicate that WRN phosphorylation at S1141 influences the ability of BRCA1 to get relocalised in response to CPT, especially during recovery, and that constitutive phosphorylation of S1141 interferes with correct formation of RAD51 foci. In addition, they demonstrate that the presence of “aberrant” RAD51 foci in the phosphomimetic S1141D-WRN mutant contributes to impede completion of long-range end-resection and prevent any pathway-switch at replication-induced DSBs.
DISCUSSION
Here, we demonstrate that ordered and hierarchic phosphorylation of WRN through ATM/ATR-dependent events is crucial to support execution of end-resection and correct RAD51-dependent recombination. We also show that ATM/ATR-dependent phosphorylation of WRN cross-talks with that mediated by CDK1, suggesting that WRN regulation may function as a molecular switch bridging together the two stages of HR at replication-dependent DSBs: end-resection and RAD51-dependent strand invasion.
CDK1 and ATM cross-talk to regulate WRN function during HR
End-resection of DSBs is a highly-regulated process in which cells need high CDK1 activity (Trovesi et al, 2013; Symington, 2016) and, consistently, WRN is phosphorylated by CDK1 to carry out its pro-resection function (Palermo et al, 2017). However, end-resection also requires ATM-dependent phosphorylation of several proteins (Symington, 2016). The WRN protein is phosphorylated by ATM on multiple residues in response to replication stress or ionizing radiation (Ammazzalorso et al, 2010; Matsuoka et al, 2007). Our data show that only two of these residues, S1058 and S1141, are important for DSBs repair by recombination, but with two distinct functional roles. Phosphorylation of WRN by ATM at S1058 is essential for long-range end-resection and, notably, requires prior phosphorylation at S1133 by CDK1. Interestingly, both events require ATM and MRE11 nuclease activity. ATM and MRE11 are essential to initiate end-resection (Williams et al, 2009; You et al, 2009; Garcia et al, 2011; Kijas et al, 2015; Nimonkar et al, 2011), thus, our findings support the possibility that WRN phosphorylation occurs only if end-resection initiates, and are consistent with the observed requirement of WRN helicase activity in the long-range phase of resection (Sturzenegger et al, 2014). Interestingly, we observe that expression of the phosphomimetic S1058D-WRN mutant is sufficient to rescue the end-resection defect associated with loss of CDK1-dependent WRN phosphorylation (Palermo et al, 2016). In contrast, the unphosphorylable S1058A mutation can confer an end-resection-defective phenotype to the S1133D phosphomimetic mutant of WRN, which is proficient for end-resection (Palermo et al, 2016). These findings, together with the enhanced ability of ATM to phosphorylate in vitro the S/TQ sites of WRN in the presence of the S1133D mutation, suggest that the primary function of S1133 phosphorylation is to promote subsequent ATM-dependent phosphorylation at S1058. Surprisingly, although phosphorylation at S1141 of WRN is also dependent on the initiation of end-resection, it is not required for the execution of long-range end-resection.
Phosphorylation of WRN at S1141 and regulation of long-range end-resection
We find that phosphomimetic mutation S1141D confers an end-resection defect, indicating that this site needs to be dephosphorylated during resection. This observation might suggest that S1141 phosphorylation of WRN contributes to signal the end of resection or initiation of strand-invasion. Noteworthy, we show that phosphorylation at S1133 temporally precedes that at S1141, which is maintained high also during recovery from CPT when pS1133 levels declines. In addition, phosphorylation at S1141 of WRN is substantially reduced in the presence of the S1133D-WRN mutation, which we previously demonstrated to enhance resection and deregulate HR (Palermo et al, 2016).
