

Summary
Choices for genome engineering and integration involve high efficiency with little or no target specificity or high specificity with low activity. Here, we describe a targeted integration strategy, called GeneWeld, and a vector series for gene tagging, pGTag (plasmids for Gene Tagging), which promote highly efficient and precise targeted integration in zebrafish embryos, pig fibroblasts, and human cells utilizing the CRISPR/Cas9 system. Our work demonstrates that in vivo targeting of a genomic locus of interest with CRISPR/Cas9 and a donor vector containing as little as 24 to 48 base pairs of homology directs precise and efficient knock-in when the homology arms are exposed with a double strand break in vivo. Given our results targeting multiple loci in different species, we expect the accompanying protocols, vectors, and web interface for homology arm design to help streamline gene targeting and applications in CRISPR compatible systems.
In Brief Wierson et al. describe a targeted integration strategy, called GeneWeld, and a vector series for gene tagging, pGTag, which promote highly efficient and precise targeted integration in zebrafish, pig fibroblasts, and human cells. This approach establishes an effective genome engineering solution that is suitable for creating knock-in mutations for functional genomics and gene therapy applications. The authors describe high rates of germline transmission (50%) for targeted knock-ins at eight different zebrafish loci and efficient integration at safe harbor loci in porcine and human cells.
Introduction
Designer nucleases have rapidly expanded the way in which researchers can utilize endogenous DNA repair mechanisms for creating gene knock-outs, reporter gene knock-ins, gene deletions, single nucleotide polymorphisms, and epitope tagged alleles in diverse species (Bedell et al., 2012; Beumer et al., 2008; Carlson et al., 2012; Geurts et al., 2009; Yang et al., 2013). A single dsDNA break in the genome results in increased frequencies of recombination and promotes integration of homologous recombination (HR)-based vectors (Hasty et al., 1991; Hoshijima et al., 2016; Orr-Weaver et al., 1981; Rong and Golic, 2000; Shin et al., 2014; Zu et al., 2013). Additionally, in vitro or in vivo linearization of targeting vectors stimulates homology-directed repair (HDR) (Hasty et al., 1991; Hoshijima et al., 2016; Orr-Weaver et al., 1981; Rong and Golic, 2000; Shin et al., 2014; Zu et al., 2013). Utilizing HDR or HR at a targeted double-strand break (DSB) allows base-pair precision to directionally knock-in exogenous DNA, however, frequencies remain variable and engineering of targeting vectors is cumbersome.
Previous work has shown Xenopus oocytes have the ability to join or recombine linear DNA molecules that contain short regions of homology at their ends, and this activity is likely mediated by exonuclease activity allowing base pairing of the resected homology (Grzesiuk and Carroll, 1987). More recently, it was shown in Xenopus, silkworm, zebrafish, and mouse cells that a plasmid donor containing short (≤40 bp) regions of homology to a genomic target site can promote precise integration at the genomic cut site when the donor plasmid is cut adjacent to the homology (Aida et al., 2016; Hisano et al., 2015; Nakade et al., 2014). Gene targeting is likely mediated by the alternative-end joining/microhomology-mediated end joining (MMEJ) pathway or by a single strand annealing (SSA) mechanism (Ceccaldi et al., 2016). In contrast, in human cell culture, linear donors with homologous ends have been reported to show inefficient integration until homology domains reach ~600 bp (Zhang et al., 2017), suggesting that different repair pathways may predominate depending on cell type. In the initial reports using short regions of homology for in vivo gene targeting in zebrafish, the level of mosaicism in F0 injected animals was high, resulting in inefficient recovery of targeted alleles through the germline (Aida et al., 2016; Hisano et al., 2015; Nakade et al., 2014).
Here, we present GeneWeld, a strategy for targeted integration directed by short homology, and demonstrate increased germline transmission rates for recovery of targeted alleles. We provide a detailed protocol and a suite of donor vectors, called pGTag, that can be easily engineered with homologous sequences (hereafter called homology arms) to a gene of interest, and a web interface for designing homology arms (www.genesculpt.org/gtaghd/). We demonstrate that 24 or 48 base pairs of homology directly flanking cargo DNA promotes efficient gene targeting in zebrafish, pig, and human cells with frequencies up to 10-fold higher than other HR strategies. Using short homology-arm mediated end joining, we can achieve germline transmission rates averaging approximately 50% across several zebrafish loci. Southern blot analysis in the F1 generation reveals that the GeneWeld strategy can yield alleles with precise integration at both 5’ and 3’ ends, as well as alleles that are precise on just one end. Finally, we present a strategy to delete and replace up to 48kb of genomic DNA with a donor containing homology arms flanking two distal CRISPR/Cas9 sites in a gene. These tools and methodology provide a tractable solution to creating precise targeted integrations and open the door for other genome editing strategies using short homology.
Design
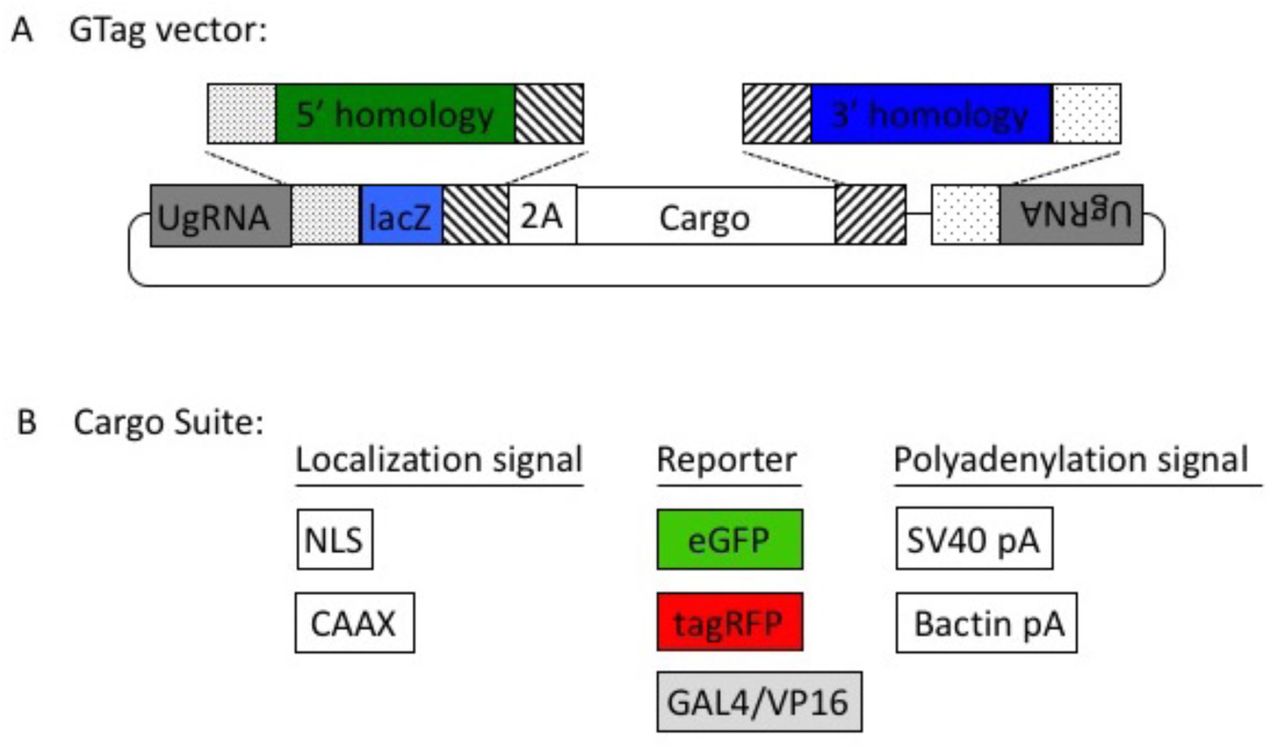
The GeneWeld strategy takes advantage of two simultaneous actions to initiate targeted integration directed by short homology (Fig. 1a). First, a high efficiency nuclease introduces a DSB in the chromosomal target. Simultaneously, a second nuclease makes a DSB in the pGTag vector integration cassette exposing the short homology arms. The complementarity between the chromosomal DSB and the donor homology arms activates a MMEJ/SSA or other non-NHEJ DNA repair mechanism, referred to as homology-mediated end joining (HMEJ). The reagents needed for this gene targeting strategy include Cas9 mRNA to express the Cas9 nuclease, a guide RNA targeting the genomic sequence of interest, a universal gRNA (UgRNA) that targets two sites in the pGTag series donor vectors to expose the homology arms, and a pGTag/donor vector with gene specific homology arms (Fig. 1a). The universal gRNA (UgRNA) has no predicted sites in zebrafish, pig, or human genomes. Alternatively, a gene specific guide RNA can be used to expose homology arms in the donor vector. For simplicity we will refer to this set of reagents as ‘GeneWeld reagents’. Using GeneWeld reagents to target various loci, we demonstrate widespread reporter gene expression in injected F0 zebrafish embryos, porcine fibroblasts, and human K-562 cells, indicating efficient and precise in-frame integration in multiple species and cell systems.

(a) GeneWeld reagent components are designed for simultaneous genome and donor nuclease targeting to reveal short regions of homology. Red arrowheads represent in vivo designer nuclease DSBs. Components include: 1 - Designer nuclease mRNA, either Cas9 to target both the genome and donor, or Cas9 to target the donor and TALEN to cut the genome; 2 - sgRNA for targeting Cas9 to genome;; 3 - Universal sgRNA to liberate donor cargo and homologous ends; and 4 - pGTag donor of interest with short homology arms. (b) Type IIs restriction endonucleases BfuAI and BspQI create incompatible ends outside of their recognition sequence, allowing digestion and ligation of both homology arms into the vector in a single reaction. Homology arm fragments are formed by annealing complementary oligonucleotides to form dsDNA with sticky ends for directional cloning into the vector. XFP = Green or Red Fluorescent Protein. pA = SV40 or β-actin 3’ untranslated region. Red and green fluorescent proteins were cloned into the pGTag vectors, and for each color, subcellular localization sequences for either nuclear localization (NLSs) and membrane localization (CAAX) are provided (c) Schematic of GeneWeld targeting in vivo. After designer nuclease creates targeted double-strand breaks in the genome and donor, end resection likely precedes homology recognition and strand annealing, leading to integration of the donor without vector backbone. Red arrowheads represent in vivo designer nuclease DSBs.
