

ABSTRACT
Streptococcus pneumoniae is a leading killer of infants and immunocompromised adults and has become increasingly resistant to major antibiotics. Therefore, the development of new antibiotic strategies is desperately needed. Targeting bacterial cell division is one such strategy, specifically targeting essential proteins for the synthesis and breakdown of peptidoglycan. One complex important to this process is FtsEX. FtsEX comprises an integral membrane protein (FtsX) and cytoplasmic ATPase (FtsE) that resembles an ATP-binding cassette (ABC) transporter. Here, we present NMR solution structural and crystallographic models of the large extracellular domain of FtsX, denoted ECL1. The structure of ECL1 reveals an upper extended β-hairpin and a lower α-helical lobe, each extending from a mixed α-β core. The helical lobe mediates a physical interaction with the peptidoglycan hydrolase PcsB, via the coiled-coil domain of PcsB (PcsB-CC). Characterization of S. pneumoniae D39 derived strains harboring mutations in the α-helical lobe shows that this subdomain is essential for cell viability and required for proper cell division of S. pneumoniae.
IMPORTANCE FtsX is a ubiquitous bacterial integral membrane protein involved in cell division that regulates the activity of peptidoglycan (PG) hydrolases. FtsX is representative of a large group of ABC3 superfamily proteins that function as “mechanotransmitters,” proteins that relay signals from inside to the outside of the cell. Here we present a structural characterization of the large extracellular loop (ECL1) of FtsX from the human opportunistic pathogen Streptococcus pneumoniae. We show a direct interaction between the peptidoglycan hydrolase PcsB and FtsX, and demonstrate that this interaction is essential for cell viability. As such, FtsX represents an attractive, conserved target for the development of new classes of antibiotics.
INTRODUCTION
Streptococcus pneumoniae is a Gram-positive, opportunistic respiratory pathogen (1–3) that has acquired antibiotic resistance worldwide (4–6). This ovococcal bacterium relies on highly conserved cell wall machinery to divide and grow (7, 8). The cell wall is primarily composed of peptidoglycan (PG), a macromolecule composed of repeating subunits of N-acetylglucosamine and N-acetylmuramic acid linked by PG peptide side chains (9, 10). Regulation of the synthesis and remodeling of PG is essential for bacterial growth and viability, due to the turgor pressure bacterial cells must withstand (10–12). One vital process for the synthesis of PG is the controlled insertion of new strands of PG. This process requires timed cleavage of the old PG matrix to allow incorporation of new nascent strands (13). PG hydrolases are the primary enzymes that carry out PG cleavage and remodeling (14, 15). Thus, regulation of these hydrolases and activation at specific times during the cell cycle is required for proper cell growth. Specific protein complexes are utilized by bacterial cells to regulate these enzymes. This work focuses on understanding the structure and function of one of these protein complexes.
From Mycobacterium tuberculosis to Caulobacter crescentus, the ABC-transporter-like protein complex FtsEX acts as a key regulator of PG hydrolysis and divisome assembly (16–19). The proposed mechanism of FtsEX activation of PG hydrolases is as follows. FtsE, upon sensing an unknown signal from inside the cell, hydrolyzes ATP to ADP. Hydrolysis causes a conformational change transmitted through the membrane via FtsX, an integral membrane protein with two extracellular loops, denoted the large (ECL1) and small (ECL2) loops. These extracellular loops interact with either the cell wall hydrolases or with effector proteins, which results in activation of PG hydrolysis via an unknown mechanism (16, 18, 20–25). In E. coli, it has been demonstrated that FtsX interacts with the effector protein EnvC to activate the PG amidases AmiA and AmiB (24, 25). In addition, FtsX interacts with other division proteins such as FtsA, where it regulates the polymerization of FtsA and recruitment of downstream division proteins (26). In other organisms including B. subtilis and M. tuberculosis, FtsEX also activates PG hydrolases (16, 23). Interestingly, FtsEX is non-essential in rod-shaped bacteria such as E. coli and B. subtilis (23, 24, 26–28). However, in S. pneumoniae FtsEX is absolutely essential (21) and depletion of FtsEX results in cell rounding and cessation of growth (20, 21).
In the case of S. pneumoniae, genetic experiments suggest that both outward-facing domains of FtsX, ECL1 and ECL2, interact with the essential PG hydrolase PcsB, via its long coiled-coil (CC) domain (20, 21). However, there is little direct biochemical evidence for this interaction. ECL1 and ECL2 are thought to allosterically activate the catalytic activity of cysteine, histidine-dependent amidohydrolase/peptidase (CHAP) domain of PcsB (20). The crystal structure of full-length PcsB including the CC domain, an alanine-rich linker region, and the CHAP domain provides insight into the mechanism of how this may occur (22). While the PcsB structure implies FtsEX activates PcsB by displacing the catalytic domain from the CC domain, the exact nature of the FtsX::PcsB interaction remains unknown.
In order to understand how FtsX activates PcsB, we determined the structure of the large extracellular loop of FtsX (FtsXECL1) by both multidimensional NMR spectroscopy and X-ray crystallography. FtsXECL1 harbors a conserved mixed α-β core and a lower α-helical lobe extending from the core identified previously in M. tuberculosis FtsX (16), and an extended β-hairpin that is unique to S. pneumoniae FtsXECL1. The N-terminal β1 and C-terminal β6 strands are adjacent in the core and connect ECL1 to the TM1 and TM2 helices, respectively, in the membrane. PcsB-CC-mediated chemical shift perturbations of 1H-15N HSQC spectra of FtsXECL1 reveal that the helical lobe consisting of the α2 helix and the α2-β5 linker (residues 107–134) of FtsXECL1 interacts with PcsB-CC. To determine if this interaction is required for FtsX function in bacterial cells, we constructed a merodiploid strain that allows for conditional expression of the mutant ftsX. We demonstrate that specific amino acid substitutions in the FtsX-PcsB interface are lethal or cause pronounced morphological defects despite the fact these FtsXECL1 mutant proteins are expressed at near wild-type levels. These findings support the model that a direct physical interaction between FtsX and PcsB is required for activation of PcsB PG hydrolytic activity.
