

Abstract
Predictions of how a population will respond to a selective pressure are valuable, especially in the case of infectious diseases, which often adapt to the interventions we use to control them. Yet attempts to predict how pathogen populations will change, for example in response to vaccines, are challenging. Such has been the case with Streptococcus pneumoniae, an important human colonizer and pathogen, and the pneumococcal conjugate vaccines (PCVs), which target only a fraction of the strains in the population. Here, we use recent advances in knowledge of negative-frequency dependent selection (NFDS) acting on frequencies of accessory genes (i.e., flexible genome) to predict the changes in the pneumococcal population after intervention. Implementing a deterministic NFDS model using the replicator equation, we can accurately predict which pneumococcal lineages will increase after intervention. Analyzing a population genomic sample of pneumococci collected before and after vaccination, we find that the predicted fitness of a lineage post-vaccine is significantly and positively correlated with the observed change in its prevalence. Then, using quadratic programming to numerically solve the frequencies of non-vaccine type lineages that best restored the pre-vaccine accessory gene frequencies, we accurately predict the post-vaccine population composition. Additionally, we also test the predictive ability of frequencies of core genome loci, a subset of metabolic loci, and naïve estimates of prevalence change based on pre-vaccine lineages frequencies. Finally, we show how this approach can assess the migration and invasion capacity of emerging lineages, on the basis of their accessory genome. In general, we provide a method for predicting the impact of an intervention on pneumococcal populations and other bacterial pathogens for which NFDS is a main driving force.
Detailed predictions of how a population will respond to a selective pressure are challenging. While evolutionary models specify how mutations with a given fitness vary in frequency over time, these are often hard to apply in practice, as we typically do not know in advance important parameters such as the fitness value of particular alleles or how this is affected by their frequency (frequency-dependent selection) or genetic background (epistasis) (1). Prediction is especially valuable in the case of infectious disease, as pathogens adapt to the interventions we use to control them (2). For example, pneumococcal conjugate vaccines (PCVs) target a fraction of the strains of Streptococcus pneumoniae, a colonizer of the human nasopharynx and common cause of bacterial pneumonia, bacteremia, meningitis, and otitis media (3). Before PCV use, there was concern that non-vaccine serotypes (NVT) could benefit from the removal of their vaccine-serotype (VT) competitors and thereby become more common in carriage and disease. Serotype replacement has indeed occurred following introduction of PCVs, with the gains from reducing VT disease partly offset by increases in NVT disease (4–6).
While serotype replacement has become evident, the scale of that replacement, NVT serotypes involved, and overall changes in the pathogen population structure were not appreciated until retrospective analysis (7–9). The apparent unpredictability of replacement is illustrated by our recent analysis of genomic data from 937 pneumococcal carriage isolates collected before and after vaccine introduction among Native American communities in the southwest United States (9). Before vaccine introduction, the population consisted of multiple lineages (often referred to as sequence clusters, SCs) including SCs comprising VT only, mixed VT and NVT, or NVT only (Figures 1 and 2). After vaccine introduction, there was non-uniform expansion of NVT SCs as well as the appearance of two previously unobserved SCs (9). There was considerable deviation from the null expectation that SCs including NVT would change in prevalence in proportion to their pre-vaccine frequency i.e., SC prevalence rank was not maintained (Figure 2, see Supplementary Information for details). Particularly, among 35 SCs, we find nine that increased significantly more than expected and five that increased significantly less. This illustrates the difficulty of prediction; even if we could be reasonably sure serotype replacement would occur, we would not have been able to say exactly which lineages would increase the most. Consequently, researchers are left playing a game of evolutionary whack-a-mole where post-vaccine pathogen surveillance is used to estimate the next epidemiologically important lineage and determine subsequent vaccine formulations; then the cycle repeats. At best, this reduces the population impact of vaccination; at worst, it could unintentionally increase the prevalence of virulent or antibiotic resistant lineages (10).