Notably, WRN S1141 phosphorylation can be similarly suppressed by ATM or ATR inhibition. However, in response to CPT, inhibition of ATM prevents formation of ssDNA, which is produced by resection and is required to recruit ATR (Zou & Elledge, 2003; MacDougall et al, 2007; Costanzo et al, 2003). Hence, our data cannot discriminate between direct vs. indirect effect of ATM inhibition on WRN S1141 phosphorylation in cellulo. Our previous in vitro experiments revealed that S1141 of WRN is targeted by ATM but not ATR (Ammazzalorso et al, 2010), however, another study has demonstrated that it is phosphorylated by ATR in cellulo, at least during extended period of recovery after CPT (Su et al, 2015). The resection-defective phenotype of the S1141D-WRN mutant, the elevated levels of pS1141-WRN during recovery from CPT treatment would support the possibility that S1141 of WRN is targeted by ATR also in our conditions. Interestingly, the negative effect of the S1133D-WRN mutation on the S1141 phosphorylation would suggest that the CDK1 site of WRN needs to be switched-off when the ATR site S1141 gets phosphorylated. The passage from the end-resection to the repair phase of recombination requires the inhibition of CDK by ATR, and a similar switch from CDK to ATR modification of BRCA1 was recently reported (Buisson et al, 2017). Thus, the multiple switch from CDK1-ATM to ATR-dependent modification of WRN reported in our study would indicate that multiple “pleiotropic” proteins change their function during recombination through this fascinating regulatory system.
Notably, pS1141WRN levels are not increased in response to CPT in the helicase-dead WRN mutant K577M. The helicase activity of WRN is essential to carry out the WRN-DNA2 branch of long-range end-resection (Sturzenegger et al, 2014). Thus, the inability to modulate pS1141 levels upon CPT treatment may indicate that WRN gets phosphorylated by ATR at this site only if it is active during long-range resection. Moreover, as S1141 is not required for resection, this result may argue for a post-resection phosphorylation. Of note, the K577M-WRN mutant shows high basal levels of pS1141. Phosphorylation at S1141 has been also linked to degradation of WRN during extended recovery from replication-dependent DSBs (Su et al, 2015). Thus, elevated basal levels of pS1141 in the K577M-WRN could correlate with the need to degrade the mutant protein. In our HEK293shWRN cells, proteins are mildly over-expressed by transient transfection and we failed to observe enhanced degradation of the K577M or S1141D mutants. However, since inhibition of the WRN helicase by small molecules interferes with proliferation (Aggarwal et al, 2013), over-expression of the helicase-dead WRN mutant could act as a dominant-negative, and be highly-phosphorylated on S1141 independently on end-resection to target the protein for degradation. Alternatively, WRN S1141 might be targeted by ATM in other pathways.
The kinetics of WRN S1141 phosphorylation, the resection phenotype of the unphosphorylable mutant and the dependency of the phosphorylation on proper execution of long-range resection support that targeting of this residue may contribute to regulate rate of long-range resection or signal its ending to the cell. Consistent with this, expression of the unphosphorylable S1141A mutation leads to a faster kinetics of accumulation of ssDNA in response to DSBs and, strikingly, a slightly delayed assembly of RAD51 foci.
How human cells regulate long-range resection is not yet precisely defined. Recently, the HELB protein has been found to regulate end-resection and its loss greatly enhanced accumulation of ssDNA after DSBs (Tkáč et al, 2016). In addition, also deregulation of the BRCA1-A complex enhances resection, although through a distinct mechanism (Tkáč et al, 2016; Dever et al, 2011; Coleman & Greenberg, 2011). Loss of S1141 phosphorylation, however, does not confer a severe hyper-resection phenotype in response to CPT treatment. The mild phenotype of S1141A-WRN suggests that HELB is functional and that other factors can contribute to a negative feedback during end-resection. Alternatively, inability to phosphorylate WRN at S1141 may affect only a subset of DNA ends as opposed to a more general effect induced by loss of HELB or the BRCA1-A complex. However, as described for HELB, the effect of a deregulated phosphorylation of WRN at S1141 does not interfere with end-resection through enhancement of 53BP1. Indeed, both the action of HELB and WRN S1141-phosphorylation take place after initial end-resection. Notably, also deregulated phosphorylation of EXO1, another nuclease involved in end-resection, is able to interfere with resection (Bolderson et al, 2010; Kijas et al, 2015), suggesting that phosphorylation by ATM/ATR of nuclease is a common mechanism to regulate resection.