Results
A single homology domain of up to 48 bp drives efficient integration of RFP into noto
To develop baseline gene targeting data, we engineered variable length homology domains to target noto. These lengths were based on observations that DNA repair enzymes bind DNA and search for homology in 3 or 4 base pair lengths (Supplementary Fig. 1a) (Conway et al., 2004; Singleton et al., 2002). Upon injection of a noto sgRNA to target both the genome and one homology domain of a 2A-TagRFP-CAAX-SV40 donor vector, efficient integration was observed as notochord-specific RFP (Supplementary Fig. 1b, c, Supplementary Table 1-3). The frequency of RFP expression increased as the homology domain was expanded up to 48 bp (Supplementary Fig. 1b), while 192 bp of homology displayed reduced integration activity (data not shown). Somatic junction fragment analysis showed precise integration efficiencies reaching 95% of sequenced alleles (Supplementary Fig. 1d). Following these initial experiments, a 3 bp spacer sequence was included in all homology arm designs to separate the donor CRISPR/Cas9 target PAM and the homology domain and prevent arbitrarily increasing the length of the targeting domain, as single base pair alterations in the homology region can affect knock-in efficiency up to 2-fold (Supplementary Fig. 2).
Using the UgRNA to Liberate Donor Homology
To simplify donor design and liberate donor cargo in vivo with reproducible efficiency, a UgRNA sequence, with no predicted targets in zebrafish, pig, or human cells, was designed based on optimal base composition (Supplementary Fig. 3a) (Moreno-Mateos et al., 2015). The target sequence for the UgRNA with a PAM sequence was cloned 5’ adjacent to the homology arm in a donor vector to direct a DSB for homology exposure. Experiments targeting noto with this UgRNA donor plasmid resulted in RFP expression in the notochord in 21% of injected embryos, indicating correct targeting of noto and demonstrating the efficacy of Cas9/UgRNA to expose the single 5’ homology arm in the donor and drive precise integration (Supplementary Fig. 2b, 3c). The high frequency of RFP+ cells following injection of GeneWeld reagents suggests that repair of the DSB preferentially utilizes the homology in the targeting construct over the NHEJ pathway.
Liberating Short Homology by Dual Targeting of Donor Vectors and Genomic Loci Directs Precise Integration in Somatic Tissue
We leveraged the activity of the UgRNA in the design of the pGTag vector series by including UgRNA target sites on both ends of the cargo (Fig. 1b). Homology arms can be simply added to the vectors using Golden Gate cloning. Cleavage by Cas9 at the UgRNA sites in the embryo or cell liberates the DNA cargo from the plasmid backbone and exposes both 5’ and 3’ homology arms for interaction with DNA on either side of the genomic DSB (Fig. 1c). Injection of GeneWeld reagents containing either 2A-TagRFP-CAAX-SV40 or 2A-eGFP-SV40 donors targeting noto resulted in 24% of embryos showing extensive notochord expression of the reporter, indicating a similar targeting efficiency compared to targeting with 5’ homology alone (Fig. 2a, e; Supplementary Fig. 1, Table 1-3).

(a-d) Live confocal images of F0 injected embryos showing fluorescent reporter expression of noto-2A-eGFP-SV40 at mid somite stage (a, a’), cx43.4-2A-tagRFP-CAAX-SV40 at 31 hours post fertilization (b, b’). Expression from UAS-RFP Tg(UAS:mRFP)tpl2 after injection with tyr-2A-Gal4VP16-β-actin at 5 days post fertilization (dpf) (c, c’), or esama-2A-Gal4VP16-β-actin at 2 dpf (d) and 3 dpf (d’). (e) Reporter gene expression following targeting with HMEJ at noto, cx43.4, tyr, and esama results in a high proportion of injected F0 embryos displaying widespread signals. Homology lengths flanking donor cargos indicated by 24/24 or 48/48, in base pairs. Data represents mean +/- s.e.m. of 3 independent targeting experiments. Scale bars, 100 μm.
To extend our results to other loci in zebrafish, we targeted different genes with 2A-TagRFP-CAAX-SV40 and varying homologies (Fig. 2, Supplementary Table 1-4). GeneWeld reagents targeting connexin43.4 (cx43.4) with 24 and 48 bp of homology resulted in broad RFP expression throughout the nervous system and vasculature in 38 to 50% of the injected embryos (Fig. 2b, e). Together, these results suggest that 24 bp of homology directs targeted integration as efficiently as 48 bp.
Targeting exon 4 of tyrosinase (tyr) with GeneWeld reagents did not result in detectable RFP signal, similar to previous reports (Hisano et al., 2015). However, PCR junction fragments from injected larvae showed the 2A-TagRFP-CAAX-SV40 donor was precisely integrated in frame into tyr exon 4 (Supplementary Fig. 5), suggesting RFP expression was below the threshold of detection. To amplify the fluorescent signal, we injected homology directed pGTag-2A-Gal4VP16-βߝactin to integrate the transactivator Gal4VP16 into the tyr exon 4 target site in transgenic zebrafish embryos carrying a 14xUAS-RFP reporter, Tg(UAS:mRFP)tpl2 (Balciuniene et al., 2013). This resulted in strong RFP signal in 64% of injected animals; however, the embryos were highly mosaic compared to targeting 2A-TagRFP-CAAX-SV40 into noto and cx43.4, with only 9% of RFP embryos displaying extensive expression throughout pigmented cells (Fig. 2c, e).
Similar to tyr, GeneWeld reagents used for targeting 2A-TagRFP-CAAX-SV40 into exon 2 of the esama gene did not result in detectable RFP expression, however, targeting pGTag-2A-Gal4VP16 in the Tg(UAS:mRFP)tpl2 transgenic background resulted in 21% of embryos displaying extensive RFP expression specifically in the vasculature (Fig. 2d, e). This approach was further extended to five additional loci, targeting 2A-Gal4VP16 to filamin a (flna), moesin a (msna), aquaporin 1a1 (aqp1a1), aquaporin 8a1 (aqp8a1), and annexin a2a (anxa2a). At these loci, transient expression of RFP was observed following injection in 4-55% of Tg(UAS:mRFP)tpl2 embryos (Supplementary Table 1 and 2). Taken together, these results show that the application of GeneWeld reagents promotes high efficiency somatic integration following injection into zebrafish embryos.
Efficient germline transmission
Three out of five (60%) noto-2A-TagRFP-CAAX-SV40 injected founder fish raised to adulthood transmitted noto-2A-TagRFP-CAAX-SV40 tagged alleles through the germline (Figure 3, Table 1, Supplementary Tables 3 and 4). For tyr-2A-Gal4VP16-β–actin injections, 8 embryos with expression in the retinal pigmented epithelia were raised to adulthood and outcrossed. Of those, three founder animals transmitted tagged alleles to the next generation (37.5%) (Figure 3, Table 1 and Supplementary Table 3 and 4). Likewise, for esama-2A-Gal4VP16-β-actin, 18 F0s displaying widespread vasculature RFP expression were raised to adulthood, and 12 (66.7%) transmitted esama-2A-Gal4VP16-β-actin alleles to the F1 generation. We have extended these results to flna, two different target sites in msna (exon 2 and 6), aqp1a1, aqp8a1, and anxa2a with a combined F0 transmission rate of 49% across all loci (Figure 3, Table 1, Supplementary Tables 3 and 4). Taken together, these results indicate GeneWeld reagents which efficiently promote targeted integration in zebrafish F0s based on reporter expression are also efficiently transmitted through the germline.

(a, a’) Tg(noto-2A-TagRFP-CAAX-SV40) embryo at mid somite stage showing expression in the notochord and floor plate. (b, b’) Tg(tyr-2A-Gal4Vp16-β-actin) displaying expression in the melanocytes in a 5 dpf larva. (c, c’) Tg(esama-2A-Gal4Vp16-β-actin) larva showing expression in the vascular system at 4 dpf. (d, d’) Tg(flna-2A-Gal4VP16-β-actin) embryo at 1 dpf showing widespread expression. (e, e’ and f, f’) Tg(msna-2A-Gal4VP16-β-actin) targeted to either exon 2 or exon 6 showed expression in the central nervous system and vasculature at 2 dpf. (g, g’ and h, h’) Tg(aqp1a1-2A-Gal4VP16-β-actin) and Tg(aqp8a1-2A-Gal4VP16-β-actin) display expression in the trunk and tail vasculature at 2 dpf. All images are lateral views, and the Gal4VP16 integrations have Tg(UAS:mRFP)tpl2 in the background for visualization of expression. Scale bars are 100 μm.
Germline transmission of alleles in zebrafish using GeneWeld.
Precise 5’ and 3’ junctions and single copy integration in F1s
Given that homology was present on both sides of the pGTag constructs used for targeting, it was expected that precise integration would occur at both 5’ and 3’ of the genomic cut site. Genomic Southern blot analyses of four F1s from two of the noto-2A-TagRFP-CAAX-SV40 founders confirmed a single copy integration in noto exon 1 (Fig. 4, a-c). Progeny from the first founder had precise integrations at the 5’ end of the target site but contained an ~400 bp deletion downstream extending into exon 2. The second founder produced offspring with an allele with precise 5’ integration and also included an insertion at the 3’ end, likely to be a segment of the vector backbone based on the size of the insert on Southern blot. Sequencing of junction fragments from the first founder confirmed that NHEJ drove integration at the 3’ end rather than a homology-based mechanism which was not expected (Fig. 4, Supplementary Fig. 4). In contrast, Southern blot analysis and sequencing of tyr-2A-Gal4VP16-β-actin F1 progeny demonstrated a single copy integration of the Gal4VP16 cassette with precise integration at both 5’ and 3’ ends (Fig. 4, d-f, Supplementary Fig. 4, 5).

(a-c) Tg(noto-2A-RFP) F1 targeted integration alleles from 2 independent F0 founders. (a) noto gene model with location of restriction enzymes used for genomic Southern blot analysis. Location of the 513 bp noto probe is indicated (dark lines). The predicted and recovered alleles are shown. (b) Southern blots of F1 Tg(noto-2A-RFP) individuals hybridized with RFP probe. F1 from founder F0#1 contain a ~2100 bp band corresponding to integration plus deletion of ~400 bp in noto. F1 progeny from founder F0#2 show two bands: a ~3700 bp band corresponding to integration of the reporter plus 2000 bp of vector backbone, and a ~1500 bp band which may represent an off-target integration. Loading controls (10, 1) correspond to 10 copies or 1 copy of RFP containing plasmid. WIK, wild type control DNA. (c) Southern blot in (b) stripped and re-hybridized with the noto-specific probe. A 1342 bp band representing the wild type allele was detected in all individuals. The integration allele in F1s from F0 #1 was not detected due to deletion of the region containing the probe. F1s from F0 #2 contain the ~3700 bp band corresponding to the noto-2A-RFP integration allele. (d, e) Molecular analysis of Tg(tyr-2A-GAL4/VP16) F1 offspring from a single targeted F0 founder. (d) Schematic of expected integration pattern for tyr targeted with pGTag-2A-GAL4/VP16. 148 bp tyr probe in Exon 3 and 583bp probe in GAL4/VP16 are indicated. (e) GAL4/VP16 and (f) tyr probed Southern blots of genomic DNA from wild type (WIK) and 4 individual GAL4/VP16 positive F1s. The expected 7400 bp band is detected with both probes, suggesting a single copy integration.