Maximum likelihood phylogeny inferred from a core genome alignment of 937 isolates obtained from a subset of three observational studies of pneumococcal carriage conducted among Native American communities in the southwest US from 1998 to 2012. Thirty-five sequence clusters (SCs), which were identified by subdividing the pneumococcal population on core genome diversity, are shaded by color on the phylogeny and labeled to the right. SCs were assigned based on core genome nucleotide diversity and are numbered such that closer numbers are more genomically similar. The text color of the SC label indicates the serotype composition. The heatmap indicates PCV7 vaccine and non-vaccine serotypes and collection period, illustrating the removal of vaccine serotypes by the introduction of PCV7.

Raw and adjusted change in sequence cluster (SC) prevalence from pre-vaccine (PCV7) to post-vaccine (PCV7). The bars represent the raw change in SC prevalence and are colored based on serotype composition: non-vaccine serotype (NVT) only, vaccine-serotype (VT) only, and mixed VT and NVT (VT-NVT). The point and whisker show the adjusted changes in prevalence and 95% confidence intervals. Adjusted estimates are the raw prevalence change minus the prevalence change expected if all VT had declined by 96.4% (the overall population frequency change) and all NVT had increased by 68.7% (the overall population frequency change) – i.e. in a null model where only the VT/NVT status of a strain determined its fitness. Confidence intervals were obtained from sampling 10,000 bootstraps from pre- and post-vaccine samples. The heatmap to the right of the plot illustrates the prevalence rank before and after the introduction of vaccine. Darker colors represent higher prevalence values. Among the most successful were SCs that contained both PCV7 VT and NVT isolates (SC22 and SC23) whose NVT component included serotypes 6C, 15C, and 35B, as well as SC24 and SC25, which were dominated by the NVT serotypes 23A and 15C, respectively. Compared to SCs comprised of solely NVT isolates, mixed NVT-VT lineages had marginally higher risk differences, indicating greater success than expected under the null model (β=0.03, SE=0.015, F(1,29)=3.67, p=0.06). Two SCs that emerged during the study period (SCs 10 and 24) were not included in this analysis as they were not present at the first time point
One clue into the post-vaccine success of pneumococcal lineages may lie in the frequencies of the loci that make up the accessory genome (i.e., those genes not found in all strains within the population) (11 ,12). Corander and colleagues recently demonstrated that while the distribution of pneumococcal SCs were not correlated across diverse geographies, the frequency of accessory clusters of orthologous genes (COGs) were (11). Further, these frequencies were restored even after significant lineage perturbation induced by the introduction of PCV7 (9 ,11). Corander et al. 2017 went on to propose negative frequency-dependent selection (NFDS) as a mechanism for maintaining intermediate frequency loci. Similar processes, driven by host immunity, have been proposed to explain the co-existence of multiple serotypes (13) and vaccine-induced metabolic shifts among pneumococci (14).
Based on the observations of Corander et al, we hypothesized that predictions of accurate post-vaccination evolutionary dynamics could be made on the premise that after vaccination, SCs with accessory genomes that could best restore COG frequencies perturbed by removal of vaccine serotypes, would increase disproportionately to other SCs. To this end, we implemented a deterministic NFDS model using the replicator equation (15 ,16) to predict pneumococcal evolution after a perturbation from near equilibrium COG frequencies (eq. 1).

Under this formulation, xi denotes the frequency of each ith sequence cluster (Sci, i = {1,…,n}).,n is the total number of SCs, and ωi denotes the predicted fitness of Sci (adapted from Ref. (11)), and φis the average population predicted fitness. In this model, we define ωi for each Sci as the dot product of two vectors whose elements correspond to the COGs: a vector Ki,l (l = {1,…,nloci}) with elements {0,1} for the absence or presence of the COGs: in SCi, respectively, and a vector containing difference between the pre-vaccine frequency el of each COGl and fl, which is the COG’s expected frequency post-vaccination based on depleting the VT from the pre-vaccine population (eq. 2).