Phosphorylation of WRN and DNA repair pathway choice
Abrogation of end-resection may induce a pathway-switch at DSBs promoting their repair by NHEJ or alt-EJ (Chapman et al, 2012). Such pathway-switch occurs also when cells cannot sustain CDK1-dependent phosphorylation of WRN (Palermo et al, 2016). However, NHEJ does not take-over HR in cells expressing the S1141D-WRN mutant although the end-resection is clearly defective. In contrast, cells expressing the 3DATM mutant of WRN, which are also end-resection-defective, show a pathway-switch from HR to NHEJ. This different phenotype between the S1141D-WRN and the 3DATM-WRN mutants might be explained by the effect of the 3DATM mutations for the relocalisation in nuclear foci of WRN. Indeed, abrogation of ATM sites on WRN increases retention in foci (Ammazzalorso et al, 2010) suggesting that reduced foci formation expected in the WRN-3DATM correlates with the ability to observed increased localisation of 53BP1 in foci. 53BP1 competes with BRCA1 at DSBs ends (Bunting et al, 2010a), and consistent with this antagonistic role, cells expressing 3DATM-WRN, but not cells expressing the S1141D-WRN mutant, show reduced BRCA1 foci on CPT.
Interestingly, expression of a WRN mimicking constitutive phosphorylation at S1141 results in elevated RAD51 foci. This is quite surprising as RAD51 requires ssDNA for recruitment and end-resection is defective in the S1141D-WRN mutant, although formation of ssDNA is not as severely compromised as in cells expressing the S1058A or S1133A mutation. These findings suggest that the presence of a deregulated phosphorylation at S1141 induces an untimely recruitment of RAD51 that is not sufficient to support repair, but may represent a physical block to further end-resection by different molecular machines, such as BLM-DNA2 (Sturzenegger et al, 2014). Consistent with this hypothesis, inhibition of RAD51 nucleofilaments restores, at least partially, end-resection in S1141D-WRN and also reverts the defective repair. Similarly, the DNA repair defect of the S1141D WRN-expressing cells can be mitigated by depletion of BRCA1. In both cases, repair is rescued but through NHEJ and not HR. Interestingly, such effect of RAD51 inhibition is reminiscent of what observed in BRCA1-deficient cells upon depletion of 53BP1 (Bunting et al, 2010b; Bouwman et al, 2010).
Since loss of WRN phosphorylation at S1141 partially delays RAD51 foci formation, but accelerates end-resection, it is tempting to speculate that WRN is targeted at S1141 also to promote recruitment of RAD51 at resected ends. Alternatively, a dephosphorylated WRN at S1141 may perform dismantling of unproductive RAD51 filaments as evidenced for the related helicases BLM and RECQ5 (Karow et al, 2000; Patel et al, 2017; Hu et al, 2007; Schwendener et al, 2010). However, the expression of phosphomimetic S1141D-WRN does not induce a hyper-recombination phenotype and, rather, decreases recombination efficiency, arguing against this hypothesis.
Absence of WRN S1141 phosphorylation reduces also BRCA1 foci and apparently prevents their modulation during recovery. BRCA1 is involved in both the initial step of HR and later on with PALB2 during RAD51 recruitment (Sy et al, 2009; Zhang et al, 2009). The absence of modulation of BRCA1 might correlate with a delayed formation of a specific BRCA1 complex promoting RAD51 assembly. Of note, WRN and BRCA1 colocalize in response to DSB-inducing treatments and form a complex (Cheng et al, 2006; Franchitto & Pichierri, 2004), suggesting that different phosphorylation of WRN may promote formation of distinct protein complexes including or not BRCA1, as observed for the BRCA1-PALB2 complex (Buisson et al, 2017).
Conclusions
Our data and published observations can be summarised in the model shown in Figure 8. In response to replication-dependent DSBs and after short-range resection, WRN is phosphorylated at S1133 by CDK1. This phosphorylation primes that at S1058 by ATM, which is the crucial regulatory event to carry out WRN-DNA2-dependent long-range resection. After long-range resection started, WRN needs to be dephosphorylated at S1133, and probably at S1058, while it is targeted by ATM or most probably ATR, at S1141 to limit end-resection. Then, phosphorylation of WRN at S1141 contributes to properly initiate HR by RAD51. Inability to properly regulate this ordered phosphorylation cascade, would result in either inability to perform end-resection, to repair DSBs by HR, or to repair at all.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(see text for details).
Thus, our study reveals a complex regulatory layer that controls the function of WRN during the repair of DSBs induced at the replication fork, and indicates that WRN may actually act as a sort of molecular switch to put together, correctly, the end-resection and the repair stages of HR.