RESULTS
The three-dimensional structure of FtsXECL1
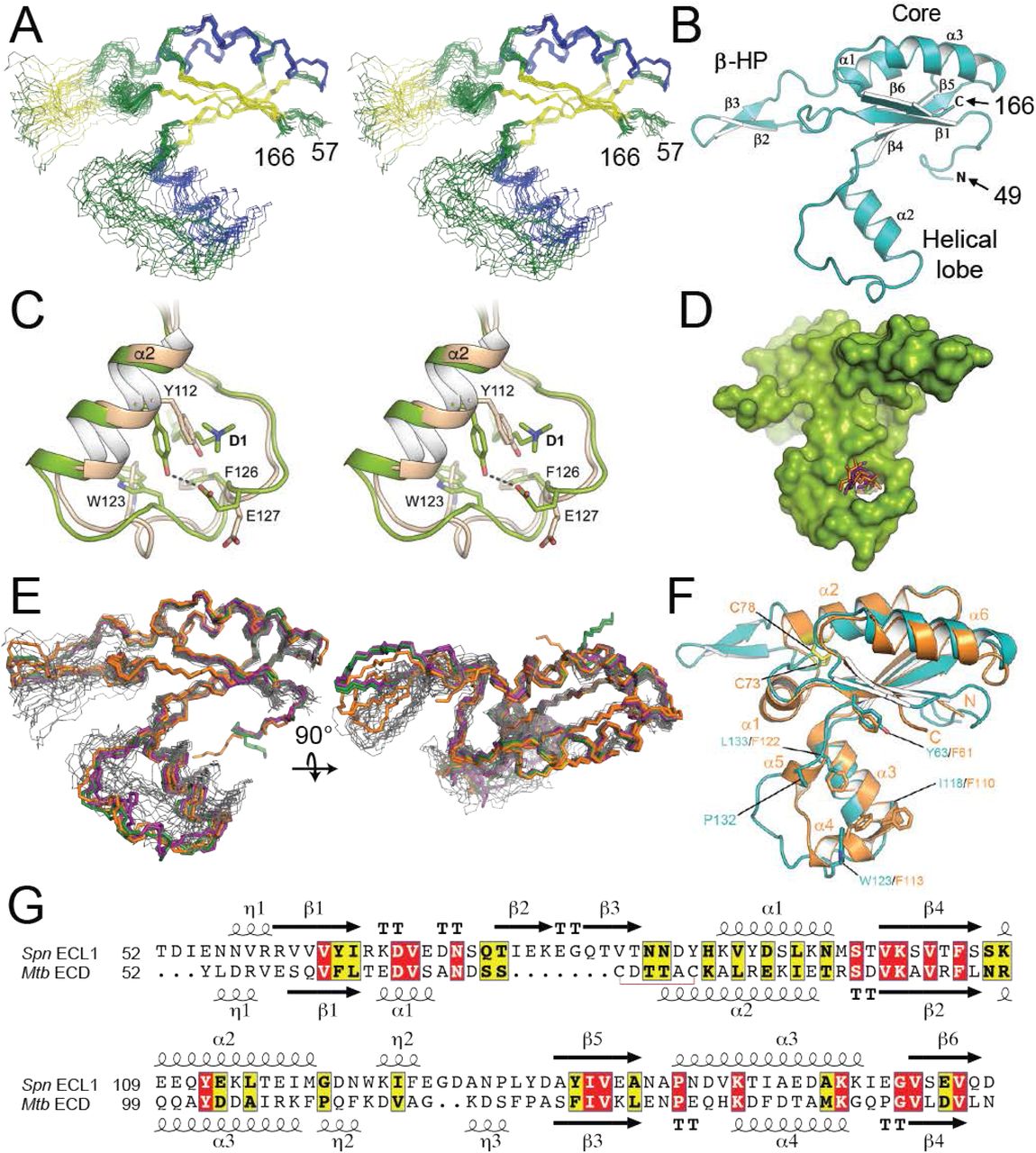
The three-dimensional structure of FtsXECL1 (residues 46–168) was solved by both NMR spectroscopy (Fig. 1A) and X-ray crystallography (Fig. 1B). The folded structure (residues 57–166) reveals a central core composed of a four-stranded antiparallel β-sheet (β1, β6, β4, β5) and two helices (α1 and α3), an α-helical lobe (residues 107–135) harboring the α2 helix, and an extended β-hairpin (β2, β3). The β-hairpin and helical lobes are connected to the central core by hinges. Details for structural determination of FtsXECL1 by NMR are presented in the methods section and structure statistics are summarized in Table 1. The solution structure shows that while the central mixed α-β core adopts a well-defined conformation, the two appended lobes are highly dynamic on multiple timescales (vide infra), presenting a range of conformations among the 20 members of the FtsXECL1 NMR structural ensemble (Fig. 1A).
Structural statistics for NMR solution structure of FtsXECL1 (From the ensemble of 20 best NMR structures).

The structure of FtsXECL1 from Streptococcus pneumoniae. A) Stereo view of the 20 conformers of the FtsXECL1 NMR structure as backbone traces, with helices shown in blue and (3-strands shown in yellow. N- and C-termini of the domain are indicated by the residue numbers 57 and 166, respectively. B) Cartoon representation of the FtsXECL1 structure obtained by X-ray crystallography (chain B in FtsXECD-D1) in which the different secondary structure elements have been numbered and labeled. N- and C-termini of the domain are indicated and correspond to residue numbers 49 and 166, respectively. C) Stereo view showing changes in helical lobe upon interaction with detergent 1. Apo-form (chain B) is colored in pale brown and holo-form (chain A) is colored in green. Relevant residues affected by the presence of the detergent are depicted in capped sticks. Polar interaction represented by dashed line. D) Surface representation of the FtsXECL1 crystal structure in which the three different detergent molecules are superimposed and shown in sticks (detergent 1, 2 and 3 are colored in green orange and purple, respectively). E) An overlay of the backbone traces for the six FtsXECL1 structures (in black) with the backbone traces of the 20 FtsXECL1 NMR conformers (in green) F) Structural superposition of the crystal structure of FtsXECL1 from S. pneumoniae obtained by crystallography (colored in cyan) and the crystal structure of FtsXECD from M. tuberculosis (PDB 4N8N, colored in orange). Secondary structure elements observed in the M. tuberculosis structure are labeled. Some residues in both structures are shown as sticks and numbered (see main text). Cys residues involved in disulfide bond formation in M. tuberculosis FtsXECD are colored in yellow. See main text for details. G) Sequence alignment of ECL1 domains from S. pneumoniae (Spn ECL1) and M. tuberculosis (Mtb ECD). Secondary structure elements are indicated and numbered.
Three different structures, with resolutions ranging from 2.0 to 2.3 Å, were solved by X-ray crystallography, each in the presence of a different detergent. The presence of detergents was critical as in their absence crystals diffracted at very low resolution (≤4 Å), suggesting significant mobility in some protein regions. Details for the crystallographic determination are provided in the methods section and structure statistics summarized in Table 2. In all cases, two protein molecules are present in the asymmetric unit (Fig. S1), yielding a total of six independent structures and sufficient electron density for both the protein structure and associated detergent molecules (Fig. S2). Different conformations were observed for the β-hairpin and the helical lobe among the six structures depending on the presence and identity of the bound detergent molecule to FtsXECL1 (Fig. S1B). The structural variations observed in these crystallographic structures, however, are less dramatic than those observed in the NMR conformer bundle, obtained in the absence of ligand, although the regions with high variability are similar.
Crystallographic data collection and refinement statistics.a
In any case, the crystal structures suggest that changes in the protein backbone and side chains of the helical lobe occur when a detergent ligand is bound (Fig. 1C). These changes create a cavity in which the detergent molecules insert (Fig. 1D), with a large part of the helical lobe affected by the interaction with detergent (from Q111 to E127) (Fig. 1C). Residues Y112, W123, E127 and F126 are strongly perturbed upon detergent ligand binding (Fig. 1C) with the hydroxyl moiety of Y112 interacting with carboxylate of E127. Changes in W123 and F126 stabilize the hydrophobic region of detergent 1. A similar interaction pattern is observed for detergents 2 and 3; additional interactions are observed with N131 with detergents 2 and 3, which are characterized by a larger hydrophilic head (maltose) (Fig. S1E-F). Although a physiological role for detergent binding to the helical lobe is unknown, many of these same residues are important for the interaction with PcsB (vide infra).
