

Abstract
Flaviviruses limit the cell stress response by preventing the formation of stress granules and utilize different proteins involved in the stress granule pathway to modulate viral gene expression. In this study, we investigated the formation of stress granules during Zika virus (ZIKV) infection and the role stress granule proteins play during the viral life cycle. Using immunofluorescence and confocal microscopy, we determined that ZIKV disrupted the formation of arsenite-induced stress granules and changed the subcellular distribution, but not the abundance or integrity of stress granule proteins. To investigate the role of different stress granule proteins in ZIKV infection we used target-specific siRNAs to deplete six proteins, namely Ataxin2, G3BP1, HuR, TIA-1, TIAR and YB-1. Depletion of TIA-1 and TIAR affected ZIKV protein and RNA levels, but viral titers did not change. Conversely, depletion of Ataxin2 and YB-1 decreased virion production despite having only a small effect on ZIKV protein and expression. Notably, however, depletion of G3BP1 and HuR decreased and increased ZIKV gene expression and virion production, respectively. Using an MR766 Gaussia luciferase reporter genome together with knockdown and overexpression assays, G3BP1 and HuR were found to modulate ZIKV replication. These data indicate that ZIKV disrupts the formation of stress granules by sequestering stress granule proteins required for replication, which is where G3BP1 functions to promote ZIKV infection, while HuR exhibits an antiviral effect. The consequence of ZIKV re-localizing and subverting select stress granule proteins might have broader consequences on cellular RNA homeostasis contributing to cellular gene dysregulation and ZIKV pathogenesis.
Importance In response to viral infection, cellular translation is stalled, and translation initiation complexes, cellular mRNAs, and RNA binding proteins aggregate in stress granules. Because the assembly of stress granules antagonize translation of viral proteins, which is a critical step for single-stranded positive-sense RNA viruses to replicate the viral genome, many viruses have developed strategies to inhibit stress granule formation. In this study, we observed that Zika virus restricts the formation of stress granules likely by re-localizing specific stress granule proteins during infection. We also determined that specific stress granule proteins function to facilitate and limit Zika virus replication. This interaction of Zika virus with stress granule proteins is interesting, as many stress granule proteins are also known to function in neuronal granules, which are critical in neural development and function. Moreover, dysregulation of different stress granule proteins in neurons has been shown to play a role in the progression of neurodegenerative diseases. The likely consequences of Zika virus modulating stress granule assembly and subverting specific stress granule proteins are alterations to cellular mRNA transcription, splicing, RNA stability, and translation. Such changes in cellular ribostasis could have profound effects on neural development and contribute to the devastating developmental and neurological anomalies observed following intrauterine Zika virus infection. Our study provides new insights into virus-host interactions and the identification of the stress granule proteins that may contribute to the unusual pathogenesis associated with this reemerging arbovirus.
Introduction
Zika virus (ZIKV) is an enveloped, single-stranded positive-sense RNA virus belonging to the Flaviviridae family, which includes Dengue virus (DENV), Yellow fever virus (YFV), and West Nile virus (WNV) (1). While ZIKV was discovered in Uganda in 1947 (2), the virus garnered renewed interest during the 2015-2016 outbreak in the America, in particular because of the observed congenital infections during the first trimester and the devastating developmental abnormalities such as severe microcephaly, decreased brain tissue, macular scarring, congenital contractures, and hypertonia (3–8). Additionally adults infected with ZIKV were reported to develop Guillain-Barré syndrome, a debilitating disorder affecting the peripheral nerves (9–12). Similar to other flaviviruses is transmitted by the Aedes aegypti and Aedes albopictus mosquitoes, although recent evidence has shown ZIKV is capable of sexual and vertical transmission (13–16). While half a century has passed since the discovery of ZIKV, little-to-no research was published on ZIKV prior to the emergence of the current strain in the Americas associated with devastating neurological pathologies. Because there is no licensed vaccine and antiviral treatments are elusive, a fundamental understanding of the molecular biology of ZIKV and virus-host interactions is critical to developing therapeutic strategies.
The ZIKV single-stranded positive-sense RNA genome contains a 5’-cap, lacks a poly(A) tail, and encodes one open reading frame (ORF) that is flanked by highly structured 5’ and 3’ untranslated regions (UTRs). Similar to other flaviviruses, translation of the ZIKV RNA results in one long polyprotein that is co- and post-translationally proteolytically processed to produce at least three structural proteins (capsid [C], premembrane [prM] and envelope [E]) and seven nonstructural proteins (NS1, NS2a, NS2b, NS3, NS4a, NS4b and NS5) (1). Although cap-dependent and cap-independent translation have been reported for DENV (17), it is presently unknown whether ZIKV employs similar translation strategies. Similarly, little is known regarding the control strategies ZIKV employs to promote translation of the viral RNA.
In response to different environmental stresses, mammalian cells rapidly halt translation via the activation of one of the four eIF2-αkinases. In particular, the presence of double-stranded RNA (dsRNA) during viral infection activates protein kinase R (PKR) (18); the accumulation of unfolded proteins in the endoplasmic reticulum (ER) and resulting stress activates PKR-like endoplasmic reticulum kinase (PERK) (19); amino acid starvation activates general control non-repressed 2 (GCN2) (20); and oxidative stress activates heme-regulated inhibitor kinase (HRI) (21). Phosphorylation of the α subunit of eIF2 by one of the four stress response kinases results in the stalling of translation initiation, and disassembly of polysomes. Stalled translation initiation complexes bound to mRNA are recognized by several RNA binding proteins, which aggregate to form RNA-protein macromolecular complexes called stress granules (SGs) (22). Once the stressor is abated, eIF2-α is dephosphorylated by protein phosphatase 1 (PPI) and the PPI cofactor growth arrest and DNA-damage-inducible 34 (GADD34), allowing for the return of sequestered mRNA transcripts to active translation (22).
SGs are dynamic nonmembrane-bound cytoplasmic structures that can rapidly assemble in response to stress and disassemble once the stress has been alleviated (22). SGs typically contain mRNAs, stalled translation initiation complexes, and numerous RNA binding proteins. Indeed, SGs may contain upwards of 260 different proteins (23), and ~50% of these are proposed to be RNA-binding proteins (24). Of these Ras-GTPase activating protein (SH3 domain binding protein 1 (G3BP1), Caprin1, T-cell internal antigen 1 (TIA-1) and TIA-1 related protein (TIAR) are proposed to be key nucleators of stress granule assembly (25–28). In addition to functioning to repress translation and sort mRNAs, SGs also amplify the innate immune response by aggregating critical antiviral factors. Because translation is a critical first step in the flavivirus life cycle, the formation of SGs presents an immediate obstacle to infection. Notably, however, during infection with different flaviviruses, such as WNV, DENV, and Japanese encephalitis virus (JEV) SGs are absent, and treatment of virus-infected cells with arsenite fails to induce SGs (29–32). While WNV, DENV and JEV all belong to the same Flavivirus genus each virus employs a unique mechanism to block SG assembly. For example, early during WNV infection PKR is activated by the appearance of exposed dsRNA replication intermediates, which results in phosphorylation of eIF2α, the stalling of translation initiation and formation of stress granules. However, at later times during infection fewer SGs were visible as dsRNA and replication complexes were masked in membranous vesicles, and PKR remained inactive (29, 33, 34). More recently, WNV was shown to also limit stress granule assembly by upregulating and activating key transcription factors to modulate the antioxidant response (31). In contrast, DENV affects p38-Mnk1 signaling and phosphorylation of the cap-binding protein eIF4E to inhibit SG formation via an eIF2α phosphorylation-independent mechanism by (32). Amorium et al., recently reported that ZIKV employs yet another mode to block stress granule formation by increasing the rate of eIF2α dephosphorylation (35). Flaviviruses also subvert specific SG proteins to promote viral gene expression. For example, the SG-nucleating proteins TIA-1 and TIAR facilitate WNV replication (36); G3BP1, G3BP2 and Caprin1 promote translation of interferon stimulated genes (ISGs) to limit DENV infection (37, 38); and JEV inhibits SG assembly by colocalizing Caprin 1 with the viral capsid protein (30). More recently, G3BP1 was shown to interact with ZIKV RNA, and G3BP1 and Caprin1 with ZIKV capsid protein (39).