Intuitively, the vector (ei – fi) represents the shape of the “hole” left in the population by vaccination, and Ki,l quantifies the ability of Sci to fill that hole. We make no explicit assumptions about carrying capacity, migration, mutation, or recombination rate, requiring only knowledge of the population structure (SCs) and COG frequencies before the intervention (e.g., vaccine); these quantities can be estimated from a pre-vaccine population survey with genome sequencing. However, there is an implicit assumption that over the study period recombination negligibly affects the frequency and distribution of COGs. Using simulated data, we first assessed the ability of a SC’s standardized fitness (ωi -φ) immediately after intervention to predict the direction of change in SC frequencies from pre-vaccine to the post-vaccine equilibrium (Figure 3A). The predicted fitness represents the SC’s ability to resolve the perturbation resulting from the vaccine-induced population bottleneck. Using this model, we show that the predicted fitness accurately estimates the direction of a simulated SC’s adjusted frequency change (positive predictive value (PPV) = 99.9%, negative predictive value (NPV) = 83.9%, 1000 simulations), independent of the initial pre-vaccine SC frequency (Figure 3B). The accuracy of prediction was also robust to varying the proportion of the population removed between 5-30% of SCs to mimic the affect of PCV7 on the pneumococcal population.

A) Descriptive representation of the Sequence Cluster (SC) prevalence at different stages. We modeled a population of VT and NVT SCs (represented as unique genotypes with alleles 1 or 0 at a locus denoting the presence or absence of a single COG) and simulated the removal of VT genotypes, following the post-vaccine population to equilibrium (details in Supplemental Information). A total of 8 SCs are present at time zero and the system is allowed to evolve until it reaches a steady state (‘pre-vaccine equilibrium’). Three SCs where then targeted to mimic a vaccine introduction, which removes them from the system. The predicted fitness was then estimated from the period just after the vaccine introduction, when the population has been depleted of VT but relative prevalence of NVT have not changed – a quantity that can be calculated from pre-vaccine data alone. Finally, the system goes to a second steady state (‘post-vaccine equilibrium’). Different shades of red represent the rank of the SC frequencies in the post-vaccine equilibrium. B) Predicted fitness from simulated data. Ten independent replicates were calculated for 2371 COGs, 35 randomly chosen SCs, and 3 vaccine types removed from 35 SCs (consistent with our empirical observations(9)). The predicted fitness accurately predicts the direction of the adjusted prevalence change 90.1% of the time (mean of 1000 simulations).
Next, we asked whether this approach could predict the post-vaccine composition of real pneumococcal populations, and more specifically, the relative contribution of lineages to serotype replacement, without the need for full forward simulation. To test this, we evaluated a pneumococcal sample from the southwest US, comprised of 937 strains collected before and after the introduction of PCV7. For each NVT SC present pre-vaccine (considering NVT taxa only, in the case of SCs that contained both) we calculated their predicted fitness based on their accessory genome. We identified COGs as detailed in supplementary materials, using the 2371 COGs found in between 5% and 95% of the population and calculated the frequency of each COG among NVT taxa in each SC before vaccination. We found the predicted fitness value was significantly and positively correlated with the adjusted prevalence change – its change in prevalence minus what would be expected if all NVT SCs increased by the same proportion from their pre-vaccine prevalence (Adjusted R2=0.44, p<<0.001, Figure 4A). More than 90% of the SCs were accurately assigned based on whether they increased or decreased after vaccine (Figure 4A-B). SCs with a positive adjusted prevalence change had substantially higher predicted fitness than those with a negative one (p=0.012, Figure 4B).