Junction fragment analysis of F1s from each of seven transmitting loci indicated precise events were primarily recovered at the 5’ for all the genes examined (30/31 or 97% across seven genes) (Supplementary Fig. 4). This result is expected as precise 5’ integrations are selected for by screening for expression of the report from the donor cassette. For esama, the 3’ junctions were also precise in 9/10 of the F1s examined from 6 different F0s, and both aqp1a1 and app8a1 had precise 3’ junctions. This is compared to msna E2 targeting with 2A-Gal4VP16-β-actin, where only one out of the 12 F1s examined had a precise 3’ junction.
Together, these results indicate that GeneWeld reagents can promote precise single copy integration at a genomic cut site without vector sequences, although events involving NHEJ at the at the 3’ end are also recovered.
Homology Engineered to Distal Genomic gRNA Sites Seeds Deletion Tagging in Somatic Tissue
To further demonstrate the utility of GeneWeld reagents, we tested whether the pGTag donors could function to bridge two CRISPR/Cas9 genomic cuts, resulting in simultaneous deletion of endogenous sequences and integration of exogenous DNA to create a “deletion tagged” allele. The pGTag-2A-Gal4VP16 donor was cloned with homology arms to two gRNA target sites in the zebrafish retinoblastoma1 (rb1) gene. The two gRNA sites were located in exons 2 and 4, which are located 394 bp apart, or in exons 2 and 25 which are separated by ~48.4 kb (Fig. 5a). The 5’ homology arm contained sequence upstream of the cut site in exon 2, while the 3’ homology arm contained sequence downstream of the cut site in either exon 4 or exon 25. Injection of GeneWeld reagents with the corresponding exon 2-exon 4 or exon 2-exon 25 pGTag-2A-Gal4VP16-β-actin donor into Tg(UAS:mRFP)tpl2 embryos resulted in 59% and 60%, respectively, of injected embryos showing broad and ubiquitous RFP expression (Fig. 5b-c, f, Supplementary Table 1,2). Somatic junction fragment analysis showed precise integration at both the 5’ (29/30 or 97% of the fragments analyzed) and 3’ ends (20/30 or 67%) of rb1 (Supplementary Fig. 6). Increasing the size of the deleted region from 394 bp to 48.4 Kb did not affect the frequency of reporter integration. One out of 16 F0 founders screened (6%) transmitted a precise 5’ junction through the germline, but the 3’ junction could not be amplified by PCR (Supplementary Table 3, 4).

(a) Schematic for Gal4VP16 reporter integration to tag a deletion allele of rb1 exons 2-4 (top) and rb1 exons 2-25 (bottom). Arrowheads designate CRISPR/Cas9 DSBs. CRISPR gRNAs in two exons are expected to liberate the intervening genomic DNA. The targeting vector to replace the liberated genomic sequence, contains a 5’ homology arm to the upstream exon and a 3’ homology arm to the downstream exon inside of uGuide sites. (b, b’) Live confocal image of F0 Tg(UAS:mRFP)tpl2 embryo deletion tagged at rb1 exons 2-4, and (c, c’) deletion tagged at rb1 exons 2-25 by integration of 2A-Gal4VP16-β-actin. (d) Schematic for Gal4VP16 deletion tagging of msna exons 2-6. (e, e’) Live confocal image of F0 Tg(UAS:mRFP)tpl2 embryo deletion tagged at msna exons 2-6 with 2A-Gal4VP16-β-actin.(f) Somatic reporter efficiency of targeted deletion tagging using 48 bp homology arms for rb1 exons 2-4, rb1, exons 2-25 and msna exons 2-6. Data represents mean +/- s.e.m. of 4 (rb1) and 5 (msna) independent targeting experiments. Scale bars in b, c, c’, e are 200 μm and b’, e’ are 100 μm.
Using the same approach, we targeted the zebrafish gene moesina (msna) at exons 2 and 6, located 7.8 kb apart, with 2A-Gal4VP16-β–actin using 48 bp of homology. This resulted in 63% of embryos displaying RFP in a pattern consistent with the expression of msna (Fig. 5d-f, Supplementary Table 1,2). Somatic junction fragment analysis showed precise integration at both the 5’ (11/13 or 85% of the fragments analyzed) and 3’ ends (9/20 or 45%) of msna (Supplementary Fig. 6). Of the 10 F0 zebrafish raised to adulthood, none transmitted a deletion tagged allele to the next generation. In contrast, targeting 2A-Gal4VP16-β–actin to exon 2 or 6 alone resulted in 2 out of 7 F0s transmitting a targeted allele to the next generation (Supplementary Table 3, 4).
Together, these results demonstrate simultaneous targeting of two distal genomic cut sites can create integration at both ends of a pGTag reporter cassette by HMEJ in somatic tissue, but these events are not easily passed through the germline. This was reinforced by attempting deletion tagging at additional loci, including kdrl, s1pr1, and vegfaa, which showed 32-81% expression in F0s, but no germline transmission to F1s (Supplementary Table 2).
Integration of Exogenous DNA Using HMEJ in Porcine and Human Cells is More Efficient than HR
To determine if HMEJ integration directed by short homology functions efficiently in large animal systems, we tested the GeneWeld targeting strategy in S. scrofa fibroblasts. The ROSA26 safe harbor locus was targeted with a cassette that drives ubiquitous eGFP expression from the UbC promoter (Fig. 6a-c). GeneWeld reagents, where the genomic sgRNA was replaced with mRNAs encoding a TALEN pair to generate a genomic DSB in the first intron of ROSA26, were delivered to pig fibroblasts by electroporation. This strategy was compared to cells electroporated with just the TALEN pair and a HR donor containing approximately 750 bp of homology flanking the genomic target site. GFP expression was observed in 23% of colonies using GeneWeld reagents, compared to 2% of colonies using the HR donor with ~750 bp homology arms. Co-occurring precise 5’ and 3’ junctions were observed in over 50% of the GFP+, GeneWeld engineered colonies while none of the GFP+, HR colonies contained both junctions. Sequencing of junctions from 8 GFP+, GeneWeld engineered colonies that were positive for both junctions showed precise integration in 7/8 colonies at the 5’ junction and 8/8 colonies at the 3’ junction.

(a) Strategy for integration using HMEJ and HR donors into intron 1 of S. scrofa ROSA26 locus. Arrow heads CRISPR/Cas9 (for HMEJ donor) and TALEN (genome) DSBs. (b) Targeting efficiency of the HMEJ donor vs the HR donor as reported by GFP positive colonies out of total colonies. (c) Percent of GFP positive colonies analyzed containing properly sized junction fragments, comparing HMEJ and HR donors. Data are from three independently targeted cell populations. Data represents mean +/- s.e.m. of 3 independent targeting experiments. (d) Diagram of HR and HMEJ strategies for targeted integration of a MND:GFP reporter cassette into the human AAVS1 locus. (e) Flow cytometry analysis of GFP expression 14 days post-electroporation for each targeting modality: HR (left), HMEJ without universal sgRNA (middle), and HMEJ with universal sgRNA (right). Stable gate was drawn to measure the uniformly expressing population formed by targeted integration and was set based on episome only controls. (f) Quantitation of stable GFP expressing population as measured by flow cytometry at day 14. Data are from three independently targeted cell populations. Data represents mean +/- s.e.m. of 3 independent targeting experiments. p values calculated using two-tailed unpaired t-test.
The GeneWeld strategy was also used to target integration of a MND:GFP reporter (Halene et al., 1999) into the AAVS1 safe harbor locus in human K-562 cells (Fig. 6d-f). Integrations were attempted with either GeneWeld reagents or an HR donor targeting the AAVS1 cut site by electroporation of K-562 cells. Cells were FACs sorted by GFP at day 14 following electroporation. With GeneWeld reagents over 50% of cells were GFP positive, compared to only 6% of cells electroporated with the HR donor. This suggests the GeneWeld strategy promoted efficient integration and stable expression of the MND:GFP cassette at the AAVS1 locus (Supplementary Fig. 7). Expression was maintained over 50 days, and 5’ precise junction fragments were observed following PCR amplification in bulk cell populations (Supplementary Fig. 8). The results above demonstrate that the GeneWeld strategy outperforms traditional HR techniques in mammalian cell systems and is effective without antibiotic selection.
Discussion
The results described here demonstrate the utility of short homology-based gene targeting for engineering precise integration of exogenous DNA and expand the potential of efficient tagging to diverse loci with differing endogenous expression levels. We show that using short homology to bridge distal ends together simultaneously creates a deletion and a reporter integration, however, these events are not easily passed through the germline. We demonstrate efficient integration of cargos up to approximately 2 kb in length in zebrafish, pig fibroblasts, and human cells. Both CRISPR/Cas9 and TALENs are effective as GeneWeld genomic editors, providing flexibility in deployment and genome-wide accessibility.
Several components of the GeneWeld strategy may lead to enhanced somatic and germline targeting efficiencies in zebrafish as compared to previous reports (Hisano et al., 2015). Canonical NHEJ is highly active during rapid cell divisions in early zebrafish embryogenesis (Bedell et al., 2012). However, given the correct sequence context surrounding the dsDNA break, MMEJ is the preferred method of non-conservative repair (Ata et al., 2018; He et al., 2015; Kent et al., 2015). GeneWeld homology arms are rationally designed based on the known homology searching activity of RAD51 and strand annealing activity of RAD52 (Conway et al., 2004; Singleton et al., 2002). In our experiments at noto, gene targeting is significantly reduced when 48 bp homology arms are altered by 1 bp to 47 or 49 bp (Supplementary Fig. 3). This suggests that optimal short homology arms should be designed in groups of 3 and/or 4 bp increments. We are currently testing this hypothesis further. Additionally, Shin et al., 2014 showed the highest rates of somatic targeting when their donor was linearized in vitro inside a ~1 kb 5’ homology arm, leaving 238 bp of homology flanking the knock-in cargo. Thus, it is tempting to speculate that gene targeting in these experiments proceeded not through HR, but through other related HMEJ DNA repair pathway more similar to the findings presented here.