It is notable that a number of the RNA-binding proteins that localize in SGs, such as Ataxin-2, G3BP1, HuR and TIA-1, are known to contribute to different neuropathologies (40, 41). Our goal in this study was to elucidate the role of different SG proteins linked to neurodegeneration during ZIKV infection. Here we show a systematic analysis of SGs during ZIKV infection; the effect of depleting six different SG proteins on ZIKV protein and RNA levels and viral titers; and elucidate the biological function of two SG proteins on ZIKV gene expression. We determined that the Cambodian ZIKV strain (160310) disrupted arsenite-induced SG assembly and that several SG markers co-localized with sites of ZIKV replication. Additionally, the SG protein G3BP1 is required for ZIKV gene expression, while HuR exhibited antiviral activity. Using a Gaussia luciferase reporter genome, our studies revealed that G3BP1 and HuR modulate ZIKV replication. This work advances our understanding of the interplay between ZIKV, the cellular stress response and cellular RNA metabolism, and demonstrates a role that specific RNA-binding proteins play in ZIKV gene expression.
Materials and Methods
Cell Maintenance and ZIKV Stocks
Huh7 hepatocellular carcinoma cells were maintained in Dulbecco’s minimal essential medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS; Seradigm), 10 mM nonessential amino acids (NEAA; Life Technologies), and 5 mM L-glutamine (Life Technologies). Vero cells (ATCC# CRL-81) were maintained in DMEM supplemented with 10% FBS and 10 mM HEPES (Life Technologies). Mammalian cell lines were grown at 37C with 5% CO2. C6/36 cells (ATCC# CRL-1660) grown at 27C with 5% CO2 were maintained in Eagle’s minimal essential medium (EMEM; Sigma) supplemented in 10% FBS, sodium pyruvate (0.055 g/L; Life Technologies), Fungizone (125 μg/L; Life Technologies), and penicillin and streptomycin (50000 units/L penicillin, 0.05 g/L streptomycin; Life Technologies). ZIKV (Cambodia-160310, Uganda MR766, and Puerto Rico PRVABC59) stocks were generated in C6/36 cells. Briefly, C6/36 cells nearing confluency were infected at a multiplicity of infection (MOI) of 0.1. Seven days post-infection supernatants from the infected cells were collected, aliquoted and stored at −80°C. RNA was extracted from cells to confirm infection via northern blotting, and viral titers were determined by plaque assay.
Small Interfering RNA (siRNA) and DNA Plasmid Transfections
Sense and antisense siRNA oligonucleotides were synthesized by Integrated DNA Technologies (IDT). The siRNA sequences are provided in Supplemental Table 1. Oligonucleotides were resuspended in RNase-free water to a 50 μM final concentration. Sense and antisense strands were combined in annealing buffer (150 mM HEPES (pH 7.4), 500 mM potassium acetate, and 10 mM magnesium acetate) to a final concentration of 20 μM, denatured for 1 minute at 95°C, and annealed for 1 hour at 37°C (42).
For siRNA and plasmid transfections, Huh7 cells seeded at 7.5×106 in a 10cm cell culture dish were transfected 24 hours later with 100 nM of the indicated siRNA duplex or 2 μg of 3×FLAG-BAP (Bacterial Alkaline Phosphatase; Sigma #C7472), HuR-FLAG (GenScript #OHu23723D), or G3BP1-FLAG (GenScript #OHu02150D) plasmids using Lipofectamine 3000 as per manufacturer’s protocol (Invitrogen). At 24 hours post-transfection, cells were infected with ZIKV.
ZIKV Infections
On the day of infection, one mock culture plate was trypsinized and counted to calculate MOI. Cells were infected at either MOI of 1 or 5, as noted in the text and figure legends. Appropriate amount of viral stocks thawed at room temperature were mixed in PBS to a final volume of 3.5 mL. Culture dishes were aspirated, the viral solution was added, and cells were returned to the incubator for 1 hour with rocking every 15 minutes. At the end of the hour, 4 mL complete media was added to the viral solution in each plate.
Immunofluorescence Analysis and Confocal Microscopy
Cells seeded in 8-chambered slides (Nunc Lab-Tek Chamber Slide system; Sigma C7182) were infected with ZIKV at MOI of 5. Two days post-infection, cells were either mock-treated or treated with sodium-arsenite (2.5 mM final concentration; Sigma) for 30 minutes. Hereafter the cells were washed three times with phosphate-buffered saline (PBS) and fixed with paraformaldehyde (4%)-PBS for 10 minutes at room temperature. Cells were washed once with PBS and permeabilized with 100% iced methanol for 15 minutes at room temperature. Cells were washed in blocking buffer (PBS-1% fish gelatin (FG); Sigma) three times for 15 minutes. Primary antibody was diluted in blocking buffer, added to the appropriate wells and incubated overnight at 4°C. Secondary antibodies diluted in blocking buffer were added for 1 hour at room temperature in the dark. Hoechst-33342 (Life Technologies) was applied for 15 minutes. Between and after application of antibodies, cells were washed with blocking buffer three times for 15 minutes. Finally, the cells were washed twice with PBS for five minutes, the 8-chamber upper structure was removed, Fluoromount (Southern Biotech) added, and a cover-slip applied. Slides were stored at 4°C if imaging was undertaken within 1 week, or at −20°C for long-term storage. Antibodies and concentrations used are listed in Supplemental Table 2. Slides were imaged on a Zeiss LM710 confocal microscope with a 63× oil objective.
Quantification of SG-positive Infected-Cells
A 40× oil objective was used to acquire images of ZIKV-infected cells stained with mouse-anti-dsRNA antibody to detect ZIKV-infected cells and one of the following anti-SG protein antibodies: rabbit-anti-Ataxin-2, rabbit-anti-G3BP1, rabbit-anti-HuR, goat-anti-TIA-1, rabbit-anti-TIAR, and rabbit-anti-YB1. A combined 200 to 300 infected cells between three or more biological replicates were counted and the ratio of infected cells presenting with or without SGs was taken. A two-tailed student T-test was performed using the mean percentage of infected cells presenting with or without SGs in mock- and arsenite-treated samples from at least three biological replicates.
Harvest of ZIKV-infected cells for Protein and RNA Analysis
At the indicated time-point, media from culture dishes was aspirated and the cells were washed once with PBS. Cells were manually scraped from plate in one ml PBS, divided between two microcentrifuge tubes, and pelleted at 14,000 RPM for 15 seconds. The supernatant was removed, and cells were resuspended in 50 μL RIPA buffer containing protease inhibitors (mini tablets EDTA-free; ThermoScientific Pierce) or 1 mL TRIzol (Invitrogen) for protein and RNA respectively. To isolate proteins, cells resuspended in RIPA buffer with protease inhibitors were incubated on ice for 20 minutes. The lysate was clarified by centrifugation at 14,000 RPM at 4°C for 20 minutes. The clarified protein lysate was collected and processed for western blot analysis. RNA from cell lysates was extracted using TRIzol per the manufacturer’s instructions.
Western Blot Analysis
Protein lysates were quantified using the DC protein assay kit (BioRad) and 20 μg lysate was fractionated for two hours at 100V through SDS-10% PAGE in running buffer (25 mM Tris-base, 190 mM Glycine, 3.5 mM SDS). Protein was transferred to a PVDF membrane at 100 V for 60 minutes at 4°C with transfer buffer (25 mM Tris-base 190 mM Glycine, and 20% methanol). Transfer efficiency was determined by Ponceau S (Sigma) staining, after which the blots were washed in buffer (0.1% Tween in PBS (PBS-T)) and blocked for 30 minutes in blocking buffer (5% powdered milk (w/v) in PBS-T). Primary antibodies diluted in blocking buffer were added to the blots and incubated at 4°C overnight. Secondary antibodies diluted in blocking buffer were incubated at room temperature for 1 hour. The assay was developed using Clarity Western ECL Blotting Substrate (BioRad) and imaged on a chemiluminescent imager (BioRad). Prior to application of additional antibody, blots were stripped using ReBlot Plus Mild (Millipore Sigma) according to manufacturer’s suggestion. Before and after application of primary/secondary antibodies and stripping, blots were washed for 15 minutes three times in wash buffer. Antibodies and concentrations are listed in Supplemental Table 2.