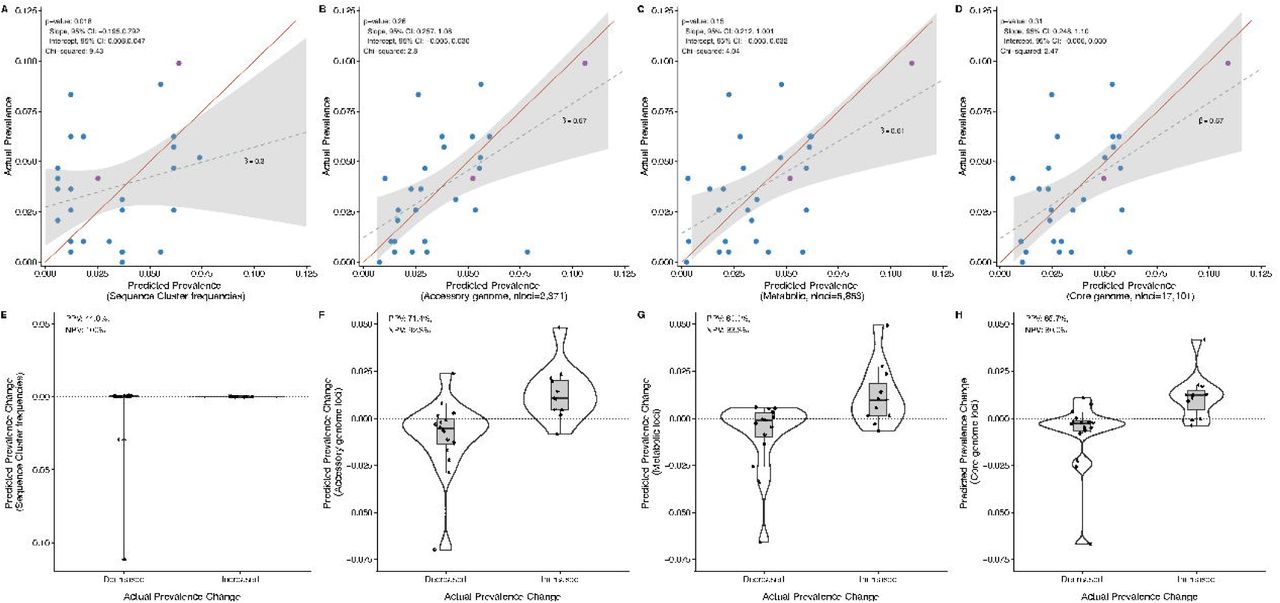
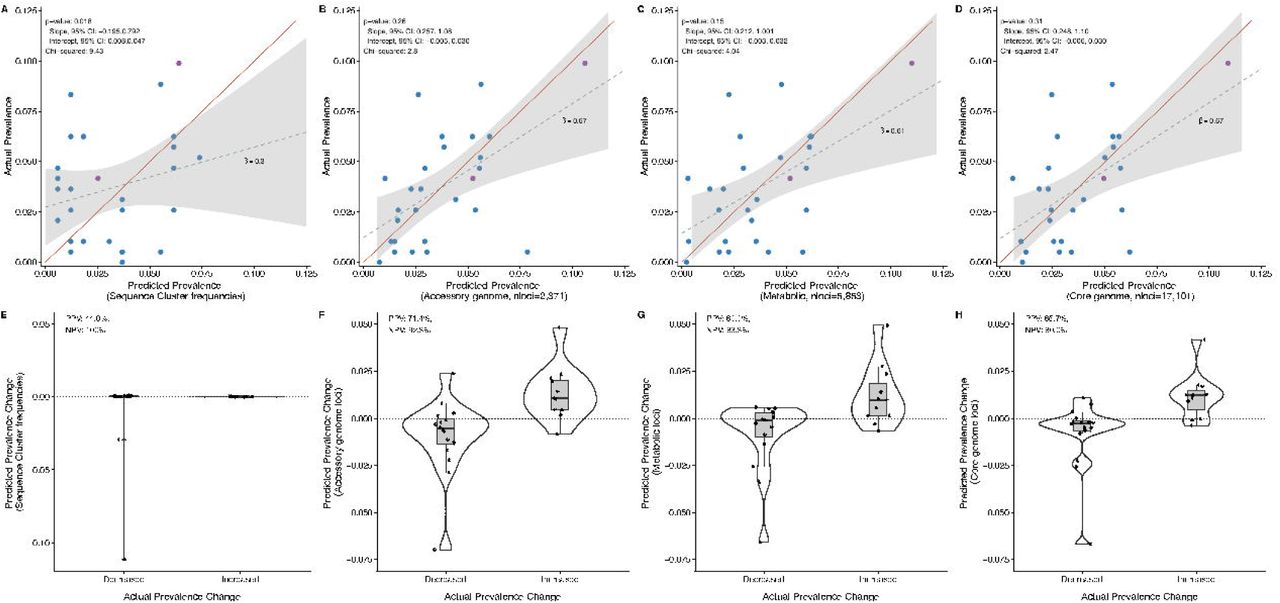
(A) Relationship between predicted fitness of a sequence cluster (SC) and its adjusted prevalence change from pre- to post-vaccine. Predicted fitness was calculated using data solely from the pre-vaccine sample, with the exceptions of SCs 04C, 10, and 22-24 for which there were no non-vaccine serotype (NVT) isolates present in the sample before the introduction of PCV7. For those SCs, data were imputed from the time point during which they were first observed (see supplemental methods). Gray circles around points are scaled to the standard errors of each adjusted prevalence change estimate. The points are colored by SC composition: NVT only (blue) and mixed vaccine serotype (VT) and NVT (purple). The grey shaded quadrants indicate regions of accurate prediction of the prevalence change direction (increased post-vaccine vs. decreased) given the predicted fitness value. Three outlier SCs are annotated. (B) Comparison of predicted fitness between SCs that increased or decreased based on their adjusted prevalence change (p=0.012). (C) Scatterplot of actual versus predicted prevalence of SCs at post-vaccine based on quadratic programming analysis. Points are colored based on SC serotype composition as described in panel A. Accessory loci frequencies pre-vaccine were regressed on the COG frequencies of NVT SCs. We tested the significance of slope β = 1 and intercept α = 0 using Student’s t test (p < .05). The line of equality (1:1 line), in red, shows the accuracy of the predicted to actual frequencies. Two outliers are annotated. (D) Comparison of the predicted prevalence change from quadratic programming analysis between SCs that increased or decreased based on their actual pre- to post-vaccine prevalence change (p=0.001).
While the predicted fitness accurately determines the direction of prevalence change, it does not provide the SC prevalence once the population has achieved post-vaccine equilibrium. To address this limitation we used quadratic programming (QP) to numerically identify the set of NVT SC prevalence that produced COG frequencies closest to those observed pre-vaccine (see Supplementary Information for more details). In short, assuming the pre-vaccine COG frequencies represent an equilibrium, we removed the VT population and then asked which combination (estimated as a proportion) of the NVT SCs present pre-vaccine best restored the equilibrium COG frequencies. QP accurately predicted SC frequencies following vaccination i.e., the 95% confidence interval of the observed vs. predicted post-vaccine SC frequencies included the line of equality (1:1 line), which denotes a perfect prediction, and the intercept and slope did not differ significantly from zero and one, respectively (p=0.26; intercept 95% CI: - 0.005, 0.030; slope 95% CI: 0.257, 1.075, Figure 4C). In addition, QP also accurately predicted which SCs would have a positive prevalence change (PPV=71.4%, NPV=92.3%, Fisher’s exact test score = 25.4, p=0.001, Figure 4D). These results were also robust to restricting the post-vaccine population to only those isolates collected in 2010 (n=119), prior to the introduction of PCV13 (Supplementary Information). In comparison, a naïve estimate based solely on pre-vaccine prevalence performed poorly (Figure 5A), as expected given the discordance in the pre- to post-vaccine rank changes illustrated in Figure 2. We further tested the predictive value of different genomic elements, finding that core genome loci (nloci= 17,101) and metabolic loci (nloci=5,853) were also capable of predicting the impact of vaccine (Figure 5). This finding must be considered in the context of recombination, selection, and the evolutionary timescale impacting the pneumococcal genome. Despite moderate levels of bacterial recombination among pneumococci, there remains appreciable linkage disequilibrium between loci nearby as well as genome-wide (1), which makes it difficult to discern the relative selective importance of any particular locus. Operationally, COGs provide accurate prediction using significantly fewer loci and are easy to obtain using widely available genomic tools.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comparison of naïve and quadratic programming predictions using varying genomic loci. A) Naïve estimate of SC prevalence based on NVT-VT composition and pre-PCV7 SC frequency, B) 2,371 COG predictions present among 5-95% of taxa, C) 17,101 biallelic polymorphic sites found in 1,111 genes in the core genome and present among 5-95% of taxa, D) 5,853 biallelic polymorphic sites found in 272 metabolic genes present in the core genome and present among 5-95% of taxa. For each model, the slope and 95% confidence interval of the actual vs. predicted prevalence values are compared to the 1:1 line using a linear test. P-values >0.05 indicate that the intercept and slope of observed versus predicted post-vaccine SC frequencies did not differ significantly from zero and one (joint model). The χ values are presented in relation to the line of equality (in red). Figures E-H show comparisons of the predicted prevalence change from quadratic programming analysis between SCs that increased or decreased based on their actual pre- to post-vaccine prevalence change. The positive predictive value (PPV, i.e. the probability that a SC predicted to increase post-vaccine truly increased) and negative predictive value (NPV, i.e., the probability that a SC predicted to decrease post-vaccine truly decreased) are also presented for each model.