A structural comparison with M. tuberculosis FtsXECL1 (16) (PDB 4N8N) reveals differences in both the overall structure (RMSD value of 2.2 Å) and in the appended lobes of the core domain (Fig. 1F). The main differences between the mycobacterial and pneumococcal FtsXECL1 domains are the presence of an extra helix (α1) and a disulfide bond in M. tuberculosis ECL1 that are absent in pneumococcal ECL1, and an extended β-hairpin (residues 71–87) unique to the pneumococcal ECL1 domain. It is worth noting that in M. tuberculosis ECL1, the upper and lower lobes form a large hydrophobic cleft with four exposed Phe residues (F61, F110, F113 and F122) and this region was suggested as a strong candidate interaction surface between FtsX and RipC (16). These phenylalanine residues are not conserved in the pneumococcal ECL1 (Fig. 1F-G) but the hydrophobic nature of this region is preserved (Y63, I118, W123 and L133).
FtsXECL1 is dynamic in solution
We next performed additional NMR experiments to explore the mobility of the FtsXECL1 domain, both to validate the heterogeneity of the structural ensemble in solution, and to elucidate function. The 1DHN residual dipolar couplings (RDCs) obtained by weak alignment in Pf1 filamentous phage correspond well to previously determined secondary structure elements (29), with uniform values for the entire length of α1 and α2, as expected for straight helices. In contrast, 1DHN values near 0 for the N-terminal tail, the very C-terminus, and the non-helical part of the helical lobe between α2 and β5, are suggestive of significant conformational disorder in solution (Fig. S3A). These regions of small or zero RDCs are regions of very high RMSD in the NMR structure bundle (Fig. S3B). As anticipated, the correlation between experimentally measured RDCs and predicted RDCs back-calculated from the structures (30) is high, but only in the core subdomain, and match poorly in the β-hairpin for most of the crystal structures (Fig. S3C-D).
We previously reported that the 15N-{1H} heteronuclear NOE (hNOE) is low or negative at the termini, indicating that they are highly flexible in solution (29). The hNOE is also smaller in the β2-β3 hairpin region, as well as in the C-terminal end of the α2 helix and the subsequent coiled region leading into β5 (29) (Fig. S3E). Mapping these dynamics data onto a representative structure from the solution NMR ensemble shows that these regions with fast-timescale dynamics correspond to the β-hairpin and the helical lobe (Fig. S3F). Information on ps-ns fast-timescale motions extracted from the R2/R1 ratio also reveals that α2-β5 linker is highly flexible, while the β1-β2 linker and β-hairpin show an elevated R2/R1 ratio, specifically indicative of millisecond-timescale slow conformational exchange (Fig. S3G-H). These findings were directly confirmed by (Carr-Purcell-Meiboom-Gill) relaxation dispersion NMR spectroscopy (31) (Fig. S3I-J). We conclude that the β-hairpin exhibits flexibility on both the sub-ns and ms timescales. Interestingly, the β1-β2 linker also shows increased B-factors that are qualitatively consistent with ms-timescale conformational exchange observed by NMR.
These complex motions observed in the solution dynamics experiments are reflected in the heterogeneity of the NMR structure, with high Cα RMSDs particularly in the α2-β5 linker but also in the β-hairpin and β1-β2 linker, thus validating the conformational spread in the ECL1 structure in solution (Fig. S3B). The dynamic nature of the helical lobe is also reflected in the heterogeneity and B-factors of the crystal structures (Fig. S3K-L). Full-length FtsX itself is likely a dimer in vivo (32), and one can speculate that the flexible helical lobe and β-hairpin regions may contribute to dimerization or to interactions with a binding partner such as PcsB.
The PcsB coiled-coil (PcsB-CC) domain interacts with FtsXECL1
The 1H-15N HSQC spectrum for ECL1 has excellent chemical shift dispersion and lends itself readily to studies of protein-protein interactions (Fig. S4A). In contrast, full-length PcsB is 42 kDa and forms a dimer, and is thus challenging to study by NMR due to its size. We therefore constructed truncation mutants of PcsB focusing on the coiled-coil domain (PcsB-CC), thus limiting the molecular weight to 23–24 kDa. 15N labeled PcsB-CC (47–267) and PcsB-CC (47–254) are both characterized by 1H-15N HSQC spectra with limited 1H signal dispersion (Fig. S6A), consistent with the high helical content of this domain. Circular dichroism confirms that these proteins are primarily α-helical, in agreement with the crystal structure (22), thus indicating that they are properly folded (Fig. S6A) and can be used for ECL1 binding studies.
To determine if PcsB physically interacts directly with FtsXECL1, we titrated unlabeled PcsB-CC (47–267) into 15N labeled FtsXECL1 at molar ratios of 1:1, 2:1, 4:1, and 6:1, respectively, and recorded 1H-15N HSQC spectra (Fig. S4A-B). Numerous crosspeaks move in response to the addition of PcsB-CC to 15N FtsXECL1. The largest changes occur in the helical lobe of FtsXECL1 (Fig. 2A, S4-S5). In particular, residues M119, W123, I125, F126, G128 exhibit the greatest changes in crosspeak position and intensity in the 1:2 FtsXECL1 to PcsB-CC sample (Fig. 2A, Fig. S4C); when additional PcsB-CC is added, these crosspeaks broaden beyond detection (Fig. S5B-C). A reciprocal 1H-15N HSQC experiment with 15N labeled PcsB-CC (47–254) further confirms an interaction with unlabeled FtsXECL1 (Fig. S6B), as multiple crosspeaks shift upon the addition of increasing FtsXECL1 (Fig. S6B). We measured the binding affinity of the FtsXECL1-PcsB-CC complex using isothermal titration calorimetry (ITC), which reveals a Ka of 3.0 × 104 M−1 (Kd ~ 34 µM) (Table 3, Fig. 2B). These data provide the first biochemical evidence for a direct physical interaction between PcsB-CC and FtsXECL1 (Fig. 2, Fig. S4-S6).
Thermodynamic parameters of wild-type and mutant FtsXECL1 from direct analysis of isothermal titration calorimetry (ITC).a

FtsXECL1 binds PcsB-CC. A) Significant chemical shifts and peak height changes upon 1H-15N HSQC titration of 50 μM 15N FtsXECL1 with 100 μM unlabeled PcsB-CC map to the lower lobe of FtsXECL1. Chemical shift (Δδ, ppm) and peak height changes are mapped as color gradients on the FtsXECL1 structure, orange to red and light grey to blue, respectively. L115 and M119 a carbons are shown as spheres on the upper image. Peaks that overlap in the1H-15N HSQC spectra are colored white. Chemical shift and peak height change upon addition of 2 molar equivalents of PcsB-CC to FtsXECL1 are mapped to the structure. B) Representative titration of PcsB-CC with wild-type (WT) FtsXECL1 or L115A/M119A FtsXECL1 as monitored by ITC. Conditions: 50 mM potassium phosphate, 50 mM NaCl, 0.5 mM EDTA, pH 7.0 at 25.0 °C. Top panel, corrected ITC data; bottom panel, kcal/mole of injection vs. molar ratio. The black line overlapping the WT data indicates the best fit to a one-site binding model. Fitting parameters are summarized in Table 3. A red line drawn through the L115A/M119A FtsXECL1 data is for reference.