Northern Blot Analysis
TRIzol-extracted RNA (10 μg) was resuspended in loading buffer (1× MOPS-EDTA-sodium acetate (MESA; Sigma), 4.5% formaldehyde, and 32% formamide), denatured for 15 minutes at 65°C, separated in a 1.2% agarose gel containing 5.92% formaldehyde (v/v) and 1× MESA (v/v), and transferred via capillary action to a Zeta-probe membrane (BioRad) overnight at room temperature. RNA was crosslinked to the membrane using a TL-2000 (UVP) and then stained with methylene blue to visualize transfer of RNA. Methylene blue was removed by washing the membranes in 1×SSC/1%SDS for 15 minutes three times, and then prehybridized in 5mL ExpressHyb (ClonTech) for 1 hour at 65°C. The hybridization buffer was changed and a 32P-labeled dsDNA probe (Invitrogen) was added for 1 hour at 65°C. Blots were then washed with 0.1×SSC/0.1%SDS three times for 15 minutes at 55°C, exposed overnight to a phosphorimager screen and, subsequently visualized on a Typhoon 9400 (GE). The ZIKV 3’UTR probe (targeting nt 10324-10808 of the viral genome) was generated by RT-PCR amplification of the 3’ UTR region from ZIKV-infected cells and cloning of the cDNA into pCR-TOPO2.1 (Invitrogen). The actin and ZIKV probes were randomly labeled with α32P dATP (Perkin Elmer) using a RadPrime kit (Invitrogen).
Isolation of ZIKV Replication Complexes
Viral replication complexes were isolated following a modified protocol adapted from Schlegel et al. (1996) and Chen et al. (2015) (43, 44). Mock- and ZIKV-infected Huh7 cells were harvested at 2 days post-infection. Specifically, cells were washed twice with cold PBS, gently dislodged using a cell lifter, collected and pelleted at 1,000 ×g for 5 minutes. The cell pellets were resuspended in 900 µL hypotonic buffer (10 mM Tris-HCl (pH 8.0), 10 mM NaCl, 1 mM MgCl2) containing protease and phosphatase inhibitors (EDTA-free; Pierce) and then incubated on ice for 10 minutes. After incubation, cells were homogenized using the tight pestle of a Dounce homogenizer approximately 50 times on ice. Samples were then centrifuged at 10,000 ×g for 10 minutes at 4°C, the supernatant collected and NaCl was added to the supernatants to a final concentration of 300 mM. The replication complexes in the collected supernatant were separated in a 10%/60% gradient. The 10%/60% sucrose gradient was prepared in Ultraclear tubes (Beckman Coulter) by first adding 5mL of 10% sucrose solution (300 mM NaCl, 15 mM Tris-HCl [pH 7.5], 15 mM MgCl2, 0.1% heparin and 10% sucrose), followed by a slow deposition of 5mL 60% sucrose (300 mM NaCl, 15 mM Tris-HCl [pH 7.5], 15 mM MgCl2, 0.1% heparin and 60% sucrose) at the bottom of the tube underneath the 10% sucrose layer. Five percent of the supernatant layered onto the sucrose gradient was reserved as the input sample. Samples were layered onto the sucrose gradients and ultracentrifuged using the SW-41 rotor at 115,915 ×g for 16 hours at 4°C. The opaque layer sedimenting between the 10 and 60% sucrose layers was collected for analysis. For western blot analysis the proteins were precipitated with methanol and resuspended in a buffer containing 8 M urea and 100 mM Tris-HCl (pH 8).
Plaque Assays
Six-well plates were seeded with 6×105 Vero cells per well. The following day, serial dilutions of virus were prepared in PBS, media was aspirated, and 300μL of appropriate dilution was added. Cultures were returned to incubator for 1 hour with rocking every 15 min. After incubation, 2.5mL of a 1:1 overlay (1.2% Oxoid agar and modified media (2×DMEM, 4%FBS, 10mM HEPES)) was added to each well. Agar was left to solidify at room temperature for 10 minutes before returning the plates to incubator. At 4 days post-infection, plaques were developed using 1% crystal violet in 20% methanol.
Luciferase Reporter Virus and Assays
To construct a ZIKV reporter virus, the Gaussia luciferase gene was cloned into the previously described plasmid pCDNA6.2 MR766 Intron3127 HDVr encoding a CMV promoter driven MR766 ZIKV (45). In particular, Gaussia luciferase was cloned between the NS1 and NS2A coding sequences, which was identified as a region of the viral genome that tolerated insertions in a transposon mutagenesis screen (46). The full-length Gaussia luciferase reporter gene, along with its signal sequence, was inserted into the viral genome as a translational fusion and flanked at both termini with 18 nucleotides encoding the NS1/NS2A junction, with the intent of providing proteolytic cleavage sites to excise Gaussia luciferase from the rest of the polyprotein. The sequences for the 5’-end and 3’-end of this cassette were 5’-AGG TCA ATG GTG ACc GCG GGG TCA ACC GAT CAT ATG ATG GGA GTC AAA GTT CTG TTT GCC-3’ and 5’-AAG ATC AAG GGG GCC GGT GGT GAC AGG TCA ATG GTG ACc GCG GGG TCA ACC GAT CA-3’, respectively, with the Gaussia luciferase gene underlined and a silent mutation to introduce a SacII restriction site in lower case. This plasmid was termed pCDNA6.2 MR766 clGLuc Intron3127 HDVr. To create a replication incompetent version of this plasmid, this reporter cassette was also cloned into the previously described pCDNA6.2 MR766 Intron3127 Pol(-) HDVr (45) to create pCDNA6.2 MR766 clGLuc Intron3127 Pol(-) HDVr. Huh7 cells were seeded into 6 cm dishes at a density of 5×105 cells/ml. The cells were transfected with siRNAs or the respective viral cDNA described above. Twenty-four hours post-transfection, cells were subsequently seeded into 24-well plates at a density of 5×104 cells/mL. The following day, 200 ng of the replication-competent and replication-incompetent pCDNA6.2 MR766 clGLuc Intron3127 HDVr, along with appropriate siRNAs (40 nM) or DNA plasmids (500 ng) were transfected using Lipofectamine 3000. At various time points post-transfection, media from wells was collected and stored at −20°C. Assays were performed using NEB BioLux Gaussia Luciferase (GLuc) kit (NEB #E3300L) following the stabilized protocol. Briefly, samples and GLuc assay solution were prepared and equilibrated to room temperature. The sample (35 μL) was added to three individual wells in a white 96-well plate. The GLuc assay solution (50 μL) was dispensed into each sample well, shaken for 5 seconds, incubated at room temperature for 30 seconds, and luminescence recorded with an integration time of 10 seconds using a BioTEK Synergy luminometer.
Statistical Analysis
Quantification of SGs between different infections was completed as follows: for each treatment, the percentage of uninfected cells with/without SGs and ZIKV-infected cells with/without SGs was calculated. Using the percentages from cells with/without SGs from three or more biological replicates, a one-tailed student T-test was carried out. For RNA quantification of northern blotting, ImageQuantTL was used to obtain the mean density of gZIKV, sgZIKV, and actin. RNA levels of ZIKV were initially divided by the mean density of actin and subsequently standardized to the control siGL2. A two-tailed student T-test comparing control RNA levels to each knockdown sample was performed using data from three or more biological replicates. Relative viral titers were determined by standardizing the plaque forming units/mL (PFU/mL) for each treatment to the control siGL2. A two-tailed student T-test was performed using relative viral titers from three biological replicates. All graphs were generated using Microsoft Excel for Mac 2011. Statistical analysis was performed using StatPlus:mac LE.
Results
ZIKV inhibits arsenite-induced SG formation
To determine whether SGs form during ZIKV infection, Huh7 cells were infected with the Cambodian-isolate (160310) at an MOI of 5, and the formation of SGs at 24 hours post-infection was visualized by immunofluorescence and confocal microscopy using an antibody to detect TIA-1, a protein known to facilitate nucleation of SGs (26). ZIKV-infected cells were identified by staining for double-stranded (ds) RNA, a marker of viral replication sites. In mock-infected cells, TIA-1 localized to the nucleus (Figure 1A). Similar to other flavivirus infected cells (29, 30), TIA-1 was mostly localized in the nucleus and we observed few-to-no SG foci in the cytoplasm of ZIKV-infected cells (Figure 1A). A lack of SGs during ZIKV-infection suggested ZIKV either inhibited the formation of stress granules or promoted the disassembly of stress granules. To investigate whether ZIKV inhibited the formation of SGs, we treated cells with 2.5 mM sodium arsenite for 30 minutes. In mock-infected cells treated with arsenite we observed ~90% of the cells containing TIA-1 SGs in the cytoplasm (Figure 1A and 1E). In contrast, TIA-1-containing SGs were observed in ~24% of ZIKV-infected treated with arsenite (Figure 1B and 1F).