Given that the predicted fitness estimation requires the pre-vaccine SC prevalence, we can only retrospectively calculate the predicted fitness of the two SCs (SC10 and SC24) that emerged over the study period and compare them with samples collected elsewhere. Comparing with a carriage dataset of 1,354 pneumococci collected in Massachusetts children (12 ,17), we found that SCs 10 and 24, observed in the present sample only post-PCV, had higher predicted fitness than any of the other potential migrant SCs found only in Massachusetts and not our southwest US sample before vaccination, and higher predicted fitness than any SC seen in both carriage collections, except for SCs 23 and 9. As such, we can use this approach to ask which lineages are most likely to successfully invade following vaccination, and given that SCs 10 and 24 were present in USA carriage samples around the time of PCV7’s introduction, they appear to have been primed for emergence.
Considering population structure and accessory genome content, post-vaccine COG frequencies may be restored by: 1) replacement by NVT SCs with varying degrees of relatedness in terms of core genome distance, or 2) clonal replacement by NVT strains belonging to SCs containing both VT and NVT taxa. In the southwest US sample, we observe both. Regarding the former, we find that the similarity of SCs in terms of COG presence-absence is only weakly associated with the phylogenetic distance between them, calculated from the core genome (see Supplemental Information; Supplemental Figures 2A and 3). Therefore, SCs may be divergent in core genome distance but share similar accessory genomes and comparable predicted fitness, as shown by the varied association between core and accessory loci and respective predicted fitness values (Supplemental Figure 2). A clear example was the post-PCV7 success of SC24, which possessed a high predicted fitness due to COG similarity with SC9. Regarding the latter, we find the NVT component of SCs containing both VT and NVT taxa (e.g., SC09 and SC23) possessed high predicted fitness, as expected under our model since these taxa are very similar to those that have been removed in both their core and accessory loci. Hence we should strongly expect the NVT part of any mixed SCs to increase post vaccination, especially since these NVT taxa are sometimes similar to their VT counterparts in terms of serotype properties such as capsule thickness and charge, which are independently correlated with prevalence (18 ,19). A good example of this is the serotype 15B/C component of SC26, which we now predict to be successful following the more recent introduction of a vaccine incorporating six additional serotypes (PCV13) and which has indeed been noted to be increasing in recent samples (20–22). This information may be relevant for current vaccine considerations.