The interaction region of FtsXECL1 with PcsB-CC is essential for cell growth and proper morphology
Having identified the interaction region between FtsXECL1 and PcsB-CC, we next sought to determine the degree to which this interaction interface mapped by NMR spectroscopy contributes to pneumococcal viability. A multiple sequence alignment of this region (residues 102–155) among bacterial species in which FtsX has been studied and in related Streptococcal species (Fig. S7A) reveals that amino acids in this region are either partially or completely conserved (Fig. S7A). We therefore decided to target E109, Q111, L115, M119, W123, F126, and N131 singly or in combination for substitution with alanine (Fig. S7A). Given the essentiality of the FtsXECL1 and PcsB-CC interaction, we predicted that mutating these residues might be lethal (20). To allow for the cross-in of potentially lethal point mutations, we employed the Janus cassette method to insert point mutations at the native site of ftsX into a strain containing an ectopic copy of ftsX+under a zinc inducible promoter (33) (Fig. S7B). We then transformed markerless mutant alleles of ftsX in the presence of zinc. This allows for expression of the wild-type ftsX+and mutant ftsX simultaneously. As long as the mutant ftsX is not dominant negative, we could obtain a strain that expresses the wild-type copy of ftsX+under zinc induction and mutant ftsX only in the absence of zinc (Fig. S7B).
Zinc toxicity has been observed to cause aberrant cell morphology and growth inhibition in S. pneumoniae, when cells are not supplemented with manganese (34, 35). To rule out any deleterious effects of the zinc/manganese (Zn/Mn) addition used to induce ftsX expression, we measured the growth of the parent and the FtsX merodiploid strain in the presence of these metals. To verify that the addition of 0.45 mM ZnCl2 and 0.045 mM MnSO4 (Zn/Mn) did not cause growth or morphological defects, cells were grown in the presence and absence of Zn/Mn and imaged at 3 h and 6 h into the growth curve (Fig. S7C-D). Wild-type cells (D39 ∆cps rpsL1) had no morphological or growth defects at these time points with or without the addition of Zn/Mn (Fig. S7C-E).
In contrast, the FtsX merodiploid strain (Pzn-ftsX ∆ftsX) had significant morphological and growth defects at 3 or 6 h in the absence of Zn/Mn (Fig. S7C-E). Cessation of growth and aberrant cell morphology was observed in 90% of cells at 3 h and 95% of cells at 6 h growth (Fig. S7D). These cells are significantly shorter and rounder than wild-type cells (Fig. S7E), and a large variability in their volumes was observed (Fig. S7E), as previously found for a strain expressing ftsX+under a fucose-inducible promoter (21). If the strain was grown in the presence of Zn/Mn, FtsX was expressed and the strain has no growth or morphological defects (Fig. S7C-E). This indicates the defects observed are solely due to the absence of FtsX.
We next constructed three classes of amino acid substitution or insertion mutants (Table 4) in an effort to disrupt the FtsXECL1-PcsB-CC interaction defined by NMR spectroscopy. These are designated Class I (single amino acid changes), Class II (multiple amino acid changes), and Class III (insertion of a (Gly3Ser)2 linker) mutants. Class I strains were made by introducing single amino acid substitutions in the merodiploid strain and measuring growth or morphology defects (Fig. S8, Table 4). Class I mutants targeted both the α2 helix and the loop region (residues 107–120, and 121–130, respectively) of the FtsXECL1 helical lobe (Table 4, Fig. 3A, S8A). Single amino acid substitutions of FtsX (E109A, L115A, M119A, W123A, F126A, N131A, N131D) exhibited morphological defects without Zn/Mn (Fig. S8C-D, Table 4), but did not induce a measurable growth phenotype (Fig. S8B and Table 4). Expression of FtsX (L115A) resulted in cell shape defects (aspect ratio, length, width, and volume) (Fig. S8C-D), while expression of FtsX (M119A) only resulted in a change in cell volume (Fig. S8D). These differences were not due to mis-expression of FtsX, as western blotting indicates that all were expressed at or near wild-type levels (Fig. 4C). Two other single amino acid substitutions (E109Q and Q111A) did not strongly affect growth, morphology, or expression (Table 4, Fig. 4D).