Huh7 cells were either mock-infected or infected with the Cambodia ZIKV-strain (160310) at a MOI of 5. Cells were mock or arsenite-treated (2.5mM) for 30 minutes 24 hours post-infection, fixed, permeabilized, and analyzed using a 63× oil immersion objective on a Zeiss LSM710 laser scanning confocal microscope. ZIKV-infected cells were detected with mouse-anti-dsRNA antibody (green). A) Distribution of dsRNA (green) and TIA-1 (red) in mock- and ZIKV-infected cells not treated with arsenite. White arrows highlight stress granules. Quantification of SGs containing Ataxin-2, G3BP1, eIF3B, HuR, TIA-1, TIAR and YB1 proteins in B) mock- and C) ZIKV-infected cells. D) Formation of TIA-1 (red) SGs in mock- and ZIKV-infected cells treated with arsenite. ZIKV-infected cells were identified by staining for dsRNA (green). Quantification of Ataxin-2, G3BP1, eIF3B, HuR, TIA-1, TIAR and YB1-containing SGs in E) mock- and F) ZIKV-infected cells. Note, in the original immunofluorescence images, TIA-1 was visualized using anti-donkey Alexa Fluor® 647 (magenta) Donkey anti-Goat IgG secondary antibody. In panels A) and D) TIA-1 has been pseudo-colored red. The immunofluorescence images are representative of at least three independent experiments. The errors bars shown on the SG quantification data (Panel 1B, 1C, 1E and 1F) are of the mean ± SD. Significance was determined by one-tailed student T-test (*p<0.05; **p<0.01; ***p<0.001; and N.S. not significant).
We similarly investigated SG formation following infection with the 1947 Uganda-isolate (MR766) and a recent ZIKV strain isolated in Puerto Rico in 2015 (PRVABC59) (43, 44). In untreated cells infected with MR766 or PRVABC59, 3-9% of cells contained SGs (Figures S1). Consistent with the number of SGs in cells infected with the Cambodia ZIKV strain, 31-47% of cells infected with MR766 or PRVABC formed SGs following treatment with sodium arsenite. These data indicate that the three ZIKV strains (Cambodia, Uganda and Puerto Rico) tested inhibit the formation of arsenite-induced SGs and supports earlier studies with WNV, DENV and ZIKV (29, 31, 32, 35, 39).
To determine whether ZIKV inhibited a particular subset of SGs, we next investigated the formation of SGs containing Ataxin-2, G3BP1, eIF3B, HuR, TIAR, and YB1 in mock and ZIKV-infected cells in the absence or presence of sodium arsenite (Figures 1B-F). Quantification of the different SG proteins in mock-infected cells showed that less than 10% of untreated cells (Figures 1B) and more than 90% of cells treated with arsenite contained SGs (Figures 1E). In contrast, analysis of SGs as visualized using different SG proteins in cells infected with the Cambodia ZIKV-isolate (160310), showed that 92-97% of untreated cells and 73-82% of arsenite-treated did not contain SGs (Figures 1B and S2). Consistent with the TIA-1 data, 18-27% of ZIKV-infected cells treated with arsenite contained the SG components G3BP1, TIA-1, Ataxin-2, HuR, TIAR, and YB1 (Figures 1B and S1 [15-30% for SG markers in S1]). While ZIKV effectively blocked the assembly of arsenite-induced SGs containing common SG components, we observed that 44% of infected cells showed translation initiation factor eIF3B-containing SGs (Figure 1F and S2). Interestingly we observed that the Cambodian ZIKV-isolate (160310), in contrast to the Ugandan and Puerto Rican strains, was particularly effective at inhibiting the formation of Ataxin-2, G3BP1, HuR, TIA-1, TIAR, and YB1-containing SGs (Figure 1 and S1). These data show that all three ZIKV inhibit the formation of SGs albeit to different extents. Interestingly, we observed a higher number of SGs containing the eIF3B (Figure 1 and S2), indicating that the SGs that are induced in ZIKV-infected cells contain translationally repressed ribonucleoprotein complexes.
ZIKV does not change the integrity or abundance of SG proteins during infection
To determine if the ability of ZIKV to inhibit arsenite-induced SG assembly is a result of promoting proteolytic cleavage of and/or degrading SG-nucleating proteins, we infected Huh7 cells with ZIKV at an MOI of 1 and 5, and investigated the abundance and integrity of select SG proteins at 1, 2, and 3 days post-infection. By western blot analysis we found that the abundance of most SG proteins remained the same between mock- and ZIKV-infected samples (Figure 2A and data not shown), and no cleavage products were identified (Figure 2A and data not shown). Interestingly, independent of MOI, the levels of Ataxin-2 increased with ZIKV infection at 1- and 2-days post-infection (Figure 2A). We similarly observed an increase in Ataxin-2 abundance and no change in other SG protein levels following infections with the Ugandan and Puerto Rican strains (Figure S3). Therefore, the ability of ZIKV to block the assembly of stress granules was not a result of proteolytic cleavage or a change in the abundance of SG proteins.
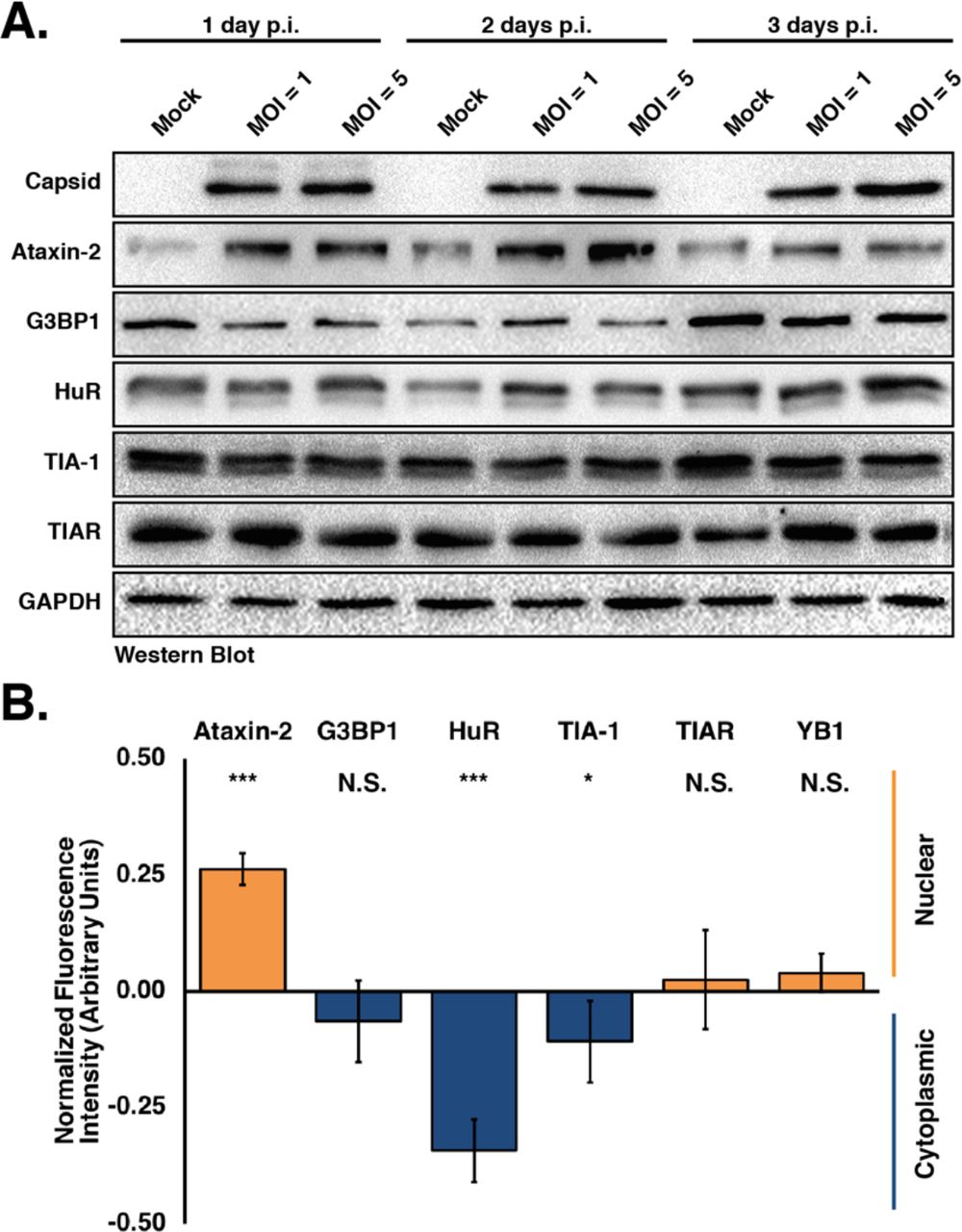
A) Huh7 cells were infected with the Cambodia strain of ZIKV (160310) at MOI of 1 or 5 were harvested at 1, 2, and 3 days post-infection. Detection of ZIKV capsid protein by western blot confirms viral infection. The abundance and integrity of Ataxin-3, G3BP1, HuR, TIA-1 and TIAR proteins was also examined at each time point. The western blot is representative of at least three independent experiments. B) The subcellular distribution of Ataxin-3, G3BP1, HuR, TIA-1, TIAR and YB1 was examined by quantifying the nuclear and cytoplasmic fluorescence intensity signal of each protein. Fluorescence intensity was derived in ImageJ using a freehand selection of the nucleus in mock- and ZIKV-infected cells. Values obtained from each SG marker were divided by the nucleus signal (Hoescht) within uninfected and infected cells. Fluorescence intensity of the infected cells were standardized against uninfected cells and arbitrarily assigned a zero value. Negative values (i.e. less than 0.00) suggest increased localization in the cytoplasm (blue bars). Conversely, positive values (i.e. greater than 0.00) suggest increased localization in the nucleus (orange bars). Two-tailed student T-tests were performed to calculate significance with *p<0.05, ***p<0.001 and N.S. not significant.