MATERIALS AND METHODS
Cell lines and culture conditions
The SV40-transformed WRN-deficient fibroblast cell line (AG11395) was obtained from Coriell Cell Repositories (Camden, NJ, USA). To produce stable cell lines, AG11395 (WS) fibroblasts were transduced with lentiviruses expressing the full-length cDNA encoding wild-type WRN (WRNWT), S1133A-WRN (WRNS1133A), S1133D-WRN (WRNS1133D), 3AATM-WRN (WRN3AATM), or 3DATM-WRN (WTN3DATM). The WRNWT and the AG11395 complemented with an helicase-dead WRN (WRNK577M) have been described elsewhere (Pirzio et al, 2008). HEK293T cells were from American Type Culture Collection. HEK293TshWRN, cells were generated after transfection with pRS-puro-shWRN (5′-AGGCAGGTGTAG-GAATTGAAGGAGATCAG-3′; sequence ID: TI333414 Origene) and puromicin selection (Palermo et al, 2016). All the cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% FBS (Boehringer Mannheim) and incubated at 37 °C in a humidified 5% CO2 atmosphere.
Chemicals
Camptothecin (ENZO Lifesciences) was dissolved in DMSO and a stock solution (10 mM) was prepared and stored at −20°C. Camptothecin was used at a final concentration of 5µM if not otherwise specified. Mirin (Calbiochem), an inhibitor of MRE11 exonuclease activity, was used at 50 µM; the B02 compound (Selleck), an inhibitor of RAD51 activity, was used at 27 µM. Roscovitine (Selleck), a pan-CDKs inhibitor, was used at final concentration of 20 µM. ATM inhibitor (KU-55933, Selleck) and ATR inhibitor (VE-821, Selleck) were used at final concentration of 10µM. NU7441 (Selleck), a DNAPKcs inhibitor, was used at final concentration of 1 µM, while the Ligase I/III L67 inhibitor (Sigma-Aldrich) was used at 6µM.
Immunoprecipitation and Western blot analysis
Immunoprecipitation experiments are performed using 2.5×106 cells. RIPA buffer (0.1% SDS, 0.5% Na-dehoxycolate, 1% NP40, 150mM NaCl, 1mM EDTA, 50mM Tris/Cl pH 8) supplemented with phosphatase, protease inhibitors and benzonase was used for cells lysis. One-milligram of lysate was incubated overnight at 4°C with 20 µl of Anti-Flag M2 magnetic beads (Sigma-Aldrich). After extensive washing in RIPA buffer, proteins were released in 2X electrophoresis buffer and subjected to SDS-PAGE and Western blotting.
Western blotting was performed using standard methods. Blots were incubated with primary antibodies: rabbit anti-WRN (Abcam); mouse anti-β-Tubulin (Sigma-Aldrich); rabbit anti-Lamin B1 (Abcam); mouse anti-DDK-Flag tag (Origene); rabbit anti-pS/TQ (Cell Signaling Technology), rabbit anti-pS1141WRN (Sigma-Aldrich); rabbit anti-pS1133WRN (Genscript-custom); rabbit anti-GST (Calbiochem). After incubation with horseradish peroxidase-linked secondary antibodies (1:20000, Jackson Immunoscience), the blots were detected using the Western blotting detection kit WesternBright ECL (Advansta) according to the manufacturer’s instructions. Quantification was performed on scanned images of blots using Image Lab software, and values shown on the graphs represent normalization of the protein content evaluated through LaminB1 or β-Tubulin-immunoblotting.
Immunofluorescence Assays
Cells were cultured onto 22×22 coverslip or 8-well Nunc chamber-slides. To detect WRN, BRCA1, RPA32-or RAD51 foci, we performed pre-extraction for 5 min on ice in CSK buffer as described elsewhere (Franchitto & Pichierri, 2004). To detect 53BP1 and EdU cells were fixed with 4% PFA for 10 min at room temperature and processed as in (Iannascoli et al, 2015). Cells were incubated for 2h at RT with specific primary antibody diluted in 1%BSA/PBS: rabbit anti-WRN (Abcam); mouse anti-RPA32 (Calbiochem); rabbit anti-BRCA1 (Bethyl); rabbit anti-RAD51(Abcam); mouse anti-53BP1 (Millipore), Detection was performed by specie-specific fluorescein-conjugated secondary antibodies (Alexa Fluor 594 Anti-Rabbit or Alexa Fluor 488 Anti-Mouse), and DNA counterstained with 0.5µg/ml DAPI. Slides were analysed with Eclipse 80i Nikon Fluorescence Microscope, equipped with a VideoConfocal (ViCo) system. For each time point, at least 300 nuclei were scored at 60×.