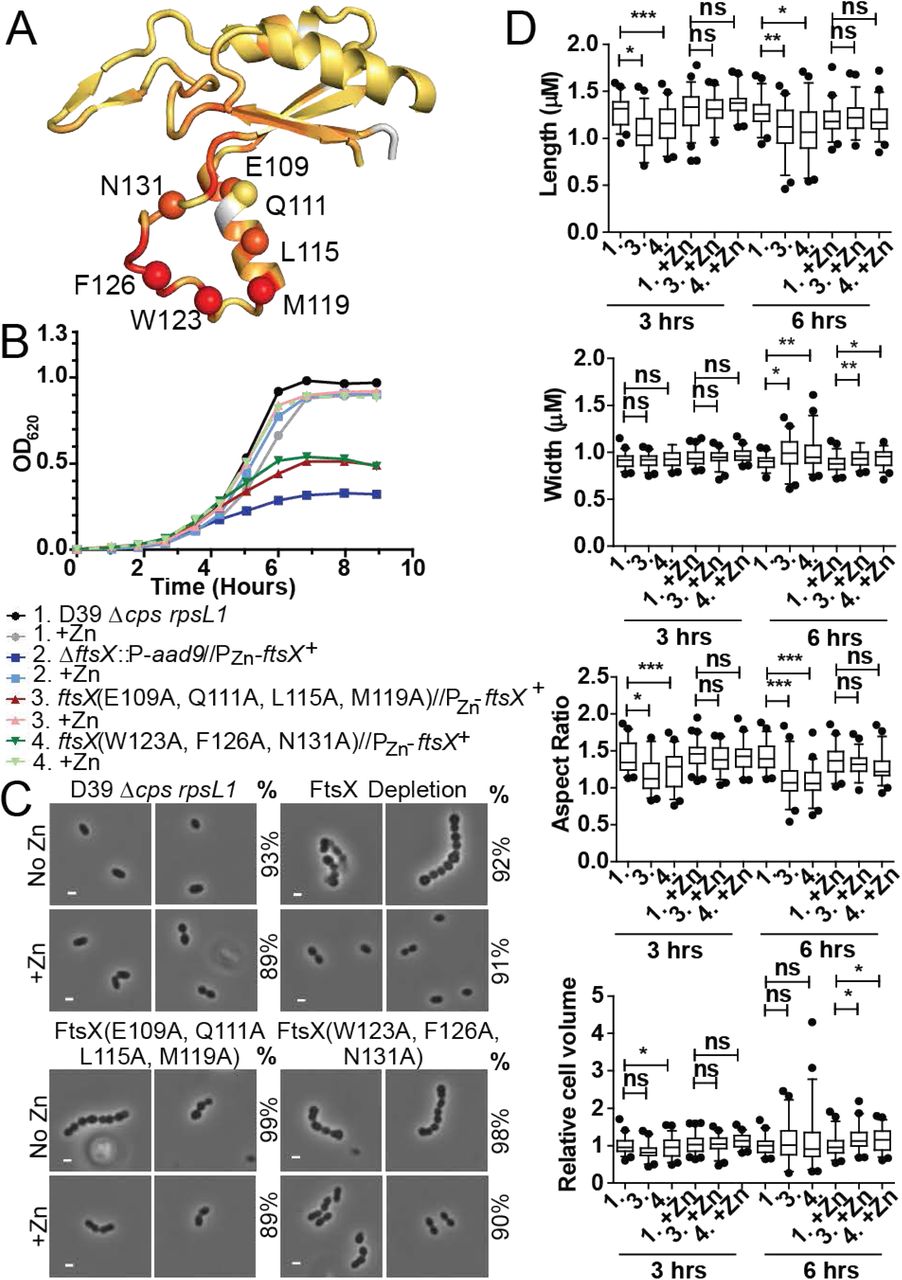
Multiple amino acid changes in the lower lobe of FtsXECL1 cause morphological and growth defects. A) Amino acid changes made mapped to the structure of FtsXECL1. The a carbon of each residue is shown as a colored sphere. The orange to red coloring on the FtsXECL1 structure represents the peak height changes from 1H-15N HSQC spectra upon addition of 2 molar equivalents of PcsB-CC to FtsXECL1 B) Representative growth curve of strains with amino acid changes in the lower lobe of FtsXECL1, compared to the growth of a FtsX depletion strain. These strains were grown in the presence or absence of 0.45 mM ZnCl2 supplemented with 0.045 mM MnS04 (indicated as +Zn). Strains shown are as follows: black circle, D39 rpsL1 Δcps wild-type parent (1, IU1824); grey circle, IU1824 +Zn; dark blue square, D39 rpsL1 Δcps ΔftsX.:P-aad9//bgaA::tet-PZn-ftsX+ (2, IU12376); light blue square, IU12376 +Zn; red triangle, D39 rpsL1 Δcps ftsX(E109A, Q111A, L115A, M119A)//bgaA::tet-PZn-ftsX+ (3, IU12861); pink triangle, IU12861 +Zn; dark green inverted triangle, D39 rpsL1 Δcps ftsX(F126A, W123A, N131A)//bgaA::tet-Pzn-ftsX+(4, IU12864); light green inverted triangle, IU12864 +Zn. This growth curve was repeated three times with similar results. C) Representative images of strains at 6 hours growth. The genotype of the strain shownis indicated above each panel. No Zn or +Zn indicates if Zn/Mn mixture was added. % indicates the percentage of cells in the population that are morphologically similar to the images shown. Greater than 50 cells per strain, condition, and experimental repeat were analyzed. These experiments were performed three times independently with similar results. Scale bar shown is equal to 1 pM. D) Length, width, aspect ratio, and relative cell volume of strains at 3 hours and 6 hours growth. Strains are indicated according to numbering in panel B. Greater than 50 cells were measured per strain and condition over two experimental replicates. For statistical analysis, a Kruskal-Wallis test (one-way AN OVA) with Dunn’s multiple comparison post-test was used to determine if length, width, aspect ratio, and relative cell volume were significantly differenl between strains and conditions. ns=non significant, * = p<0.05, ** = p<0.005, *** = p<0.0005
Amino acid changes made in vivo to disrupt the FtsXECL1-PcsB interaction.a
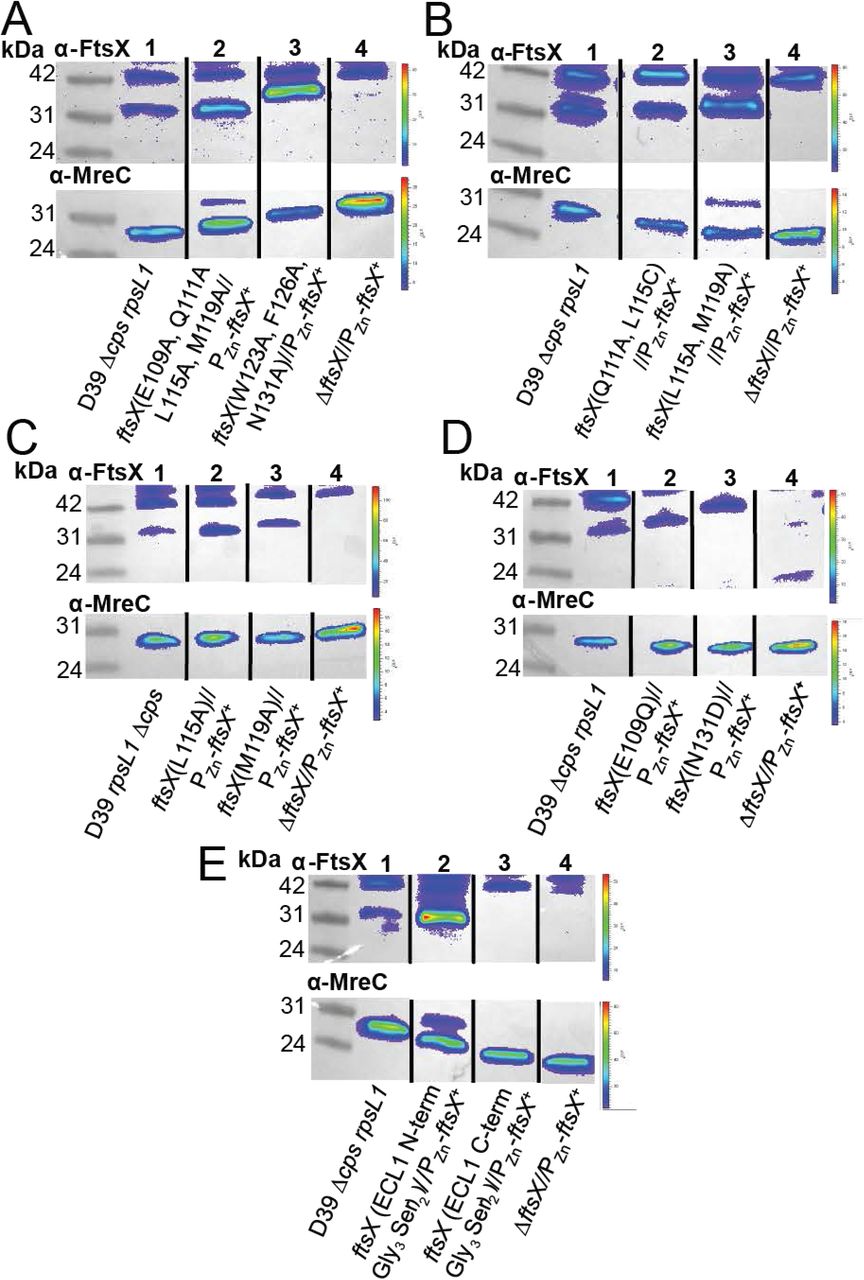
In contrast to the somewhat modest physiological impact of Class I substitutions, selected Class II mutants (Table 4) exhibited severe morphological and growth defects (Fig. 3 and S9, Table 4). In strains targeting the α2 helix, FtsX(E109A, Q111A, L115A, M119A), or the coil, FtsX(F126A, W123A, N131A), ≥98% of cells had severe growth and morphology defects. The growth and morphology of these strains was similar to ftsX depleted cells (Fig. 3B-D). Cells expressing these ftsX alleles became significantly rounder and shorter (Fig. 3D), and growth was inhibited without zinc (Fig. 3B). Importantly, in the presence of zinc the cells were indistinguishable from wild-type at 3 h (Fig. 3D). At 6 h with zinc, these cells exhibited changes in width and volume, which could be due to overexpression of FtsX at this time point or expression of wild-type and mutant FtsX simultaneously (Fig. 3D). Western blotting confirmed that FtsX(E109A, Q111A, L115A, M119A) and FtsX(F126A, W123A, N131A) were expressed in the absence of zinc at 6 h post-depletion (Fig. 4A). The triple FtsX(F126A, W123A, N131A) mutant migrated slightly higher on an SDS-PAGE gel compared to wild-type FtsX in the absence of zinc, but was expressed (Fig. 4A). Taken together, these results reveal that both the α2 helix and loop in the helical lobe of FtsXECL1 are important for FtsX function in vivo and confirm the functional importance of the physical interaction of FtsX and PcsB mapped by NMR.