SG proteins are re-localized during ZIKV infection
To investigate whether SG proteins are re-localized during ZIKV infection, we examined the relative distribution of different SG markers in the cytoplasm versus nucleus. In particular, the fluorescence signal of Ataxin-2, G3BP1, HuR, TIA-1, TIAR, and YB1 from the nucleus and cytoplasm in confocal microscopy images was quantified. The relative fluorescence intensity signal of each SG protein in mock-infected cells was set to 1, where values in ZIKV-infected cells greater than 1 indicated that the SG protein was localized more in the nucleus, versus values less than 1 indicating that the SG protein showed increased cytoplasmic localization (Figure 2B). These analyses revealed that the distribution of G3BP1 and TIAR between the cytoplasm and nucleus did not change compared to mock-infected cells (Figure 2B). Interestingly, during ZIKV infection we observed that Ataxin-2 and YB1 showed increased nuclear localization (Figure 2B). Conversely, more HuR and TIA-1, two RNA-binding proteins predominantly localized in the nucleus, was localized to the cytoplasm during ZIKV infection, although TIA-1 was re-localized to a lesser extent than HuR (Figure 2B). Therefore, the change in the distribution of select SG proteins between the nucleus and the cytoplasm may contribute to the absence of SGs during ZIKV infection.
G3BP1 and HuR modulate ZIKV gene expression
The re-localization of SG proteins during ZIKV infection raised the possibility that ZIKV subverts select SG proteins to modulate viral gene expression. To elucidate the role of SG proteins during ZIKV infection we transfected Huh7 cells with siRNAs targeting six different SG proteins and infected with the Cambodian-isolate (160310) at an MOI of 5. Two days post-infection cells were harvested for protein and RNA analyses by western and northern blot, and media was collected for plaque assays. Western blot analysis showed that the siRNAs depleted the abundance of the targeted SG proteins (Figure 3A). Additionally, depletion of individual SG proteins did not impact the levels of other SG markers (Figure 3A). Although the abundance of ZIKV capsid protein (Figure 3A, top) did not change following depletion of Ataxin-2, we observed a dramatic decrease in viral protein when G3BP1 was depleted. In contrast, depletion of HuR and TIA-1 substantially increased ZIKV capsid protein levels. Depletion of TIAR and YB1 similarly increased the levels of ZIKV capsid protein, albeit not to the same extent as depletion of HuR and TIA-1 (Figure 3A).
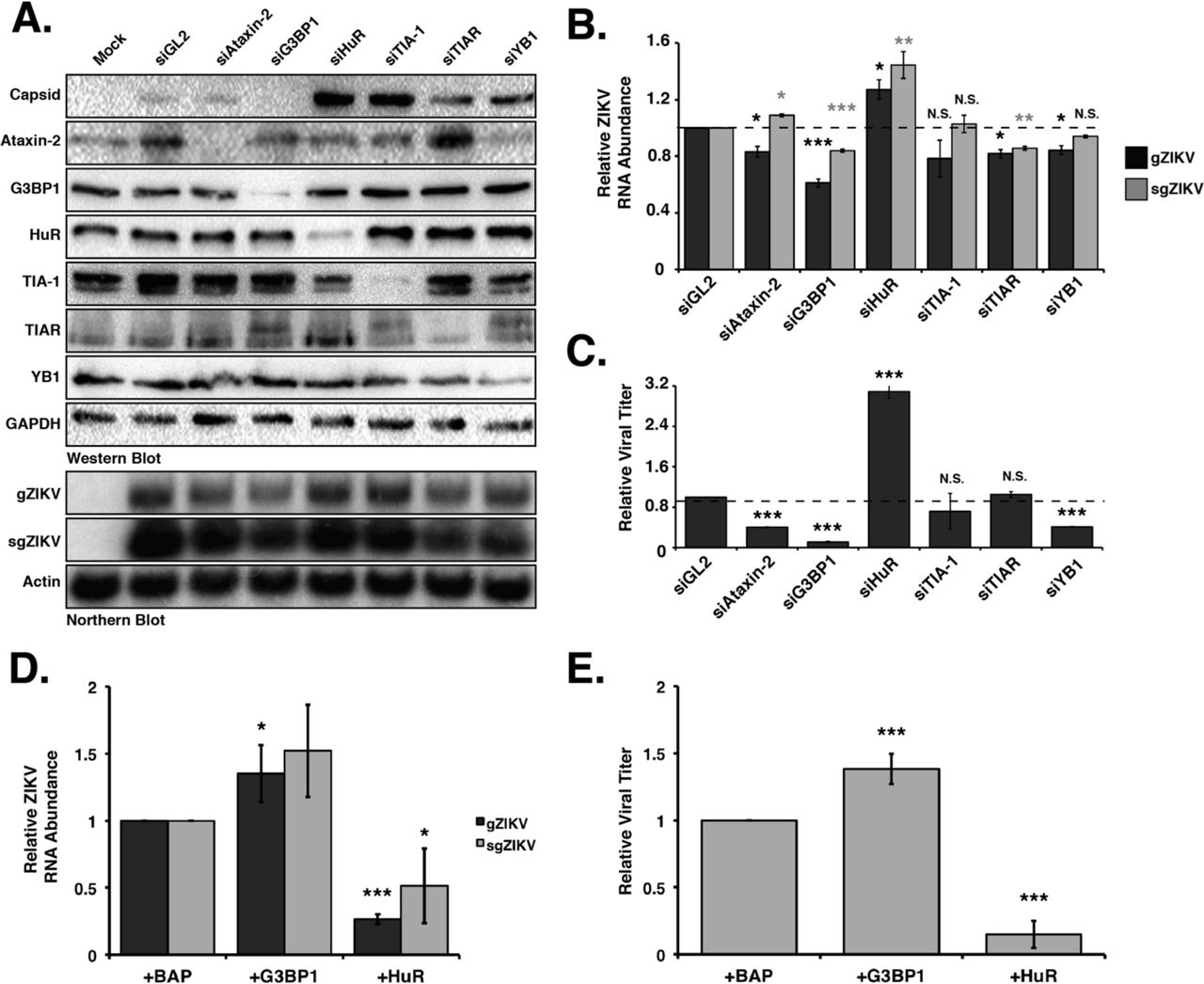
Ataxin-2, G3BP1, HuR, TIA-1, TIA-1, TIAR and YB1 proteins were depleted in Huh7 cells using target-specific siRNAs, and then infected 24 hours later at a MOI of 5 with the Cambodia strain of ZIKV (160310). A siRNA targeting Gaussia luciferase (siGL2) was used as a control, non-targeting siRNA. Protein, RNA and media were harvested 48 hours post-infection. A) Western blot shows expression of ZIKV capsid, and depletion of each stress granule protein. Expression of GAPDH was used as a loading control. The northern blot shows the effect of depleting each stress granule protein on the abundance of the ZIKV genomic (gZIKV) and subgenomic (sgZIKV) RNA. Actin mRNA levels were evaluated as a loading control. B) Quantification of ZIKV gRNA and sgRNA. ZIKV RNA levels were normalized to actin mRNA, and then represented relative to siGL2 control. C) Viral titers in the extracellular media were determined by plaque assay and relative viral titers were calculated by normalizing titers relative to siGL2 transfected cells. D) Quantification of ZIKV RNA levels following overexpression of G3BP1-Flag and HuR-Flag. Huh7 cells were transfected with plasmids expressing 3xFlag-Bacterial Alkaline Phosphatase (BAP; control), G3BP1-Flag or HuR-Flag, and then infected with ZIKV (Cambodia (160310) 24 hours post-infection. The effect of overexpressing G3BP1-Flag and HuR-Flag on gZIKV and sgZIKV RNA was evaluated by northern blot. E) Effect of overexpression G3BP1-Flag and HuR-Flag on viral titers. Data present in Figure 3 are from at least three independent experiments. Error bars show mean ± SD, and statistical significance was determined using a two-tailed student T-test. *p<0.05; **p<0.01; ***p<0.001; N.S. not significant).