The potential of NFDS to structure a pathogen population is consistent with findings from environmental microbiology research on multiple bacterial species (23). Among pneumococci, changes in population dynamics after the introduction of vaccine have been explained by metabolic types, antibiotic resistance, carriage duration, recombination rates, and serotype competition, which may involve NFDS as well as other types of selection (10 ,14 ,24 ,25). In the case of three distinct pneumococcal carriage samples, COG frequencies consistently rebounded after being perturbed by PCV (11). Our approach, which does not require forward simulation, is predicated on the relatively simple hypothesis that SCs whose COG frequencies best resolve the PCV perturbation will be more successful in the post-vaccine evolutionary landscape. Indeed, we find a significant linear relationship between predicted fitness and the adjusted prevalence change of a SC. By optimizing NVT SC prevalence conditional on the pre-PCV7 COG frequencies equilibrium, we are able to recover observed post-PCV7 SC prevalence. Both the predicted fitness and numerical approximations of the post-vaccine equilibrium by QP robustly predicted SC trajectory after PCV7, and the same rationale leads us to predict that serotype 15BC from SC26 will now increase in carriage prevalence following the use of PCV13.
It should be noted that we do not have a full mechanistic account of how selection produces the equilibrium of COG frequencies, which we have used to predict the consequences of vaccination. It is quite conceivable that a minority of COGs or other loci are involved (e.g., polymorphic protein antigens (26)), or SNPs in the core genome, which also show a correlation (see Supplementary Information and (11)), and the correlations that we have leveraged to predict the impact of vaccination are due to the amount of genetic linkage that persists in the pneumococcus despite appreciable transformation and recombination. Further, as evidenced by two outliers to QP predictions (SC18 and SC25 in Figure 3C), we acknowledge that the model does not currently capture how the ordering of strain invasions may affect the emergence of genotypes in the years post-PCV or other types of selection acting on pneumococci. Stochasticity resulting from patterns of strain migration will inevitably have an effect. This model is also restricted to the period in which recombination likely plays a limited role in transferring COGs and affecting their frequencies, although previous modeling suggests this period lasts several years (27). Ultimately, expanding the model to include immigration of other SCs and disentangling the relative contribution of selection on various loci is likely to be a fruitful area for future research. One area worth exploring is the degree to which recombination acts to maintain COG frequencies on the timescale of population level shifts in lineage composition.
Predicting evolution is a central goal of population genomics especially when related to human health. While evolutionary theory provides an understanding of bacterial population processes including the relative success of lineages, distribution of phenotypes, and ecological niche adaption, these analyses are often conducted retrospectively. Here, we demonstrate a method for predicting the impact of vaccination on the pneumococcal population and make future predictions based on the PCV13-era data. By incorporating information on invasive capacity, these predictions could be extended to inform changes in invasive disease rates. These dynamics may suggest novel vaccine strategies in which one could target not only prevalent serotypes but also those serotypes whose removal would result in a predicted re-equilibration that favors the least virulent or most drug-susceptible lineages. As NFDS appears to be pervasive among bacterial populations, future studies should assess extant pathogen genomic samples for this signal in both the core and accessory genomes.
Footnotes
Pamela P Martinez pmartinez{at}hsph.harvard.edu, Brian J Arnold brianjohnarnold{at}gmail.com, Lindsay R Grant lgrant10{at}jhu.edu, Jukka Corrander jukka.corander{at}medisin.uio.no, Christophe Fraser christophe.fraser{at}bdi.ox.ac.uk, Nicholas J Croucher n.croucher{at}imperial.ac.uk, Laura Hammitt lhammitt{at}jhu.edu, Raymond Reid rreid2{at}jhu.edu, Mathuram Santosham msantosham{at}jhu.edu, Robert R Weatherholtz rweathe1{at}jhu.edu, Stephen D Bentley sdb{at}sanger.ac.uk, Katherine L O’Brien klobrien{at}jhu.edu, Marc Lipsitch mlipsitc{at}hsph.harvard.edu, William P Hanage whanage{at}hsph.harvard.edu
↵* Co-senior authors
References