The dramatic shift of DNA repair at genomic DSBs from cis-NHEJ to trans-HMEJ using GeneWeld donors likely also influences enhanced editing of the germline. Across all zebrafish experiments with germline transmission, 49% of founders transmitted tagged alleles, with 17.4% of gametes carrying the edited allele of interest (Supplementary Table 3, 4), demonstrating decreased germline mosaicism and increased germline transmission from previous reports. Given that our somatic knock-in and germline transmission rates are higher than published reports, we conclude that GeneWeld is a more effective homology-based method for generating precisely targeted knock-in alleles in zebrafish.
While targeting noto with 5’ only homology shows an increase in targeting efficiency with longer homology, increasing homologies on both ends of the cargo DNA did not increase targeting efficiency (Fig. 2e). Positive events are selected only by fluorescently tagged alleles, indicating precise 5’ integration patterns. We speculate that inclusion of homology at the 3’ end of our cargo creates competition for the donor DNA ends, as not all editing events are precise at both 5’ and 3’ junctions (Fig. 4 and Supplementary Fig. 4). Thus, it is conceivable that precise events at the 3’ end could preclude precise integration at the 5’ end during some editing events, and vice versa. It is tempting to speculate that this data hints at synthesis dependent strand annealing (SDSA) as a possible DNA repair mechanism for pGTag donor integration (Ceccaldi et al., 2016). After strand invasion using either of the homology domains and replication through the reporter, second DNA end capture may abort before or after replication through the opposing homology domain, resulting in imprecision, as greater than or equal to 150 bp is required for proper second end capture in yeast (Mehta et al., 2017). Experiments to address this hypothesis by varying homology arm lengths flanking the donor cassettes and including negative selection markers are of note for future work in determining the genetic mechanisms that promote efficient integration.
Timing and turnover of Cas9 during the genomic editing event can influence cut efficiency and somatic mosaicism/germline transmission rates (Clarke et al., 2018; Zhang et al., 2018), increasing the interest of using RNP during all precision gene editing applications. However, we were unable to observe fluorescence following injection of GeneWeld components with RNPs or detect targeted integrations at a high frequency (unpublished data). We hypothesize this is due to Cas9 and UgRNA locating and binding to the UgRNA sites on the pGTag donors during dilution of the injection mixture. This heteroduplex either activates DSBs on the donor in vitro, or directly after injection, before the genomic gRNA can locate and cut the genome. Thus, the stochastics of DNA end availability are altered using RNPs and integration activity is greatly reduced. Injection of the GeneWeld plasmid donor and RNPs in separate injection mixtures does not produce integration in zebrafish embryos (unpublished data). Further experiments could address these limitations through the use of inducible nuclease systems.
Targeting genes with lethal phenotypes, such as tumor suppressors or other genes required for embryogenesis, is of interest to the zebrafish community. However, using fluorescence to screen for targeted events can be misleading. For example, the RFP signal is dramatically reduced or lost upon biallelic inactivation of noto, likely when notochord cells transfate to muscle cells (Melby et al., 1996; Talbot et al., 1995). Additionally, though deletion tagging using two target sites in the genome seems to be robust in somatic tissue, germline transmission of deletion tags is rare. This suggests that edited germ cells may be lost to apoptosis due to the additional cut in the genome, or that heterozygous deletion tagged alleles are recognized during homologous chromosome pairing and are repaired or lost as germ cells mature. Similar susceptibility of stem cells to apoptosis following gene editing has been previously observed (Ihry et al., 2018; Li et al., 2018). In both of these cases, it may be necessary to modulate GeneWeld reagent concentrations in order to avoid biallelic inactivation of the genomic target, or to ensure homozygous deletion tagging.
Amplification of the fluorescence signal using GAL4/VP16 allowed us to target several genes for which we did not observe a fluorescence report from integration of a fluorescent protein directly. While this approach is advantageous for selecting correctly targeted embryos to examine for germline integration, GAL4/VP16 may have toxic effects as reported previously (Ogura et al., 2009). For example, we found dominant phenotypes in the F1 generation for both msna and flna which could reflect toxicity from high levels of expression of GAL4/VP16. Alternatively, these gene could also display haploinsufficiency or express a partial protein product that functions in a dominant manner. Heterozygous msna mutants targeting exon 5 in the F1 generation display phenotypes similar to morpholino targeting of this gene (Wang et al., 2010) (data not shown), suggesting haploinsufficiency or a dominant negative peptide is a likely explanation.
GeneWeld is also an effective strategy to precisely control exogenous DNA integration in mammalian cell lines. While our data shows an approximate 10-fold increase in targeted integration using 48 bp of homology to drive HMEJ versus HR, Zhang et al. (2017)concluded that targeted integration did not appreciably increase until homology arms of ~600 bp were used (Zhang et al., 2017). However, this could reflect differences in the experimental design or cell types used and suggest different DNA repair pathways may be more prevalent in certain conditions. Deciphering the DNA repair pathway used for HMEJ in zebrafish and mammalian cells is paramount to increasing editing efficiencies in basic research and for gene therapy.
Given the high efficiency and precision of GeneWeld, additional applications to efficiently introduce other gene modifications, such as single or multiple nucleotide polymorphisms, by exon or gene replacement is possible using the deletion tagging method. Further, GeneWeld could be used to create conditional alleles by targeting conditional gene break systems into introns (Clark et al., 2011). In conclusion, our suite of donor vectors with validated integration efficiencies, methods, and web interface for pGTag donor engineering will serve to streamline experimental design and broaden the use of designer nucleases for homology-based gene editing at CRISPR/Cas9 and TALEN cut sites in zebrafish. We also demonstrate an advanced strategy for homology-based gene editing at CRISPR/Cas9 and TALEN cut sites in mammalian cell lines. Our results open the door for more advanced genome edits in animal agriculture and human therapeutics.
Methods
Contact for reagent and resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeffrey Essner (jessner{at}iastate.edu).
Experimental model and subject details
Zebrafish were maintained in Aquatic Habitats (Pentair) housing on a 14 hour light/10 hour dark cycle. Wild-type WIK were obtained from the Zebrafish International Resource Center. The Tg(miniTol2/14XUAS:mRFP, γCry:GFP)tpl2, shortened to Tg(UAS:mRFP)tpl2, was previously described (Balciuniene et al., 2013). All experiments were carried out under approved protocols from Iowa State University IACUC.
The human K-562 chronic myelogenous leukemia cell line (ATCC CCL-243) used in gene targeting experiments was cultured at 37°C in 5% CO2 in RPMI-1640 (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) and Penicillin/Streptomycin. Electroporation was conducted with 1.5 × 105 cells in a 10 μl tip using the Neon electroporation device (Thermo Fisher Scientific) with the following conditions: 1450V, 10ms, 3x pulse. Nucleic acid dosages were as follows: 1.5 μg Cas9 mRNA (Trilink Biotechnologies), 1 μg each chemically modified sgRNA (Synthego), and 1 μg donor plasmid.
Fibroblasts were cultured in DMEM (high glucose) supplemented to 10% vol/vol FBS, 20 mM L-glutamine and 1X Pen/ Strep solution and transfected using the NeonTM system (Invitrogen). Briefly, 1 × 106 fibroblasts were transfected with 1 ug of polyadenylated ROSA TALEN mRNA, 1 μg of universal gRNA mRNA, 1 μg of polyadenylated Cas9 mRNA and 1 μg of donor plasmid. Transfected cells were cultured for 3 days at 30°C before low density plating, extended culture (10 days) and colony isolation. Individual colonies were aspirated under gentle trypsanization, replated into 96-well plates and cultured for 3-4 days.
pGTag series vectors
To build the pGTag vector series, 2A-TagRFP, 2A-eGFP, and 2A-Gal4/VP16 cassettes were assembled from a 2A-TagRFP-CAAX construct, p494. To clone the eGFP cassette, the plasmid p494 was amplified with primers F-p494-XhoI and R-p494-SpeI to generate unique enzyme sites in the backbone. The eGFP coding sequence (Clontech Inc.) was amplified with the primers F-eGFP-SpeI and R-eGFP-XhoI to generate the corresponding enzyme sites on the eGFP coding sequence. Fragments were digested with SpeI-HF and XhoI (NEB) and following column purification with the Qiagen miniprep protocol, were ligated to the plasmid backbone with T4 ligase (Fisher).
The Gal4/VP16 coding sequence and zebrafish β-actin 3’ untranslated region was amplified from vector pDB783 (Balciuniene et al., 2013) with primers F-2A-Gal4-BamHI and R-Gal4-NcoI to add a 2A peptide to the 5’ end of the Gav4Vp16 cDNA. The resulting PCR product was then cloned into the intermediate Topo Zero Blunt vector (Invitrogen) and used for mutagenesis PCR with primers F and R ‘-gal4-Ecofix’ to disrupt the internal EcoRI restriction site. The resulting Gal4/VP16 sequence was cloned into the BamHI and NcoI sites in the p494 backbone.
The 5’ universal/optimal guide site and lacZ cassette were added to pC-2A-TagRFP-CAAX-SV40, pC-2A-eGFP-SV40, and pC-2A-Gal4VP16-β-actin with the following steps. The lacZ was first amplified with primers F-lacZ and R-lacZ, which add the type IIS enzyme sites to either end of the lacZ. The resulting PCR product was then cloned into an intermediate vector with the Zero Blunt® TOPO® PCR Cloning Kit (Invitrogen). This intermediate was used as a template in a nested PCR to add the Universal guide sequence GGGAGGCGTTCGGGCCACAGCGG to the end of the lacZ sequence. The nested PCR used primers F-lacZ-universal-1 and R-lacZ-universal-BamHI to add the first part of the universal guide to one end and a BamHI site to the other. This was used as template for PCR with the primers F-lacZ-universal-EcoRI and R-lacZ-universal-BamHI to add the remainder of the universal guide and an EcoRI site. The fragment was column purified as above, digested with EcoRI-HF and BamHI-HF and cloned into the appropriate sites in the three vectors.
The 3’ universal guide and type 2 restriction enzyme sites were cloned into each vector in two steps. A segment from a Carp beta-actin intron containing a 99 bp spacer flanked by two BspQI sites was amplified using the primers F-3’-uni-1 and R-3’-uni-1 to add the universal site to one side of the spacer. This product was column purified as above and used as template for the second amplification with primers F-3’-uniNco1 and R-3’-uniEagI to add cloning sites. This product was column purified and cloned using the Topo zero blunt kit. This intermediate was digested with NcoI-HF and EagI, and the BspQI fragment purified and cloned into the three vectors as above. Ligations were grown at 30°C to reduce the possibility of recombination between the two universal guide sites.