We next examined the abundance of ZIKV genomic RNA (gRNA). By northern blot we observed significant reductions in genomic ZIKV RNA (gZIKV) levels following depletion of G3BP1 compared to cells transfected with the control siRNA, siGL2 (Figure 3B). Depletion of Ataxin-2, TIAR and YB1 also decreased ZIKV genomic RNA (Figure 3B). Depletion of TIA-1 did not alter the levels of viral RNA (Figure 3B). Consistent with the increase in ZIKV capsid protein, depletion of HuR significantly increased gZIKV levels (Figure 3B). During flaviviral infections a small noncoding viral RNA (sgRNA), corresponding to the 3’ UTR, is produced by incomplete degradation of the ZIKV gRNA by the cellular 5’-to-3’ exonuclease Xrn1 (47). By northern blot of the gRNA we also detected the sgZIKV RNA (Figure 3B). Overall a decrease in ZIKV gRNA coincided with a decrease in sgRNA, although the abundance of sgRNA was greater than the gRNA, likely because of Xrn1 degradation of both translating and replicating ZIKV RNA (Figure 3B).
We next examined the effect of depleting SG proteins on viral titers by plaque assay (Figure 3C). While, depletion of TIA-1 and TIAR had no effect on the production of infectious virus, depletion of Ataxin-2, G3BP1 and YB1 significantly reduced viral titers. Consistent with the increase in ZIKV protein and RNA following depletion of HuR, we also observed an increase in viral titers (Figure 3C).
To confirm the effect of G3BP1 and HuR on ZIKV gene expression, we overexpressed G3BP1-Flag and HuR-Flag in ZIKV-infected cells and examined viral RNA levels and viral titers (Figure 3D and 3E). Analysis of viral RNA abundance showed that overexpression of G3BP1 and HuR significantly increases and decreases ZIKV RNA respectively (Figure 3D). Consistent with the effects on ZIKV RNA, we observed an increase and decrease in viral titers following overexpression of G3BP1 and HuR, respectively (Figure 3E). Together these data indicate that TIA-1, TIAR and YB1 promote translation of the ZIKV polyprotein, but limit replication of the viral genome. YB1 may additionally have a role in virus assembly. Ataxin-2 and G3BP1 are proviral cellular factors as depletion of Ataxin-2 and G3BP1 decreased ZIKV RNA levels, and overexpression of G3BP1 increased viral RNA, which in turn affected the production of infectious particles (Figure 3). Finally, HuR exhibits antiviral activity as depletion of this RNA binding protein significantly increased ZIKV protein and RNA levels and viral titers, and overexpression of HuR-Flag showed a reciprocal effect on ZIKV gene expression (Figure 3).
G3BP1 and HuR abundance modulates ZIKV replication
Depletion of G3BP1 and HuR significantly decreased and increased ZIKV gene expression respectively, suggesting a role for these SG proteins in ZIKV translation or replication. To decipher the function of G3BP1 and HuR in ZIKV gene expression, we used the replication-competent (WT) and -deficient (Pol-) Gaussia luciferase (GLuc) ZIKV reporter plasmid (pCDNA6.2 MR766 clGLuc Intron3127 HDVr Figure 4A). These pCDNA6.2 MR766 clGLuc Intron3127 HDVr constructs were derived from the cDNA clone containing the full-length genome of the Uganda MR766 strain under the control of the cytomegalovirus (CMV) promoter (Figure 4A) (45). Within the ZIKV coding region, the GLuc gene was cloned as a translational fusion between the NS1 and NS2A genes. The NS1 and NS2A junction was duplicated on both sides of the reporter gene to provide a means for GLuc to be cleaved out of the polyprotein. Following transfection of this reporter construct, transcription and 5’-end capping of ZIKV genomic RNA are directed by the host; hepatitis D virus ribozyme (HDVr) at the end of the 3’ UTR trims the ZIKV genomic RNA to create an authentic 3’ end (Figure 4A) (45). Using expression of GLuc as a proxy for viral RNA, translation of capped ZIKV RNA is detected 6 hours post-transfection. Translation of the ZIKV polyprotein, including the synthesis of NS5, the RNA-dependent-RNA polymerase (RdRp), directs replication of the reporter genome such that the increase in viral RNA as a result of replication is observed at 48- and 96-hours post-transfection. We also expressed a ZIKV GLuc reporter genome that contained a mutation in the RdRp active site (GDD-to-GNN; pCDNA6.2 MR766 clGLuc Intron3127 Pol(-) HDVr) which rendered this mutant GLuc reporter genome replication-deficient (Pol-). Using the Pol(-) reporter genome we were clearly able to show a difference between replicating WT genomes and ZIKV RNA transcribed from the transfected plasmid at 48- and 96-hours post-transfection (Figure 4B-4E).
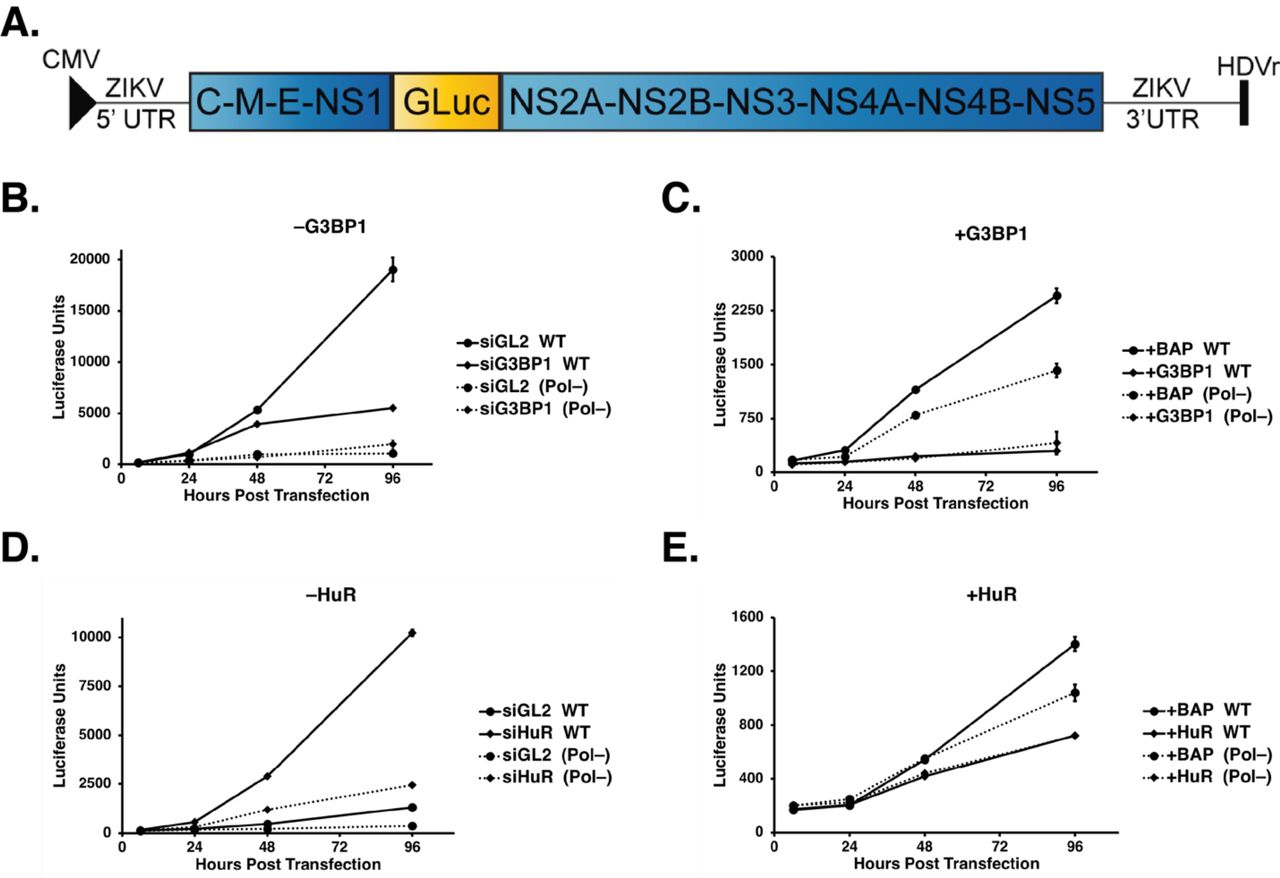
A) Schematic of the ZIKV MR766 Gaussia luciferase reporter genome (pCDNA6.2 MR766 clGLuc Intron3127 HDVr) showing the positions of the 5’-end cap, 5’ and 3’ UTRs, the mature viral proteins within the single-open reading frame, as well as the position of the Gaussia luciferase gene within the MR766 genome. Two elements within the plasmid namely the CMV promoter and the HDVr, which creates an authentic 3’ UTR in the genome, are also denoted. B-E) Huh7 cells were first transfected with either the control or target-specific siRNAs, or p3×Flag-BAP, pG3BP1-Flag or pHuR-Flag plasmids. These cells were subsequently transfected with the pCDNA6.2 MR766 clGLuc Intron3127 HDVr WT replication-competent or Pol(-) replication-deficient plasmids, and GLuc activity in the media of transfected cells was assayed at 6, 24, 48, and 96 hours post-transfection. B) Effect of G3BP1 knockdown on ZIKV-GLuc genome expression. C) Effect on ZIKV-GLuc reporter genome expression following overexpression of 3×Flag-BAP (control) and G3BP1-Flag. D) Effect of depleting HuR on ZIKV-GLuc genome expression. E) Effect of overexpressing 3×Flag-BAP and HuR-Flag on ZIKV-GLuc genome expression. The data shown in Figure 4B-4E are from a single experiment and are representative of at least three independent experiments. Error bars indicate mean ± SD.