FtsX with amino acid changes are expressed at near wild-type levels. Representative blots of a-FtsX and α-MreC (Western blotting control for loading) are shown, with the genotype indicated under each lane. Expected molecular weight (MW) for FtsX is 34.2 kDa, expected MW for MreC is 29.7 kDa. Samples were grown without Zn and harvested at 6 hours growth. Westerns were imaged as described in Supplemental Materials and Methods. A) FtsX(E109A, Q111A, L115A, M119A) and FtsX(W123A, F126A, N131A) are expressed at or above wild-type levels without zinc. Lane 1, D39 rpsL1 Δcps (IU1824); lane 2, D39 rpsL1 Δcps ftsX(E109A, Q111A, L115A M119A)//bgaA::tet-Pzn-ftsX+ (IU12861); lane 3, D39 rpsL1 Δcps ftsX(W123A, F126A, N131A)//bgaA:.tet-Pzn-ftsX+ (IU12864); lane 4, D39 rpsL1 Δcps ΔftsX//bgaA:.tet-Pzn-ftsX+ (IU13461). B) FtsX(Q111 A, L115C) and FtsX(L115A, M119A) are expressed at near wild-type levels without zinc. Lane 1, D39 rpsL1 Δcps (IU1824); lane 2, D39 rpsL1 Acps ftsX(Q111A, L115C)//bgaA::tet-Pzn-ftsX+ (IU13064); lane 3, D39 rpsL1 Δcps ftsX(L115A, M119A)//bgaA::tet-Pzn-ftsX+ (IU13066); lane 4, D39 rpsL1 Δcps ΔftsX//bgaA::tet-Pzn-ftsX+. C) FtsX (L115A) and FtsX (M119A) are expressed at near wild-type levels without zinc. Lane 1, D39 rpsL1 Δcps (IU1824); lane 2, D39 rpsL1 Δcps ftsX(L11A)//bgaA::tet-Pzn-ftsX+ (IU12521); lane 3, D39 rpsL1 Δcps ftsX(M119A)//bgaA::tet-Pzn-ftsX+ (IU12637); lane 4, D39 rpsL1 Δcps ΔftsX//bgaA::tet-Pzn-ftsX+ (IU13461). D) FtsX (E109Q) and FtsX (N131D) are expressed at near wild-type levels without zinc. Lane 1, D39 rpsL1 Δcps (IU 1824); lane 2, D39 rpsL1 Δcps ftsX(E109Q)//bgaA::tet-Pzn-ftsX+ (IU13088); lane 3, D39 rpsL1 Δcps ftsX(N131D)//bgaA::tet-Pzn-ftsX+ (IU13089); lane 4, D39 rpsL1 Δcps ΔftsX//bgaA::tet-Pzn-ftsX+ (IU13461). E) FtsX with (Gly4Ser)2 after residue 51 is expressed, whereas FtsX with (Gly4Ser)2 after residue 173 is not expressed. These are referred to as FtsX N-term ECL1 (Gly4Ser)2 and FtsX C-terminal ECL1 (Gly4Ser)2, respectively. Lane 1, D39 rpsL1 Δcps (IU 1824); lane 2, D39 rpsL1 Δcps ftsX N-term ECL1 (Gly4Sery)2//bgaA::tet-Pzn-ftsX+ (IU12629); lane 3, D39 rpsL1 Δcps ftsX C-term ECL1 (Gly4 Ser)2//bgaA.:tet-Pzn-ftsX+ (IU12869); lane 4, D39 rpsL1 Δcps ΔftsX/l bgaA:.tet-Pzn-ftsX+ (IU 13461). These experiments were performed two to three times independently.
Some of the Class II mutants we characterized had just two amino acid changes in the α2 helix or the extended loop of the helical lobe (Table 4, Fig. S9). We observed that FtsX (L115A, M119A) exhibited a strong growth and morphology defect in the absence of zinc in 99% of cells at 6 h (Fig. S9B-C, Table 4), and these cells displayed a decrease in length, width, and volume relative to wild-type cells (Fig. S9D). This mutant was expressed in the absence of zinc at near wild-type levels (Fig. 4B). These data confirm that the tandem L115A, M119A substitution disrupts FtsX function, even though as individual single mutants they result only in slight morphological defects (Fig. S8B-D, Fig. S9B-D, Table 4). Another double mutant FtsX(Q111A, L115C) induced the formation of long chains and a “boxy” cell morphology (Fig. S9C, Table 4). This mutant resulted in shorter cells with a significantly different aspect ratio from wild-type (Fig. S9D), but this strain had no growth phenotype (Fig. S9B). In contrast, another double substitution Class II mutant, e.g., FtsX(E109A, N131A) (Table 4) exhibited no strong morphology or growth defects.
Finally, Class III insertion mutants (Table 4) were constructed and used to evaluate if other regions of FtsXECL1 were important for the FtsXECL1-PcsB interaction or for FtsX function. We inserted a (Gly3Ser)2 flexible linker approximately where the FtsXECL1 is predicted to enter (residue 51) or exit (residue 173) the membrane bilayer, or in the β-hairpin, which exhibits significant conformational disorder over a range of timescales (Fig. 1A). An insertion after residue 51 in FtsXECL1 was detrimental to both growth and morphology (Table 4), and this insertion did not disrupt FtsX expression (Fig. 4E). The insertion after residue 173 in FtsXECL1 also caused growth and morphology defects, but this FtsX allele was not expressed in cells (Table 4, Fig. 4E). The β-hairpin (Gly3Ser)2 insertion (Fig. 1B, Appendix) was introduced after amino acid 78 of FtsXECL1, which corresponds to the tip of β-turn in the β-hairpin (Fig. 1B). This strain also exhibited no growth defect, but these cells were significantly smaller, but only at the 3 h time point (Appendix). We conclude that the β-hairpin does not play a major role in FtsX-PcsB interaction, consistent with the NMR mapping experiments.