Correct clones for pU-2A-TagRFP-CAAX-U, pU-2A-eGFP-U, and pU-2A-Gal4/VP16-U were selected and used as template for mutagenesis PCR with KOD to remove extra BspQI sites from the backbone with primers F/R-BBfix, digested with DpnI (NEB), and ligated with T4 ligase. A correct pU-2A-TagRFP-CAAX-U clone was used as template for PCR with F/R-TagRFPfix to interrupt the BspQI site in the TagRFP coding sequence as above. A correct clone of pU-2A-Gal4/VP16-U was selected and used as template with primers F/R-Bactfix to remove the BspQI site in the Beta-actin terminator, the product was re-cloned as above. All constructs were sequence verified.
Homology arm design and donor vector construction
For detailed methods, see Supplementary gene targeting protocol. In brief, homology arms of specified length directly flanking a genomic targeted double strand break were cloned into the pGTag vector, in between the UgRNA sequence and the cargo. A three nucleotide buffer sequence lacking homology to the genomic target site was engineered between the donor UgRNA PAM and the homology arms, in order to maintain the specified homology arm length. See Supplementary Table 4 for all homology arms, gRNA target sites, and spacers.
Zebrafish embryo injection
pT3TS-nCas9n was a gift from Wenbiao Chen (Addgene plasmid # 46757). XbaI linearized pT3TS-nCas9n was purified under RNase-free conditions with the Promega PureYield Plasmid Miniprep System. Linear, purified pT3TS-nCas9n was used as template for in vitro transcription of capped, polyadenylated mRNA with the Ambion T3TS mMessage mMachine Kit. mRNA was purified using Qiagen miRNeasy Kit. The genomic and universal sgRNAs were generated using cloning free sgRNA synthesis as described in (Varshney et al., 2015) and purified using Qiagen miRNeasy Kit. Donor vector plasmid DNA was purified with the Promega PureYield Plasmid Miniprep System. noto, cx43.4, tyrosinase, and moesina, were targeted by co-injection of 150 pg of nCas9n mRNA, 25 pg of genomic sgRNA, 25 pg of UgRNA (when utilized), and 10 pg of donor DNA diluted in RNAse free ddH2O. The rb1 targeting mixture contained 300 pg nCas9n mRNA. 2 nl was delivered to each embryo.
Recovery of zebrafish germline knock-in alleles
Injected animals were screened for fluorescence reporter expression on a Zeiss Discovery dissection microscope and live images captured on a Zeiss LSM 700 laser scanning confocal microscope. RFP/GFP positive embryos were raised to adulthood and outcrossed to wildtype WIK adults to test for germline transmission of fluorescence in F1 progeny. tyr, esama, rb1 and msna embryos targeted with Gal4VP16 were crossed to Tg(UAS:mRFP)tpl2.
DNA isolation and PCR genotyping
Genomic DNA for PCR was extracted by digestion of single embryos in 50mM NaOH at 95°C for 30 minutes and neutralized by addition of 1/10th volume 1M Tris-HCl pH 8.0. Junction fragments were PCR-amplified with primers listed in Supplementary Table X and the products TOPO-TA cloned before sequencing.
Southern blot analysis
Genomic Southern blot and copy number analysis was performed as described previously (McGrail et al., 2011). PCR primers used for genomic and donor specific probes are listed in Supplementary Table 6.
Junction fragment analysis in pig fibroblasts
Individual colonies were scored for GFP expression and prepared for PCR by washing with 1X PBS and resuspension in PCR-safe lysis buffer (10 mM Tris-Cl, pH 8.0; 2 mM EDTA; 2.5% (vol/vol) Tween-20; 2.5% (vol/vol) Triton-X 100; 100 μg/mL Proteinase K followed by incubation at 50°C for 60 min and 95°C for 15 min. PCR was performed using 1X Accustart Supermix (Quanta) with the primers: 5’ junction F-5’ TAGAGTCACCCAAGTCCCGT-3’, R-5’- ACTGATTGGCCGCTTCTCCT-3’;; 3’ junction F-5’- GGAGGTGTGGGAGGTTTTT-3’, R-5’- TGATTTCATGACTTGCTGGCT-3’. ROSA TALEN sequences are: TAL FNG NI NI HD HD NG NN NI NG NG HD NG NG NN NN; TAL RHD NN NG NI HD NI HD HD NG NN HD NG HD NI NI NG.
K-592 Flow Cytometry
K-562 cells were assessed for GFP expression every 7 days for 28 days following electroporation. Flow cytometry was conducted on an LSRII instrument (Becton Dickinson) and data was analyzed using FlowJo software v10 (Becton Dickinson). Dead cells were excluded from analysis by abnormal scatter profile and exclusion based on Sytox Blue viability dye (Thermo Fisher Scientific).
Junction PCR to detect targeted integration was conducted using external genomic primers outside of the 48bp homology region and internal primers complementary to the expression cassette. PCR was conducted using Accuprime HIFI Taq (Thermo Fisher Scientific). PCR products from bulk population were sequenced directly.
Quantification and statistical analysis
Statistical analysis was performed using GraphPad Prism software. Data plots represent mean +/- s.e.m. of n independent experiments, indicated in the text. p values were calculated with two-tailed unpaired t-test. Statistical parameters are included in the figure legends.
Data and software availability
The webtool GTagHD was developed to assist users in designing oligonucleotides for targeted integration using the pGTag vector suite. GTagHD guides users through entering: 1) the guide RNA for cutting their cargo-containing plasmid; 2) the guide RNA for cutting their genomic DNA sequence; (3) the genomic DNA sequence, in the form of a GenBank accession number or copy/pasted DNA sequence; and 4) the length of microhomology to be used in integrating the plasmid cargo. If the user is utilizing one of the pGTag series plasmids, GTagHD can also generate a GenBank/ApE formatted file for that plasmid, which includes the user’s incorporated oligonucleotide sequences. GTagHD is freely available online at http://genesculpt.org/gtaghd/ and for download at https://github.com/Dobbs-Lab/GTagHD.
Key resources table


Author Contributions
W.A.W., J.M.W., M.M. and J.J.E conceived the study; W.A.W., J.M.W., M.M. and J.J.E wrote the manuscript with input from S.C.E. and K.J.C; W.A.W., J.M.W., M.P.A., M.E.T., K.C.M.s, Z.M., A.W. and J.A.H. designed and performed the zebrafish experiments; J.M.W. and T.J.W. designed and created the vector suite with input from W.A.W., K.J.C., S.C.E., K.M.K., C.-B.C., D.B., M.M., and J.J.E.; C.M.M. created the web interface with input from J.M.W., W.A.W., C.S.M. and D.L.D.; S.L.S., D.A.W., M.K.V. and D.F.C. designed and performed the pig cell experiments. B.S.M. and B.R.W. designed and performed the human cell experiments.
Declaration of interests
J.J.E., M.M., S.L.S., K.J.C., D.W., and D.F.C. have a financial conflict of interest with Recombinetics, Inc.; J.J.E. and S.C.E. with Immusoft, Inc.; K.J.C. and S.C.E. with ForgeBio and Lifengine; B.S.M. and B.R.W. with B-MoGen Biotechnologies, Inc.
Gene Targeting Protocol for Integrations with pGTag Vectors using CRISPR/Cas9
Targeting strategy (Figure 1):
Selection of a CRISPR/spCas9 target site downstream of the first AUG in the gene of interest
Synthesize sgRNA and spCas9 mRNA
Injection of sgRNA and spCas9 mRNA
Testing for indel production/mutagenesis
Design short homology arms
One Pot Cloning of Homology Arms into pGTag Vectors
Injection of GeneWeld reagents (spCas9 mRNA, Universal sgRNA (UgRNA), genomic sgRNA and pGTag homology vector) into 1-cell zebrafish embryos
Examine embryos for fluorescence and junction fragments

Targeting integration of the pGTag vectors into the 5’ region of a gene. Upon CRISPR/Cas9 targeting and cutting of both the genome (with a sgRNA) and plasmid donor (with UgRNA), the genomic and plasmid DNA likely undergo end resection mediated by the MRN complex and ExoI, resulting in annealing of complementary homology arms. This promotes precise homology-directed integration of cargo DNA at the CRISPR/Cas9 double-strand break.
A. Selection of a CRISPR/spCas9 target site downstream of the first AUG in the gene of interest
To select a CRISPR/Cas9 target site in a 5’ exon, find and download the targeted gene’s genomic and coding sequences.
At <ensemble.org> Search for the gene name of interest for the species of interest and open the Transcript page.
In the left-hand side bar click on “Exons” to find the first coding exon and initiation ATG. If there are alternative transcripts for the gene, make sure there are not alternative initiation ATGs. If there are alternative start codons, target the first 5’ exon that is conserved in all transcripts to generate a strong allele.
Download the transcript and 5’ exon to be targeted as separate sequence files.
Using ApE: <http://biologylabs.utah.edu/jorgensen/wayned/ape/> annotate the coding sequence with the exons.
Use CRISPRScan (http://www.crisprscan.org/) (Moreno-Mateos et al., 2015) to efficiently identify target sites and generate oligos for sgRNA synthesis for the target gene.
Select the “Predict gRNAs” on the lower right-hand side of the home page of the CRISPRScan website.
Paste the 5’ exon sequence into the indicated box. If the exon is very large, start with a small amount of sequence. Ideally exon sequence of ~200 bp near the desired target site. Do not design CRISPRs to intron/exon borders. If there are problems with the copy and pasting of exon sequence, first paste the sequence into a new ape file, save, then copy and paste from the new file.
Select “Zebrafish (Danio rerio)” as the species
Select “Cas9 – nGG” as the enzyme.
Select “In vitro T7 promoter”.
Click on “Get sgRNAs.” Examine the output. The generated targets are ranked by CRISPRScan from high to low. Select a target site (the 20 bp that are capitalized in the oligo column) from those given by CRISPRScan using the following criteria (The best gRNAs will have all of these):
An exact match to the genomic locus., When an oligo is clicked on the page will display additional information to the right. In the section called “Site Type” any mismatches in the oligo are displayed. Exact matches including 5’GG- are ideal for in vitro transcription and 100% genomic target match.
The target is in the desired location of the gene.
The Target is on the reverse (template) strand. Reverse strand guides are more favorable, but either will work
A high CRISPRScan score, and a lower CFD score. However, lower score sgRNA targets may work fine.
Annotate the selected target sequence in the transcript sequence files.
For sgRNA synthesis the entire oligo sequence from CRISPRscan containing the selected target will need to be synthesized. This oligo is represented as “Oligo A” in Figure 2.