To elucidate effects on translation and replication we examined expression of the ZIKV GLuc reporter genome following siRNA depletion or overexpression of G3BP1 and HuR (Figure 4B-E). RNAi depletion or overexpression of G3BP1 and HuR did not significantly affect GLuc expression at 6- and 24-hours post-transfection of both the ZIKV WT and Pol- reporter genomes (Figure 4B-E), indicating that these two SG proteins do not affect translation of the ZIKV genome (Figure 4B-E). At 96 hours post-transfection of the WT reporter genome however, siRNA depletion and overexpression of G3BP1 significantly decreased and increased GLuc expression respectively, showing that G3BP1 facilitates ZIKV replication (Figure 4B and 4C). Conversely, at 96 hours post-transfection depletion of HuR dramatically increased GLuc expression (Figure 4D). Reciprocally, overexpression of HuR significantly decreased luciferase units compared to expression of the 3×Flag-BAP control (Figure 4E), indicating that HuR negatively impacts the ZIKV replication. Together these data indicate a role for G3BP1 and HuR in replication of the ZIKV genome.
Select SG proteins localize at ZIKV replication complexes
To further demonstrate a role for G3BP1 and HuR in ZIKV replication, we next examined the localization of these two SG proteins at replication complexes (Figure 5). We first examined the localization of G3BP1 and HuR at replication complexes by immunofluorescence analysis using dsRNA and ZIKV NS5 protein as markers for replication sites (Figure 5A and 5C). We observed strong colocalization of G3BP1 with dsRNA (Figure 5A). Although quantification of the fluorescence signal intensity showed that HuR in ZIKV-infected cells was re-localized into the cytoplasm (Figure 2B), HuR did not co-localize with dsRNA (Figure 5A). NS5 is critical for the replication of the ZIKV RNA as it functions as the RdRp and the methyltransferase that caps the 5’ end of the ZIKV genome (1). Although visualization of ZIKV replication complexes using the anti-dsRNA antibody showed clear cytoplasmic localization of replication complexes (Figure 5A), strong nuclear localization and weak cytoplasmic localization was observed for the ZIKV NS5 protein (Figure 5B). The nuclear localization of ZIKV NS5 is not surprising considering that flaviviruses RdRps contain functional nuclear localization signals (NLS) (48–50), and that Dengue NS5 modulates splicing of cellular mRNA (51). We observed co-localization of NS5 and HuR in the nucleus, although no HuR-NS5 co-localization was observed in the NS5 nuclear foci (Figure 5B). It is therefore possible that HuR might affect NS5 activities in the nucleus to negatively impact ZIKV gene expression (Figure 3 and Figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A-B) Huh7 cells infected with ZIKV at a MOI of 5 were fixed, permeabilized, and prepared for confocal imaging at 24 hours post-infection. ZIKV-infected cells were visualized using antibodies against ZIKV dsRNA and NS5 to identify sites of viral replication, while the envelope antibody was used to visualize possible viral assembly sites. White arrows highlight areas of colocalization. A) Localization of ZIKV dsRNA, NS5 and envelope proteins with G3BP1 in Huh7 cells. B) Localization of ZIKV dsRNA, NS5 and envelope proteins with HuR in Huh7 cells. Immunofluorescence images are representative of at least three independent experiments. C) Western blot analysis of the enrichment of Ataxin-2, G3BP1, HuR, TIA-1 and YB1 on replication complexes isolated from mock- and ZIKV-infected Huh7 cells. The western blot is representative of two independent experiments.
To corroborate the immunofluorescence analyses we also isolated replication complexes from mock- and ZIKV-infected Huh7 cells by sucrose gradient ultracentrifugation, and then investigated the localization of select SG proteins with replication complexes. Western blot analysis showed that G3BP1 and HuR were isolated with the replication complexes (Figure 5C). Although TIA-1 and YB1 have previously been shown to affect WNV replication (29, 36), we only observed a small amount of these proteins in the enriched membranes (Figure 5C). While we noted an increase in the Ataxin-2 levels in ZIKV infected cells (Figure 2A), Ataxin-2 did not sediment with replication complexes (Figure 5C). That G3BP1 and HuR were isolated with replication complexes (Figure 5C) further indicates a role for G3BP1 and HuR in ZIKV replication (Figure 4).
To investigate whether G3BP1 and HuR also co-localized with ZIKV assembly sites, we examined the co-localization of both proteins with ZIKV envelope protein by immunofluorescence analysis (Figure 5A and 5B). Interestingly, we observed that G3BP1 showed co-localization with the envelope protein (Figure 5A). HuR, however, did not co-localize with the envelope protein (Figure 5B). Therefore, G3BP1 might function more broadly than previously recognized, to promote replication of the ZIKV genome as well as assembly of new virions.
Discussion
The formation and sequestration of components of the translation machinery in SGs function as an intrinsic antiviral mechanism to restrict viral translation and/or replication. Flaviviruses have been shown to limit stress granule assembly by affecting the antioxidant response (31), changing the phosphorylation of the cap-binding protein eIF4E by altering p38-Mnk1 signaling (32), and increasing the rate of eIF2α dephosphorylation (35). We similarly observed fewer SGs in ZIKV-infected cells, and that the assembly of SGs was inhibited following arsenite-treatment. In contrast to previous studies we systematically investigated the effect of six different SG proteins on ZIKV protein and RNA abundance, production of infectious particles, the co-localization with ZIKV and effects on replication. Our study reveals that G3BP1 may function more broadly in the infectious cycle than previously recognized. Excitingly, our data show that HuR functions to limit ZIKV replication, which to our knowledge has not previously been shown for this cellular RNA binding protein. This systematic analysis of SG proteins during ZIKV infection is significant particularly as G3BP1, HuR and other SG proteins have an intimate role in neurodevelopment and function, which raises the possibility that ZIKV modulation of SGs and SG proteins may have a more significant impact on normal gene regulation that contributes to ZIKV pathogenesis.