Detection of nascent single-stranded DNA
To detect nascent single-stranded DNA (ssDNA), cells were plated onto 22×22 coverslips in 35mm dishes. After 24h, the cells were labelled for 15 min before the treatment with 250µM IdU (Sigma-Aldrich), cells were then treated with CPT 5µM for different time points. Next, cells were washed with PBS, permeabilized with 0.5% Triton X-100 for 10 min at 4°C and fixed wit 2% sucrose, 3%PFA. For ssDNA detection, cells were incubated with primary mouse anti-IdU antibody (Becton Dickinson) for 1h at 37°C in 1%BSA/PBS, followed by Alexa Fluor488-conjugated goat-anti-Mouse, and counterstained with 0.5µg/ml DAPI. Slides were analysed with Eclipse 80i Nikon Fluorescence Microscope, equipped with a VideoConfocal (ViCo) system. For each time point, at least 300 nuclei were scored at 40×. Parallel samples either incubated with the appropriate normal serum or only with the secondary antibody confirmed that the observed fluorescence pattern was not attributable to artefacts. Fluorescence intensity for each sample was then analyzed using ImageJ software.
Generation of the GST-WRN fragment
DNA sequence corresponding to aa 940–1432 (C-WRN) of WRN was amplified by PCR from the pCMV-FlagWRN plasmid. The PCR product were subsequently purified and sub-cloned into pGEX4T-1 vector (Stratagene) for subsequent expression in bacteria as GST-fusion proteins. The resulting vectors were subjected to sequencing to ensure that no mutations were introduced into the WRN sequence and were used for transforming BL21 cells (Stratagene). Expression of GST and GST-fusion proteins were induced upon addition of 1 mM isopropyl-D-thiogalactopyranoside (IPTG) for 2 h at 37°C. GST, and GST-C-WRN were affinity-purified using glutathione (GSH)-magnetic beads (Promega).
In vitro Kinase assay
For kinase assay, 2 μg of immunopurified GST-tagged WRN fragment was phosphorylated in vitro by immunopurified FLAG-ATM in the presence or not of 5μM ATP for 30 minutes at 37°C. After the incubation, WRN fragments were separated from the beads and phosphorylation levels were assessed by SDS-PAGE followed by Coomassie staining and densitometric analysis by Phosphorimaging or by WB using rabbit anti-pS/TQ antibody.
Neutral comet assay
After treatment, cells were embedded in low-melting agarose and spread onto glass slides as described elsewhere (Murfuni et al, 2012). DNA was stained with GelRed (Biotium) and examined at 20× magnification with an Olympus fluorescence microscope. Slides were analysed with a computerized image analysis system (Comet IV, Perceptive UK). To assess the amount of DNA DSB breaks, computer generated tail moment values (tail length × fraction of total DNA in the tail) were used. A minimum of 200 cells was analysed for each experimental point. Apoptotic cells (smaller comet head and extremely larger comet tail) were excluded from the analysis to avoid artificial enhancement of the tail moment.
Homologous recombination and end-joining Reporter Assay
HEK293TshWRN cells (Palermo et al, 2016) were seeded in 6-well plates at a density of 0.5 million cells/well. The next day, pCMV-FlagRnai-resWRNWT or other pCMV-FlagRnai-resWRN mutant constructsD were cotransfected with the I-SceI expression vector pCBASceI and the pHPRT-DRGFP plasmid reporter using DreamFect (OZBioscience) according to the manufacturer’s instruction. pHPRT-DRGFP and pCBASceI were a gift from Maria Jasin (Addgene plasmids # 26476 and #26477).
Protein expression levels were analyzed by Western Blotting 72 h post-transfection. Cells were subjected to flow cytometry analysis at 72 h after transfection to determine the percentage of GFP-positive cells.
Statistical analysis
All the data are presented as means of at least three independent experiments. Statistical comparisons of WS or WRN-mutant cells to their relevant control were analyzed by ANOVA test. P < 0.05 was considered as significant.