L115A/M119A FtsXECL1 is stably folded and unable to bind PcsB-CC
We reasoned that if the defects observed in Class I and Class II mutants were due to the disruption of the FtsXECL1-PcsB-CC interaction, this should impact the affinity of this interaction as measured by ITC. We first characterized the L115A/M119A mutant by 1H-15N NMR and CD spectroscopies to confirm its structural integrity. The CD spectrum resembled wild type, as did the 1H-15N HSQC spectrum, with clear chemical shift perturbations only among those resonances in the immediate vicinity of the double substitution (Fig. S10B). Both pieces of data suggest a local rather than global perturbation of α2-loop lobe in the FtsXECL1 structure upon introduction of the double L115A/M119A substitution. As anticipated, titration of PcsB-CC (47–267) with L115A/M119A FtsXECL1 reveals no detectable binding (no observable heat) (Fig. 2B, Table 3) compared to wild-type FtsXECL1. These data confirm that the helical lobe of FtsXECL1 interacts with PcsB-CC, and that this interaction is required for viability and proper cell shape.
In contrast to the L115A/M119A double mutant, severely functionally compromised representative triple mutant W123A, F126A, N131A FtsXECL1 and quadruple mutant E109A, Q111A, L115A, M119A FtsXECL1 exhibited more pronounced structural perturbations that nonetheless map only to the helical lobe. The triple mutant was indistinguishable from the L115A/M119A and wild-type FtsXECL1 derivatives by CD spectroscopy while the quadruple mutant exhibited less molar ellipticity, or secondary structure (Fig. S10D). Inspection of their 1H-15N HSQC spectra reveal that although the core and β-hairpin domains essentially resemble wild-type, each exhibits considerable perturbation of resonances throughout the entire helical lobe (Fig. S10A, S10C). Since these mutants are functionally compromised, these structural findings strongly support the conclusion that the structural integrity of the lower lobe is essential for the physical interaction with PcsB, and the function of FtsX in pneumococcal cells.
DISCUSSION
This study presents a comprehensive analysis of the solution and x-ray structures of the outward-facing large extracellular loop of FtsX (FtsXECL1) from S. pneumoniae and defines a physical interaction site with the coiled-coil domain of peptidoglycan hydrolase PcsB (PcsB-CC). Our FtsXECL1 structures reveal a globular fold that while similar to the large extracellular loop of FtsX from M. tuberculosis (16) is characterized by unique features. The upper β-hairpin distinguishes S. pneumoniae FtsXECL1 from that of M. tuberculosis, and despite being characterized by significant conformational dynamics on a range of timescales, is not required for the interaction of S. pneumoniae FtsXECL1 with PcsB. The function of this domain is not well-defined by our data, but could play a role in another process, e.g., FtsX dimerization, interaction with the small loop ECL2, or an interaction with another domain of PcsB. On the other hand, the helical lobe of FtsXECL1, common to both S. pneumoniae and M. tuberculosis FtsX structures, is vital for the interaction PcsB in vitro, and that this interface is functionally important in vivo. Increasing numbers of Ala substitutions tested here increasingly disrupt this interaction and ultimately cause dramatic growth and morphology defects, indicating that the helical lobe of FtsXECL1 is essential for regulation of PcsB during cell division. Interestingly, this region of FtsXECL1 corresponds with the region shown to be important for the interaction of M. tuberculosis FtsXECD with its PG hydrolase, RipC (16). This suggests that the helical lobe could be a conserved functional determinant for the interaction of FtsX with cognate hydrolases or adaptor proteins across many species of bacteria.
We propose that the helical lobe of FtsXECL1 is important for the activation of cognate hydrolase activity either directly or indirectly through their adaptors (Fig. 5). The exact role of the second extracellular loop of FtsX (FtsXECL2) is unknown, but may also regulate this process, as temperature-sensitive mutations in pcsB were found to be suppressed by mutations in the coding region for FtsXECL2 (20). Previous work suggests that FtsEX forms a dimer (32), as dimerization of the FtsE ATPase domain is likely a necessary condition for ATP hydrolysis (36, 37) (Fig. 5A). After formation of the complex, ATP hydrolysis by FtsE results in a conformational change in FtsX, releasing PcsB from what we anticipate is an inhibited state (22) (Fig. 5B-C). This interaction is mediated by the helical lobe of FtsXECL1, although the membrane, and possibly lipid binding by FtsXECL1 and FtsXECL2 itself also may play a role as well. We propose that the interaction of FtsXECL1 with the PcsB coiled-coil domain communicates release of the PcsB CHAP domain from an inhibited state and thus is important for modulating PG hydrolysis by PcsB (Fig. 5B-C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model for the activation of PcsB by FtsXECL1. A) FtsEX dimerizes to form the active complex. PcsB is secreted into the extracellular milieu. Attraction of PcsB to the area of active FtsX complexes might be mediated by its propensity to interact with membranes (Bajaj et. al, 2017). The ECL1 and ECL2 loops are indicated on FtsX. FtsXEECL1 is shown in green, with the β-hairpin and α-helical lobe shaded in yellow and blue, respectively. B) After formation of the active complex, ATP hydrolysis by FtsE causes a conformational change in FtsX. PcsB interacts with FtsXECL1 via its coiled coil domain, and this interaction causes activation of the peptidoglycan hydrolytic activity of PcsB. PcsB, along with other factors in the cell, allow for cell division to proceed normally. Functional FtsX, FtsE, and PcsB are all required for efficient cell division.
Recently, the structure of A. actinomycetemcomitans MacB was reported and suggested to be a structural paradigm for the ABC3 transporter superfamily that includes FtsX (38). MacB was proposed to function as a mechanical pump to drive enterotoxin transport through TolC in E. coli (38). Crow et. al found that MacB itself did not transport enterotoxin but drove TolC to transport it instead, due to the lack of a central cavity in the MacB structure (38). As such, they proposed that MacB as a model for other “mechanotransmitters” belonging to this same ABC3 transporter superfamily. While this proposed function of mechanotransmission may well characterize MacB and FtsX, the overall structure of FtsXECL1 from S. pneumoniae is clearly distinct from the periplasmic domain of MacB, thus revealing that MacB does not readily provide a structural basis for understanding FtsX-dependent peptidoglycan hydrolases. Future work using reconstituted FtsEX and PcsB complexes in membranes will allow for understanding how this common mechanotransmission principle extends throughout the ABC3 superfamily.
MATERIALS AND METHODS
NMR Spectroscopy
Spectra of 15N-or 15N13C-labeled FtsXECL1 were recorded at 298 K on Varian (Agilent) DDR 600 or 800 MHz spectrometers equipped with cryogenic probes in the METACyt Biomolecular NMR Laboratory at Indiana University Bloomington. NMR samples contained 50 mM potassium phosphate, pH 7.0, 50 mM NaCl and 10% v/v D2O, with 0.2 mM 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) for chemical shift referencing. Typical concentrations of FtsXECL1 for 15N HSQC spectra were 50 µM, and 400 µM for triple-resonance and dynamics experiments. 1JHN splittings for residual dipolar couplings (RDCs) were measured using 2D IPAP [15N, 1H]-HSQC spectra (39), recorded on an isotropic sample and on a sample aligned with 20 mg/mL phage Pf1 (ASLA Biotech). 1DHN was calculated from 1DHN =1JHN (anisotropic) – 1JHN (isotropic). Aromatic sidechains were assigned using the HBCBCGCDHE and HBCBCGCDCEHE experiments (40). For experiments detecting PcsB-CC binding, 15N FtsXECL1 was kept at 50 µM concentration, and 1H-15N HSQC spectra were recorded with the following concentrations of PcsB-CC (47–267): 0, 50 µM, 100 µM, 200 µM, and 400 µM. nmrPipe, Sparky, CARA (http://cara.nmr.ch), CCPNMR, and NMRbox (41–44) were used for data processing and analysis. Resonance assignments and dynamics data are available in the BMRB under accession code 30523. These NMR data were used to calculate and refine the solution structure of ECL1 (see Supplemental Materials and Methods) with the ensemble of 20 lowest energy structures (see Table 1 for structure statistics) deposited in the Protein Data Bank (accession code 6MK7).