 Figure 2.
Figure 2.Cloning-free gRNA synthesis. Oligo A is composed of the T7 promoter at the 5’ end, target sequence for gRNA, and gRNA overlap sequence for gRNA synthesis. CRISPRScan provides direct output for Oligo A. The strategy for gRNA production using Oligo A is based on (Varshney et al., 2015).
Alternative to CRISPRScan: Designing “CRISPR Oligo A” from a genomic target sequence. Skip this section if Oligo A was designed with CRISPRScan.
If the target sequence was identified using tools other than CRISPRScan, Oligo A can be designed manually. (Note: CRISPRScan will use a shorter overlap region but this does not affect template production). Add T7 and Overlap sequences (see Figure 2) to the 20 bp of target sequence without the PAM. Oligo A for the targeted gene will look like the example below:
5’-TAATACGACTCACTATAGGNNNNNNNNNNNNNNNNNNGTTTTAGAGC TAGAAATAGC-3’
The sequences in blue (first 17 characters) are the T7 promoter, the grey GG are part of the T7 promoter and ideally are part of the target sequence (see below), the Ns are the target sequence, and the sequence in green (last 20 characters) are the overlap region to synthesize the non-variable part of the sgRNA. The T7 promoter works optimally with the two grey GGs, however, these GGs will be transcribed by T7 and thus become a part of the sgRNA. Target sequences that contain the GGs may work better, but there are differing reports in the literature on the importance of this (Moreno-Mateos et al., 2015). If possible, select a target that starts with GG. Refer to Moreno-Mateos et al., 2015 for other gRNA architectures with variations on the 5’GG motif.
If the target sequence did not have two Gs at the beginning, additional G’s will need to be added to the start of the target sequence for efficient transcription as outlined below:
* The lower case ‘g’ is an extra ‘G’ not in the genomic sequence; the upper-case G is in the genomic. Lower case gs will not base pair with the genomic target.
without GG: ggN NNN NNN NNN NNN NNN NNN N (22 bp) – 2 bases are added,
with one G: gGN NNN NNN NNN NNN NNN NNN (21 bp) – one base is added, G is part of the target sequence.
with two G: GGN NNN NNN NNN NNN NNN NN (20 bp) – no bases are added, GG is part of the target sequence.
Oligo A is made by taking this target sequence with 5’GG and pasting it into a clean file.
Copy and paste the T7 promoter sequence to the 5’ end of the target sequence:
TAATACGACTCACTATA
Copy and paste the Overlap sequence to the 3’ end of the target sequence:
GTTTTAGAGCTAGAAATAGC
Check the sequences to ensure they are correct and that the PAM is NOT present in this oligo.
Oligo B design (Figure 2) contains the conserved guide RNA sequence: All Oligo Bs will be the same and can be ordered in large quantities.
5’-GATCCGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTT AACTTGCTATTTCTAGCTCTAAAAC-3’
To increase yield of the sgRNA synthesis the primers “T7 primer” (5’-TAATACGACTCACTATA-3’) and “3’gRNA primer” (5’- GATCCGCACCGACTCGGTG-3’) are also required.
For checking for mutagenesis at the target site, design ~20 bp DNA primers for PCR amplification to amplify at least 130 bp of DNA surrounding the target site. Mutagenesis is estimated through comparison of PCR products from injected and uninjected embryos, by visualizing small insertions and/or deletions (Indels) using electrophoresis, or by sequencing.
Primer 3 is used for primer design: http://biotools.umassmed.edu/bioapps/primer3_www.cgi
Paste DNA sequence surrounding the target site into the web interface. It is recommended to use 160 – 300 bp of exon sequence centered on the cut site for primer design. Intron sequence can be used, but this often contains polymorphisms that can lead to amplification failure.
Locate the target sequence, including the PAM sequence (italicied below), and predict the cut site (3 bp into the target sequence from the PAM represented here by the ‘x’). Mark the targeted exon sequence approximately 65-150 bp on both sides of the cut site by putting [square brackets] around it. Primer3 will design primers outside this sequence. This design allows the primers to be used for both checking of mutagenesis and for junction fragment analysis when checking for integration.
Example: CGGCCTCGGGATCCACCGGCC[AGAATCGATATACTACGATGAAC AGAGCAAATTT GTGTGTAATACGGTCGCCACCATGGCCTxCCTCGGTTTGCTACGATGCATTTGCAC CACTCTCTCATGTCCGGTTCTGGG]AGGACGTCATCAAGGAGTTCATGCGCTTCAA GGTGCGCATGGAGGGCTCCGTGAAC
Set the “Primer Size” variables to Min = 130, Opt = 170, and Max = 300. Everything else can be left at the defaults.
Click on “Pick Primers”
Select primers from the output. Note the “product size” expected and the “tm” or melting temperature of each primer/pair. Smaller product sizes are easier to visualize mutagenesis.

B. Synthesize the sgRNA
General guidelines and good laboratory practices for working with DNA and RNA. DNA, RNA and the enzymes are sensitive to contamination from dust and skin. Following these guidelines will prevent the degradation of the DNA and RNA you are trying to make:
Be clean. Clean the workbench, pipetmen, racks, and centrifuges with RNase Away or something equivalent.
Wear gloves and change when contaminated. Contamination will occur when gloves contact hair, face, skin, or the floor.
Keep everything on ice unless the protocol indicates otherwise.
Centrifuge components to the bottom of the tube before use, after mixing, after use, and after incubation steps.
Do not vortex enzymes. Gently flick the tube or pipet up and down to mix samples.
Avoid touching the walls of the tube when pipetting.
Use a new pipette tip for each new dip.
Dispense solutions from a pipet to the bottom of the tube, or into the liquid at the bottom of the tube when setting up reactions.
Only remove 1.5 ml centrifuge tube and PCR tubes from their package while wearing gloves. Reseal the tube package after tubes are removed.
Assembly of CRISPR Oligos A + B into a Transcription Template
For synthesis of the gRNA from Oligo A and B, make a 100 μM freezer stock and 1 μM working stock for each oligo. All oligos are described in Section A starting on page 7.
Centrifuge ordered oligos briefly before opening, to move all dried DNA flakes to the bottom of the tube.
Add a volume (x μL) of RNase-free water to make a 100 μM stock. The tubes should be labeled with the gene name as well as the number of nmol in the tube. The amount of water to be added will need to calculated based on the nanomoles of material contained within.
Vortex for 30 seconds.
Centrifuge briefly.
Make a 100-fold dilution of each 100 μM stock Oligo A and B in separate 1.5 ml tubes.
Label one 1.5 mL centrifuge tube per Oligo A with name of oligo, date, and “1 μM” to indicate working stocks.
1 μL of 100 μM Oligo A stock or Oligo B
99 μL of RNF-water
100 μL total
Vortex.
Briefly centrifuge.
Store all stocks in freezer at −20 °C for long-term storage.
Set up the following reaction in PCR tubes. The next two steps will generate a short segment of DNA (gDNA or guideDNA Template) which will be used as a template for synthesis of RNA:
12.5 μL 2X KOD Master Mix 1
μL Oligo A (1 μM)
1 μL Oligo B (1 μM)
1 uL T7 primer (10 μM)
1 uL gRNA 3’ primer (10 μM)
8.5 μL RNF-water
25 μL total
Run PCR under the following conditions:
Denature at 98°C for 2 minutes
Denature at 98 °C for 30 sec.
Anneal at 50 °C 30 sec.
Extend at 70 °C 30 sec.
Go to (step 2) nine times.
Extend at 70 °C 2 min
Hold 4 °C forever.
Run 1.2% agarose gel in 1X TAE to check that the template was synthesized:
Remove 3 μL of the reaction and place in a 1.5 ml tube.
Mix in 1 μL of 6x loading buffer.
Load all 4 μL of the sample on the gel. Run the gel at 125 V for 30 minutes. Be sure to load a molecular weight marker.
Check on the transilluminator and image the gel.
A single 120 bp band should be detected when 3 μL is loaded on gel.
In vitro transcription (IVT) using the gRNA template
Use the Ambion T7 Megascript Kit for transcription reagents, but follow the instructions below.
Thaw the T7 10X Reaction Buffer and RNF-water at room temperature, and thaw the ribonucleotides solutions on ice.
Vortex the T7 10X Reaction Buffer to make sure all DTT is solubilized. No white flecks should be visible.
Microcentrifuge all reagents briefly before opening to prevent loss of reagents and/or contamination by materials that may be present around the rim of the tube(s).
Keep the T7 Enzyme Mix on ice or in a −20 °C block during assembly of the reaction.
Make a master mix for each reaction. Assemble the reaction at room temperature on the bench. Add reagents from largest to smallest volume, adding the 10X Reaction Buffer second to last and the T7 Enzyme Mix last.
Note: Components in the transcription buffer can lead to precipitation of the template DNA if the reaction is assembled on ice. If the reaction precipitates, the synthesis reaction will not fully occur.
Reagent list:
10 μL of RNF-water
5 μL of gDNA template (100 to 500 ng total) 4 μL of NTP (1 μl of each;; A, U, C, G)
1 μL of 10x transcription buffer – must be fully resuspended at room temp
1 μL of T7 polymerase enzyme mix
Incubate at 37 °C for 4 to 16 hours. Longer incubations result in considerably better yields.
Add 1 μL of Turbo DNAse and incubate for 15 min at 37 °C. This will digest the template DNA in the sample.
Optional quality control step: Run 2 μL of sample on a 1.2% gel in 1X TAE.
Clean the gel box, comb and tray with RNase Away, rinse with DI water.
Remove 2 μl of sample into a clean 1.5 ml (Keep RNA on ice!)
Add 3 μL of RNF-water and 5 uL of Ambion RNA loading buffer with formamide.
Vortex briefly.
Spin down samples briefly.
Run all of this mixture on a 1.2% agarose gel/1X TAE, at 100 V for 1 hour.
Image gel. 2 bands should be visible at ~100 and 120 bp.
Purification of guide RNA
Use the miRNeasy Qiagen kit for purification of gRNAs according to the manufacturer’s instructions.
After Purification verify presence of RNA by running a 1.2% gel in 1X TAE.
Clean the gel box, comb and tray with RNase Away, rinse with DI water. Run on a 1.5% agarose gel/1X TAE, at 100 V for 1 hour as above.
Image gel. 2 bands should be visible at ~100 and 120 bp.
Nanodrop the RNA sample to determine the concentration.
Store RNA at −20 °C.
Preparation of SpCas9 mRNA
Digest ~5-10 μg pT3TS-nCas9n plasmid with Xba1 (plasmid Addgene #46757 (Jao et al., 2013)).