AUTHOR CONTRIBUTIONS
V.P. performed experiments to analyse WRN phosphorylation, end-resection, protein relocalisation and interaction. E.M. performed analysis of DNA damage. P.P. performed in vitro ATM kinase assays. M.S. performed flow cytometry analysis. V.P., E.M., and M.S. analysed data and contributed to designing the experiments and writing the paper. A.F. and P.P designed experiments, analysed data and wrote the paper. All authors approved the paper.
CONFLICT OF INTEREST
The authors declare that they do not have any conflict of interest.
Extended View Figure Legends
Figure Extended View 1. Phosphorylation of WRN requires MRE11 nuclease activity Cells were treated with 50μM Mirin and then with 5μM CPT for 4 hours. Cells were lysed and WRN protein was immunoprecipitated with anti-Flag-conjugated beads. Nine-tenth of IPs were analysed by WB with the anti-pS1133WRN antibody and the anti-pS/TQ antibody, while 1/10 was detected by anti-Flag antibody, as indicated. One-fiftieth of the lysate (input) was blotted with an anti-Flag antibody to verify transfection. An anti-LaminB1 antibody was used as loading control.
Figure Extended View 2. Analysis of ssDNA formation
A) WS-derived SV40-trasformed fibroblasts stably expressing the wild-type form of WRN or the 3AATM, 3DATM or S1133A mutant were treated with CPT as indicated. Anti-RPA32 immunofluorescence, an indirect readout of ssDNA, was performed and the graph shows the percentage of RPA32-foci positive cells as obtained from three independent experiments (n=200, each biological replicate), data are presented as mean±SE. B) The panel shows representative images from CPT-treated cells. DAPI was used to counterstain nuclei. C) WS-derived SV40-trasformed fibroblasts stably expressing the wild-type form of WRN, the 3AATM, or S1133A mutant were treated with CPT as indicated. The presence of ssDNA was analysed by non-denaturing IdU/ssDNA assay. The graph shows the mean intensity of IdU/ssDNA staining for single nuclei measured from three independent experiments (n=300, each biological replicate), data are presented as mean±SE. Representative images of IdU/ssDNA-stained from CPT-treated cells are shown. DAPI was used to counterstain nuclei. D) WS-derived SV40-trasformed fibroblasts were transiently transfected with the indicated Flag-tagged WRN forms and the IdU/ssDNA assay was performed. The dot plot shows the mean intensity of ssDNA staining for single nuclei measured from two independent experiments (n=300, each biological replicate), data are presented as mean±SE. Representative images of IdU/ssDNA-stained from CPT-treated cells are shown. Statistical analysis was performed by the ANOVA test (**** = P<0.0001; ** = P<0.01; ns = not significant).
Figure Extended View 3. Kinetics of ssDNA formation in wild-type cells
A) WS cell line complemented with WRN wild-type (WRNWT) was treated with CPT at different time points, as indicated. The graph shows the mean intensity of IdU/ssDNA staining for single nuclei measured from three independent experiments (n=300, each biological replicate), data are presented as mean±SE. Representative images of IdU/ssDNA-stained from CPT-treated cells are shown. DAPI was used to counterstain nuclei. Statistical analysis was performed by the ANOVA test (*** = P<0.001; * = P>0.01; ns = not significant).
Figure Extended View 4. Analysis of BRCA1 and RAD51 recruitment in multisite phosphorylation mutants of WRN
A) WS-derived SV40-trasformed fibroblasts stably expressing the wild-type form of WRN, 3AATM or 3DATM-WRN mutant were treated as indicated and then BRCA1-foci were analysed by IF. The graph shows the percentage of BRCA1-foci positive cells as obtained from two independent experiments (n=200, each biological replicate), data are presented as mean±SE. B) Cells were treated as in “A” and analysed for RAD51-foci staining by IF. The graph shows the percentage of RAD51-foci positive cells as obtained from two independent experiments (n=200, each biological replicate), data are presented as mean±SE.
ACKNOWLEDGMENTS
We are grateful to Prof. Achille Pelliccioli and Dr. Federica Marini at the University of Milan for the siBRCA1 oligos. We thank Prof. Achille Pelliccioli, the members of his group and all members of our labs for insightful scientific discussion. This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) to PP (IG17383) and to AF (IG IG119971), and by Telethon IG12144 to PP.
REFERENCES