X-Ray Crystallography
First crystallization screenings were performed by high-throughput techniques in a NanoDrop robot and Innovadyne SD-2 microplates (Innovadyne Technologies Inc.), screening PACT Suite and JCSG Suite (Qiagen), JBScreen Classic 1–4 and 6 (Jena Bioscience) and Crystal Screen, Crystal Screen 2 and Index HT (Hampton Research). Positive conditions in which crystals grew were optimized by sitting-drop vapor-diffusion method at 291 K by mixing 1 µL of protein solution and 1 µL of precipitant solution, equilibrated against 150 µL of precipitant solution in the reservoir chamber. The best crystals were obtained in a crystallization condition containing 0.1 M sodium citrate pH=5.6, 0.2 M potassium-sodium tartrate and 2 M ammonium sulfate. These crystals were further optimized by cocrystallization in the presence of detergents Dodecyltrimethylammonium chloride (detergent 1, 46 µM), n-Undecyl-β-D-maltoside (detergent 2, 0.59 mM), and n-Decyl-β-D-maltoside (detergent 3, 1.8 mM). Crystals were cryo-protected in the precipitant solution supplemented with 25% (v/v) glycerol, prior to flash cooling at 100 K. Diffraction data were collected in beamline XALOC at the ALBA synchrotron (CELLS-ALBA, Spain), using a Pilatus 6M detector and a wavelength of 0.979257 Å. Crystals diffracted up to 2.0–2.3 Å resolution and belonged to the P 43 21 2 space group. The collected datasets were processed with XDS (45) and Aimless (46). Two FtsXECL1 molecules were found in the asymmetric unit, yielding a Matthews coefficient of 2.49 Å3/Da (47) and a solvent content of 50.73%. Structure determination was performed by molecular replacement using the online server Morda (http://www.biomexsolutions.co.uk/morda/). Refinement and manual model building were performed with Phenix (48) and Coot (49), respectively. The data presented translational non-crystallographic symmetry that were treated with Phenix (48). The stereochemistry of the final model was checked by MolProbity (50). Data collection and processing statistics are shown in Table 2. The atomic coordinates of FtsXECL1 determined by co-crystallization in the presence of detergents 1, 2 and 3 have been deposited in the Protein Data Bank with codes 6HE6, 6HEE and 6HFX, respectively.
Bacterial strains, plasmids, and growth conditions
Bacterial strains and plasmids used in this study are listed in Table S1. S. pneumoniae strains were derived from IU1945, an unencapsulated derivative of serotype 2 S. pneumoniae strain D39 (51). Strains were grown on trypticase soy agar II with 5% (vol/vol) defibrinated sheep blood (TSAII-BA) plates or in Becton-Dickinson brain heart infusion (BHI) broth at 37°C in an atmosphere of 5% CO2. E. coli strains for protein expression were derived from BL21(DE3) (NEB, C2527H). E. coli strains were grown in Luria-Bertani (LB) broth or in M9 minimal media supplemented with 15NH4Cl at 37°C, with shaking at 150 rpm. When required, tetracycline (0.25–2.5 µg/mL), kanamycin (250 µg/mL), spectinomycin (150 µg/mL), streptomycin (250 µg/mL), ampicillin (100 µg/mL) and/or isopropyl β-D-1-thiogalactopyranoside (IPTG, 1 mM) were added to S. pneumoniae or E. coli culture media. S. pneumoniae strains requiring zinc for expression of essential genes were grown with 0.45 mM ZnCl2 and 0.045 MnSO4.
Growth curves and phase-contrast microscopy of strains
For physiological and morphological analyses of strains, cells were inoculated from frozen glycerol stocks into BHI broth, serially diluted, and incubated for 10–12 hours statically at 37°C in 5% CO2 overnight. If zinc was required for growth of cultures, 0.45 mM ZnCl2 and 0.045 MnSO4 was added to overnight tubes. The next day, cultures from OD620 ≈ 0.05 to 0.4 were diluted into fresh BHI to OD620 ≈ 0.003 in 4 mL volumes, and two identical cultures for each strain were prepared, one with 0.45 mM ZnCl2/0.045 MnSO4 and one without. These cultures were grown under the same growth conditions as previously described in Materials and Methods. Growth was monitored turbidimetrically every 45 min to 1 hour with a Genesys 2 spectrophotometer (Thermo Scientific). For microscopic analyses, samples (1–2 μL) were taken at 3 hours and 6 hours and examined using a Nikon E-400 epifluorescence phase-contrast microscope with a 100X Nikon Plan Apo oil-immersion objective (numerical aperture, 1.40) connected to a CoolSNAP HQ2 charged-coupled device (CCD) camera (Photometrics). Images were processed using NIS-Elements AR software (Nikon), and measurements and calculation of cell width, length, volume, and aspect ratio were performed as described previously (52, 53). Statistical significance was determined using GraphPad Prism (GraphPad Software, Inc) by comparing values for cell width, length, volume, and aspect ratio measured for at least 50 cells over two experimental replicates. To determine if values were significantly different between strains and conditions, a Kruskal-Wallis (one-way ANOVA) with Dunn’s multiple comparison post-test was used.
For additional materials and methods, please see Supplemental Materials and Methods.
ACKNOWLEDGEMENTS
We thank the members Winkler and Giedroc labs for their helpful discussion and insight, specifically Dr. Tiffany Tsui, Dr. Julia Martin, Melissa Lamanna and Dr. Hui Peng. We acknowledge the Indiana University Bloomington Department of Chemistry Mass Spectrometry Facility, specifically Dr. Jonathan Karty and Angela Hansen for their help with training and setting up mass spectrometry experiments and the MetaCyt Biomolecular NMR Laboratory. We also thank Dr. Daiana Capdevila for her help in acquiring and analyzing the isothermal titration calorimetry data, the Indiana University Biological Mass Spectrometry Facility and Dr. Giovanni Gonzales-Gutierrez in the Physical Biochemistry Instrumentation Facility at Indiana University Bloomington. We thank the staff from ALBA synchrotron facility for help during crystallographic data collection. This work was supported by NIH grants R01GM114315 and R01GM127715 to M.E.W., and NIH grant R35GM118157 to D.P.G. and by predoctoral Quantitative and Chemical Biology (QCB) NIH institutional training grant T32 GM109825 (to B.E.R.). The work in Spain was supported by grant from the Spanish Ministry of Science, Innovation and Universities BFU2017-90030-P to J.A.H.
REFERENCES