Purify digested DNA with Qiagen PCR cleanup kit or Promega PureYield Plasmid Miniprep System. Elute in RNF-water.
Run 100-500 ng on 1.2% agarose gel in 1X TAE to confirm the plasmid is linearized.
Use 100 ng to 1 μg DNA as template for in vitro transcription reaction.
Use mMESSAGE mMACHINE T3 kit Life Technologies (AM1348) and follow the instructions in the manual.
Use the miRNeasy Qiagen kit for purification of nCas9n mRNA according to the manufacturer’s instructions.
Verify mRNA integrity by mixing 1 uL of purified Cas9, 4 μL of RNF water, 5 μL glyoxl dye (Ambion).
Heat mixture at 50 °C for 30 minutes, then place on ice.
Clean the gel box, comb and tray with RNase Away, rinse with DI water.
Run all 10 μL of RNA mixture on 1.2% agarose gel in 1X TAE at 100 V for 1 hour as above. One band should be visible at 4.5 kb.
Nanodrop the RNA sample to determine the concentration. Concentrations between 0.45 and 1 μg/μL are expected.
Aliquot and store RNA at −80 °C.
C. Injection of sgRNA and spCas9 mRNA
The injections here are designed to deliver 25 pg of gRNA and 300 pg of Cas9 mRNA in 2 nL of fluid to embryos at one-cell stage.
Injection trays are cast with 1.2% agarose with 1X embryo media (Zebrafish Book;; zfin.org) in polystyrene petri dishes (Fisher No. FB0875713). Injection trays can be used multiple times and stored at 4*C for up to three weeks between use.
Trays are pre-warmed to 28.5 °C prior to injection by placing them in the 28.5 °C incubator. Try to mitigate tray cooling while not in use.
Glass needles are pulled from Kwik-Fil borosilicate glass capillaries (No. 1B100-4) on a Flaming/Brown Micropipette puller (Model P-97).
Injection samples are made to contain the following diluted in RNF water or injection buffer (final concentration: 12.5 mM HEPES pH 7.5, 25 mM Potassium Acetate, 37.5 mM Potassium Chloride, 0.0125 % glycerol, 0.025 mM DTT ph 7.5)
12.5 ng/μL of genomic gRNA
150 ng/μL of mRNA for Cas9
Needles are loaded with 1.5 to 2.5 μL of sample, and then loaded onto a micro-manipulator attached to a micro injector (Harvard Apparatus PLI - 90) set to 30-40 PSI with an injection time of 200 msec.
Needles are calibrated by breaking the end of the tip off with sterile tweezers, ejecting 10 times to produce a droplet of fluid, collecting the droplet into a 1 μL capillary tube (Drummond No. 1-000-0010), and measuring the distance from the end of the capillary to the meniscus of the droplet. This distance is converted to volume (where 1 mm = 30 nL) and adjusted to achieve an effective injection volume of 2 nL. Volumes are adjusted by changing the injection time. There is a linear relationship between volume and time at a set pressure. Avoid injection times less than 100msec and over 400 msec.
One cell embryos that have been collected from mating cages are pipetted from collection petri dishes to the wells on the injection tray.
Use the micro-manipulator and microscope to pierce the one-cell of embryos on the injection tray at an angle of 30° with the needle. Inject 2 nL of sample in the one-cell near the center of the cell-yolk boundary.
After embryos have been injected, wash them from the injection tray into a clean petri dish with embryo media.
Keep 20 - 40 embryos separate as uninjected controls. Treat and score the control embryos in the same way as the injected embryos.
At 3 - 5 hrs post injection remove any unfertilized or dead embryos from the dishes. This will prevent death of the still developing embryos.
D. Testing for indel production/mutagenesis
Phenotypic scoring of embryos
The gRNA itself may be toxic to the developing embryos. Injection toxicity can be estimated by the number dead embryos from a round of injection compared to the un-injected control dish. Count and remove any brown/dead embryos from injected and un-injected dishes. If there are significantly more dead embryos in the injected dish then the guide may be toxic, impure, or very effective at disrupting a required gene. Reducing the amount of guide or Cas9 mRNA injected may help reduce toxicity.
Score and document embryonic phenotypes on days 1 - 4 post fertilization (dpf). Under a dissection microscope examine the un-injected controls and injected embryos, and sort the embryos into categories.
Scoring categories
○ Severe-These embryos have some parts that look like a control embryos, but are missing key features. Examples: embryos that lack their head, eyes, or tail, or embryos that have an unnaturally contorted shape or are asymmetric.
○ Mild-These embryos appear mostly normal, but have slight defects such as small eyes, pericardial edema, shortened trunk/tail, or curled/twisted tails.
○ Normal-appears normal and similar to controls.
Digestion of embryos for isolation of genomic DNA for mutagenesis analysis
Genomic DNA (GDNA) can be isolated from zebrafish embryos aged between 1 and 5 dpf using this protocol. Embryos can be analyzed as individuals or as pools (maximum 5) from the same injection.
Dechorionate embryos, if they have not emerged from the chorion.
It is recommended to screen a minimum of 3 embryos from each scoring category for mutagenesis. Place each embryo, including controls, into separate PCR tubes. Remove as much of the fish water as possible. If needed, spin briefly and remove additional water.
Add 20 μL of 50 mM NaOH per embryo.
Heat the embryos at 95°C in a thermocycler for 15 minutes.
Vortex samples for 10 seconds. Be sure that the tubes are sealed to prevent sample loss while vortexing.
Spin samples down and heat for an additional 15 min at 95 °C in a thermocycler.
Vortex samples and then spin the tubes down again. The embryos should be completely dissolved.
Neutralize the samples by adding 1 μL of 1 M Tris pH 8.0 per 10 μL NaOH. Mix by vortexing then spin down.
Genomic DNA should be kept at 4 oC while in use and stored at −20°C.
Analysis of CRISPR/Cas9 mutagenesis efficiency at targeted gene locus
Set up the following PCR reactions for each tube of digested embryos using the primers designed at the end of section A, page 10.
12.5 μL of 2x GoTaq Mastermix
1 μL of Forward Primer (10 uM)
1 μL of Reverse Primer (10 uM)
1 μL of gDNA template (digested embryos)
9.5 μL of nuclease-free water
25 μL total
Vortex and briefly spin down the PCR reactions.
Run the following PCR program to amplify the targeted locus. 95°C 2 minutes 95°C 30 seconds] 55°C* 30 seconds] × 35 cycles 72°C 30 seconds] 72°C 5 minutes 4°C hold
* if the primers were designed with higher or lower tm’s than the annealing temperature in line three, then that temperature will need to be adjusted to 2°C below the designed primer tm.
Run up to 7 μL of PCR product on a 3.0% agarose gel, 1X TAE, for 1 hr at 80-100V.
Analyze the gel for DNA bands that appear diffuse or different in size from the control lane. This indicates that the presence of indels in the gene of interest
Alternatively clone and sequence PCR products or sequence them directly to verify the presence of indels.
E. Design short homology arms
Homology directed gene targeting allows the integration of exogenous DNA into the genome with precision to the base pair level. However, designing and cloning individual targeting vectors and homology arms for each gene of interest can be time consuming. The pGTag vector series provides versatility for ease of generation of knockout alleles (Figure 3). The vectors contain BfuAI and BspQI type II restriction enzymes for cloning of short homology arms (24 or 48 bp) using Golden Gate cloning. The pGTag vectors require in-frame integration for proper reporter gene function. The reporter gene consists of several parts. First, a 2A peptide sequence causes translational skipping, allowing the following protein to dissociate from the locus peptide. Second and third, eGFP, TagRFP, or Gal4VP16 coding sequences for the reporter protein have a choice of sequence for localization domains, including cytosolic (no) localization, a nuclear localization signal (NLS), or a membrane localization CAAX sequence. Finally translation is terminated by one of two different polyadenylation sequences (pA); a β-actin pA from zebrafish or the SV40pA.
For many genes, the signal from integration of the report protein is too weak to observe. In these cases the Gal4VP16 vector allows for amplification of the report to observable expression levels in F0s and subsequent generations. A 14XUAS/RFP Tol2 plasmid is provided to make a transgenic line for use with the Gal4VP16 vector.
Sequence maps for these plasmids can be downloaded at www.genesculpt.org/gtaghd/
The pGTag vectors allow one step cloning of homology arms.
All vectors can be obtained through Addgene (www.addgene.org). Because the pGTag plasmids contain repeated sequences, they may be subject to recombination in certain strains of bacteria. It is strongly recommended that they are propagated at 30°C to reduce the possibility recombination.
The web tool, GTagHD www.genesculpt.org/gtaghd/, allows for quick design of cloning ready homology arm oligos for a gene of interest.
To use the tool, choose the "Submit Single Job" tab. Follow the instructions in the tab.
There should be 4 oligos (two pairs that will be annealed) generated that should be ordered for cloning. If there are any problems with the sequences and values that were entered, the web page will display the errors and give advice on how to fix them.
The following protocol describes how to design homology arm oligos manually:
*Note* In the following section when orientation words are used, they are used in the context of the reading frame of the genetic locus of interest. Example: A 5’ template strand CRISPR means that the target site for the CRISPR is on the template strand at the locus and is toward the 5’ end of the gene. Upstream homology domains are 5’ of the CRISPR cut and downstream homology domains are 3’ of the cut with respect to the gene being targeted. Also note: Upper case and lower case bases are not specially modified; they are typed the way they are as a visual marker of the different parts of the homology arms.
For the Upstream Homology Domain
1) Open the sequence file for the gene of interest and identify the CRISPR site. (In this example it is a Reverse CRISPR target in Yellow, the PAM is in Orange, coding sequence is in purple)
Copy the 48 bp 5’ of the CRISPR cut (the highlighted section below) into a new sequence file; this is the upstream homology.
2) Observe the next three bases immediately upstream of the 48 bp of homology, and pick a base not present to be the 3 bp spacer between the homology and the Universal PAM in the vector. (Here the three bases are “GGA” so “ccc” was chosen for the spacer) Add the spacer to the new file 5’ (in front) of the homology, see below. The spacer acts a non-homologous buffer between the homology and the eventual 6 bp flap from the universal guide sequence that will occur when the cassette is liberated and may improve intended integration rates over MMEJ events.
3) Determine where the last codon is in the homology. Here the 3’ G in the homology domain is the first base in the codon cut by this CRISPR target. Complete the codon by adding the remaining bases (called padding on GTagHD) for that codon from your sequence to ensure your integration event will be in frame.
4) Add the BfuAI enzyme overhang sequences for cloning, to the ends of the homology domain. 5’-GCGG and 3’-GGAT. (Here both overhangs are added to prevent errors in copying sequence for the oligos in the next two steps.)
5) The Upstream Homology Oligo A will be this sequence from the beginning to the end of the last codon (see highlighted below). Copy and paste this sequence into a new file and save it. In this example this oligo sequence is 5’-GCGGcccGTTTTCTTACGCGGTTGTTGGATGAAATCTCCAACCACTCCACCTTCGtg-3’.
6) The Upstream Homology Oligo B will be the reverse compliment of this sequence from beginning of the spacer to the end of the sequence (see highlighted below). Copy the reverse compliment, paste it into a new file, and save it. In this example this oligo sequence is 5’-ATCCcaCGAAGGTGGAGTGGTTGGAGATTTCATCCAACAACCGCGTAAGAAAACggg-3’.

For the Downstream Homology Domain
7) Open sequence file for the gene of interest and identify the CRISPR site. (Reverse CRISPR target in Yellow, PAM in Orange, coding sequence is in purple) Copy the 48 bp 3’ of the CRISPR cut into a new sequence file; this is the downstream homology.
8) Observe the next three bases downstream of the 48 bp of homology, and pick a base not present to be the 3 bp spacer between the homology and the Universal PAM in the vector. (Here the bases are “CTG” so “aaa” was chosen for the spacer.) Add the spacer to the new file 3’ of (after) the homology.
9) Add the BspQI enzyme overhang sequences for cloning, to the ends of the homology domain. 5’-AAG and 3’-CCG. (Here both overhangs are added to prevent errors in copying sequence for the oligos in the next two steps.)
10) The Downstream Homology Oligo A will be this sequence from the beginning of the sequence to the end of the spacer (see highlighted below). In this example this oligo sequence is 5’-AAGTGGGCAAGATATGGCTCACGTTATTCATCATCTTCCGCATTGTTTTGAaaa-3’.
11) The Downstream Homology Oligo B (will be the reverse compliment of this sequence from the beginning of the homology to the end of the sequence (see highlighted below). In this example this oligo sequence is 5’-CGGtttTCAAAACAATGCGGAAGATGATGAATAACGTGAGCCATATCTTGCCCA-3’

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
F. One Pot Cloning of Homology Arms into pGTag Vectors
**Note if the homology arm oligos contain either the sequence “5’-ACCTGC-3’” or “5’-GAAGAGC-3’” (or their compliments) the cloning reaction will be less efficient.
*Note some sequences just don’t work very well. Ligation is more efficient with annealed homology arms and the purified ~1.2 kb and ~2.4kb fragments from vectors that have been digested with BfuAI and BspQI. If problems are encountered, one homology arm can also be cloned sequentially.
Homology Arm Annealing
Anneal upstream and downstream homology oligo pairs separately:
4.5 μL oligo A at 10 uM
4.5 μL oligo B at 10 uM
4 μL 10x Buffer 3.1 from NEB
27 μL dH20
total = 40 μL
Incubate at 98°C for 4 min, 98°C 45 sec x 90 steps decrementing temp 1°C/cycle, 4°C hold
(Alternatively heat in 95-98°C water for 5 minutes, and then remove the boiling beaker from the heat source and allow it to cool to room temp for 2 hours, before placing samples on ice.)
1-Pot Digest
Assemble the following:
4.0 μL dH2O
2 μL Plasmid at 50 ng/uL
1 μL 10x Buffer 3.1 from NEB
1 μL 5’ annealed homology arm
1 μL 3’ annealed homology arm
0.5 μL BfuAI enzyme from NEB
0.5 μL BspQI enzyme from NEB
10 uL total
Incubate at 50°C for 1 hr, place on ice.
Ligation
Add the following:
3 uL 5x T4 quick ligase buffer
1.5 uL dH2O
0.5 uL T4 quick ligase
15 uL total
Incubate 8-10 min at room temperature (to overnight). Store at −20 °C,
Transformation
On ice, thaw 1 (one) vial competent cells (50 μL) for every 2 ligation reactions. (approx. 5 min). It is recommended to use NEB Stable Competant E. coli (C3040H) cells to limit recombination.
While cells are thawing, label the microcentrifuge tubes for each ligation and put on ice.
Once the cells are thawed, use a pipette to transfer 25 μL of the competent cells into each labeled tube.
Add 1.5 μL of a ligation reaction into competent cells to transform.
Amount of ligation reaction added should be less than 5% of volume of competent cells.
Mix by tapping the tube several times or gently mixing with the pipet tip.
Do NOT mix by pipetting, this will lyse the cells.
Incubate on ice for 5 to 20 minutes.
Heat shock the cells by submerging the portion of the tube containing the cells in a 42°C water bath for 40 - 50 seconds.
Incubate cells on ice for 2 minutes.
Add 125 μL of room temperature LB to each transformation.
Incubate cells at 30°C for 1- 1.5 hour(s) in a shaking incubator.
While the transformed cells are recovering, spread 40 μL of X-Gal solution, and 40 μL IPTG 0.8 M on LB Kanamycin selection plates.
X-Gal is lethal to cells while wet, it is recommended to first label the plates and then place them in a 30°C incubator to dry.
After recovery and the X-Gal is dry, Plate 150 μL of each transformation on the corresponding correctly labeled plate.
Incubate plates overnight at 30°C.
Growing colonies
Pick 3 white colonies from each plate and grow in separate glass culture tubes with 3 mL LB/Kanamycin.
Or to pre-screen colonies by colony PCR:
Pick up to 8 colonies with a pipet tip and resuspend them in separate aliquots of 5 μL dH2O. Place the tip in 3 ml of LB/Kan, label, and store at 4°C.
Make a master mix for your PCR reactions containing the following amounts times the number of colonies you picked.
7.5 μL 2x Gotaq mastermix
5.5 μL dH2O
0.5 μL primer at 10 uM “F3’-check” 5’-GGCGTTGTCTAGCAAGGAAG −3’
0.5 μL primer at 10 uM “3’_pgtag_seq”5’-ATGGCTCATAACACCCCTTG-3’
14 μL total
Aliquot 14 μL of mixed master mix into separate labeled PCR tubes.
Add 1 μL of colony to each reaction as template.
or 20 ng purified plasmid as control.
Cycle in a thermocycler 95°C 2 minutes 95°C 30 seconds] 57°C 30 seconds] × 35 cycles 72°C 30 seconds] 72°C 5 minutes 4°C hold
Run 5 μL of PCR product on a 1% agarose gel. You should get bands that are a different size than the control.
Mini Prep Cultures
Follow Qiagen Protocol
Sequencing of Plasmids
The 5’ homology arm can be sequenced by the 5’_pgtag_seq primer:
5’-GCATGGATGTTTTCCCAGTC-3’
The 3’ homology arm can be sequenced with the “3’_pgtag_seq”primer:
5’-ATGGCTCATAACACCCCTTG-3’.
G. Injection of GeneWeld Reagents (spCas9 mRNA, Universal sgRNA (UgRNA), genomic sgRNA and pGTag homology vector) into 1-cell zebrafish embryos
Prepare and collect the following reagents for injection
Prepare nCas9n mRNA from pT3TS-nCas9n (Addgene #46757 from (Jao et al., 2013)) as described above (page 14).
Synthesize UgRNA and purify as described above (page 11) using the following oligo A:
5’-TAATACGACTCACTATAGGGAGGCGTTCGGGCCACAGGTTTTAGAGCTAGAAATAGC-3’
Corresponding to the universal target sequence: GGGAGGCGTTCGGGCCACAG
Alternatively, the UgRNA can be directly ordered form IDT and resuspended in RNF water.
5’-GGGAGGCGUUCGGGCCACAGGUUUUAGAGCUAGAAAUAGCAAGUUAAAAUAAGG CUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCGGAUC-3’
The pGTag homology vectors should be purified a second time prior to microinjection under RNase free conditions with the Promega PureYield Plasmid Miniprep System beginning at the endotoxin removal wash and eluted in RNF water.
Embryo Injections for Integration of pGTag vectors
Injections are performed as previously described in 2 nl per embryo with the addition of the UgRNA and targeting pGTag DNA.

H. Examine embryos for fluorescence and junction fragments
Embryos are examined for fluorescence under a Zeiss Discovery dissecting microscope with a 1X objective at 70-100X magnification. If weak signals are observed, embryos are manually dechorionated, and viewed on glass depression well slides. If no or weak signals were observed, Gal4VP16 integrations are attempted in a 14XUAS-RFP background. Embryos displaying widespread fluorescence in expression domains consistent with the targeted gene are examined for junction fragments or raised to adulthood for outcrossing.
F0 Junction fragment analysis between the genomic locus and the targeting vector is carried out by isolating DNA from embryos followed by PCR. The following primers are used for junction fragment analysis and must be paired with gene specific primers (5’ to 3’):

PCR amplification of junction fragments can be a result of artifacts (Won and Dawid, 2017), so it is important to carryout control amplifications with injected embryos that lack the genomic gRNA. F0 analysis by PCR of junction fragments is carried out to examine correct targeting. F-Gal4-3’juncM and F-Gal4-3’juncJ are two alternate primers for amplification of junction fragments from the Gal4 cassette due to gene specific mis-priming depending on the target loci.
7.5 μL 2x Gotaq mastermix
5.5 μL dH2O
0.5 μL primer at 10 uM genomic primer
0.5 μL primer at 10 uM pGTag primer
14 μL total
Aliquot 14 μL of mixed master mix into separate labeled PCR tubes.
Add 1 μL of genomic DNA to each reaction as template.
Cycle in a thermocycler with the following steps: 95°C 2 minutes 95°C 30 seconds] 55°C 30 seconds] x 35 cycles 72°C 30 seconds] 72°C 5 minutes 4°C hold
Run 5 μL of PCR product on a 1.2 % agarose gel in 1XTAE. Putative junction fragments should give bands that are of predicted size.
F0 animals that are positive for the reporter gene are raised to adults then outcrossed and examined for fluorescence as above. The Gal4VP16 system can lead to silencing resulting in mosaic patterns in F1 embryos. F1 embryos displaying fluorescence are examined for junction fragments as above, raised to outcross to make F2 families or sacrificed at 3 weeks post fertilization for Southern-Blot analysis of integrations. F0 and F1 identified fish can be incrossed or backcrossed to get an initial impression of the homozyogous phenotypes. It is recommended that lines are continuously outcrossed once established.
Acknowledgements
The following NIH grants supported this work: 5R24OD020166 to J.J.E., M.M., D.L.D, K.J.C, and S.C.E.; GM088424 to J.J.E.; and GM63904 to S.C.E.
References