Ataxin-2 is a cytosolic SG protein that is associated with the neurodegenerative disorder spinocerebellar ataxia type-2 (SCA2) (52, 53). During ZIKV infection we observed an increase in the abundance of Ataxin-2 (Figure 2A) and more Ataxin-2 localized in the nucleus (Figure 2B). Furthermore, RNAi studies showed that ZIKV gene expression requires Ataxin-2 (Figure 3B and 3C). Ataxin-2 levels have been shown to affect the abundance of PolyA binding protein (PABP) (53), where elevated levels of Ataxin-2 decreased the amount of PABP (53). Such changes in PABP would be expected to negatively affect translation of cellular mRNAs and promote ZIKV gene expression. Moreover, Ataxin-2 interacts with the cellular RNA DEAD-box helicase DDX6 (53), which we have previously shown to be required for ZIKV gene expression (54). Therefore, the impact on ZIKV RNA and production of infectious particles (Figure 3B and 3C) following RNAi depletion of Ataxin-2 might also be the result of changes to DDX6. Analysis of SCA2 brains showed an increase in Ataxin-2 levels and that Ataxin-2 localized in intranuclear inclusions in 1-2% of neurons (55, 56). Our data showing that Ataxin-2 modulates ZIKV gene expression in the hepatocellular carcinoma cell line Huh7 is exciting and additional studies in neuronal cells would illuminate the role of this protein in cellular dysfunction and neurodegeneration following intrauterine ZIKV infection.
The RNA-binding proteins TIA-1 and TIAR are broadly expressed in cells and tissues (57). These proteins are predominantly localized in the nucleus (Figure 1 and S1), have been shown to modulate translation of specific cellular mRNAs (58) and are critical nucleators of stress granules in the cytoplasm (26). During WNV and DENV infection TIA-1 and TIAR colocalize at replication sites, and bind to the 3’ stem loop region in the negative-strand of WNV RNA to promote replication (29, 36, 59). We determined by western blot analysis that TIA-1 localized in isolated replication complexes (Figure 5C). Because TIA-1 and TIAR were previously shown to promote WNV replication we also investigated the role of TIA-1 and TIAR during ZIKV infection. Similar to Hou et al. (39), we found that decreasing the levels of TIA-1 had no effect on viral titers (Figure 3C). Although Hou et al. showed that depletion of TIAR decreased ZIKV titers (39), we observed no effect (Figure 3C). Our RNAi studies also demonstrated that following the reduction of TIA-1 and TIAR levels, the abundance of ZIKV capsid protein increased (Figure 3A), and depletion of TIAR decreased ZIKV RNA levels (Figure 3A and 3B), suggesting that, similar to tick-borne encephalitis virus, TIA-1 and TIAR are re-localized to perinuclear regions of replication to modulate translation of the ZIKV polyprotein (60). While the role of TIA-1 and TIAR in our studies in Huh7 differs from the Hou et al studies undertaken in A549 cells (39), the impact of TIA-1 and TIAR might be better elucidated in neuronal cells were a link between TIA-1, the microtubule protein Tau and the neurodegenerative tauopathies has been described (61, 62).
Y box-binding protein-1 (YB1) modulates translation and stabilizes cellular mRNAs (58) and also localizes in processing bodies and stress granules (63). YB-1 has been shown to bind the DENV 3’ UTR to decrease DENV translation (64). In our study, siRNA depletion of YB-1 increased the levels of ZIKV capsid protein (Figure 3A) indicating that YB-1, similar to the role in DENV infection, negatively regulates ZIKV translation. We also observed a small decrease in ZIKV RNA levels (Figure 3A and 3B). Notably however, the modest effects of YB-1 depletion on ZIKV protein and RNA levels significantly decreased viral titers (Figure 3C).
G3BP1, similar to TIA-1, is an important nucleator of SGs (25, 27, 65). Interestingly, the role of G3BP1 varies in viral infections from limiting alphavirus replication (66–68) to interacting with the HCV RdRp NS5B and enhancing HCV infectious virus production (42, 69, 70). In DENV-infected cells, G3BP1 interacts with the small flaviviral RNA (37, 71), and sequestration of G3BP1 by the sfRNA was shown to inhibit ISG translation (38). Hou et al., showed that siRNA depletion of G3BP1 decreased ZIKV titers and that G3BP1 also bound ZIKV RNA (39). We observed a decreased ZIKV protein and RNA levels and a dramatic reduction in ZIKV titers following depletion of G3BP1 (Figure 3). G3BP1 was also identified at isolated replication complexes, and co-localized with dsRNA and ZIKV NS5 (Figure 5A and 5C). We further demonstrated a role for G3BP1 in replication using the ZIKV MR766 GLuc reporter genome (Figure 4D and 4E). Interestingly, G3BP1 co-localized with the ZIKV envelope protein (Figure 5A), which together with the decrease or increase in viral titers following G3BP1 depletion or overexpression respectively (Figure 3C and Figure 4B) indicate that G3BP1 might also function in ZIKV assembly.
Human antigen R (HuR) is a member of the ELAVL RNA-binding protein family and is ubiquitously expressed (72). Notably, the three additional members of the ELAVL family (HuB/C/D) are highly expressed in the brain (72). HuR binding to poly-U and AU-rich elements within the 3’ UTR increases mRNA stability (73, 74). During infection with the single-stranded positive-sense alphaviruses Sindbis virus (SINV) and Chikungunya virus (CHIKV), HuR is dramatically re-localized from the nucleus into the cytoplasm (75, 76). By quantification of the fluorescence intensity signal of HuR between mock and ZIKV-infected cells, and the nucleus and cytoplasm we similarly observed that HuR is re-localized into the cytoplasm (Figure 2B), albeit not to the same extent as during alphavirus infection (75, 76). HuR is recruited to alphavirus RNA via the U-rich elements within the 3’ UTR and this interaction stabilizes and protects the viral RNA from the cellular mRNA decay machinery (76). Although HuR has been shown to promote alphavirus mRNA stability (75, 76) and HCV IRES-mediated translation (42, 77) our study indicates an antiviral role for HuR during ZIKV infection (Figure 3). In particular siRNA-depletion of HuR significantly increased ZIKV protein and RNA levels and viral titers (Figure 3). Although we did not observe co-localization of HuR with dsRNA, and only modest co-localization with cytoplasmic NS5 (Figure 5B), HuR was detected in isolated replication complexes (Figure 5C). A role for HuR in ZIKV replication was further supported by siRNA depletion and overexpression of HuR-Flag that increased and decreased replication of the MR766 GLuc reporter expression respectively (Figure 4F and 4G). The mode by which HuR limits ZIKV replication remains to be elucidated. A bioinformatic analysis of the ZIKV 3’ UTRs identified putative HuR binding sites (data not shown), raising the possibility that similar to SINV (76), HuR interaction with ZIKV RNA alters the subcellular localization of the HuR, and directly affects ZIKV replication, possibly by destabilizing, rather than stabilizing, the viral RNA. Alternatively, the consequence on ZIKV replication might be indirect where HuR re-localization affects cellular RNA homeostasis or ribostasis. Indeed, re-localization of HuR by SINV into the cytoplasm was shown to decrease cellular mRNA stability and alter mRNA splicing and nuclear polyadenylation (78). Regardless, determining the mechanism by which HuR functions during ZIKV will provide new insights into viral-host interactions, particularly for a protein that exhibits antiviral functions, which to our knowledge has not previously been shown.
Beyond aggregating stalled translation complexes, RNA binding proteins in stress granules also localize in neuronal granules. These granules regulate neuronal growth and synaptic plasticity (79, 80). Interestingly, a number of stress granule proteins such as Ataxin-2, G3BP1, TIA-1 and HuR are known to contribute to different neuropathologies (40, 80). While transcriptional changes have been reported to contribute to the neurological and developmental defects observed following intrauterine ZIKV-infection (81–84), the role of stress granules and granule proteins should not be overlooked. By limiting the formation of stress granules, and re-localizing and subverting specific stress granule proteins, alterations in RNA homeostasis, such as changes in RNA splicing, RNA stability and translation, could also contribute to ZIKV neuropathologies. To this end, elucidating the interactions of stress granule proteins and the cellular consequences particularly in neuronal cells could provide new insights into the pathomechanisms underlying ZIKV congenital disease.
Funding
This work was supported by start-up funds from University at Albany-SUNY and New York State, the University at Albany Presidential Initiatives Fund for Research and Scholarship (PIFRS) to CTP; and NIH grants (R21 AI133617-01 and R01 GM123050) to CTP.
Acknowledgements
We thank Dr. Brett Lindenbach (Yale University) for ZIKV Cambodian-isolate (160310) and Ugandan-isolate (MR766), and Dr. Laura Kramer (Wadsworth Center) and the CDC for the Puerto Rican-isolate (PRVABC59). We also thank members of the Pager lab, Marlene Belfort, Gabriele Fuchs and Ing-Nang Wang for valuable comments and suggestions on the manuscript.
References
- 1.↵
- 2.↵
- 3.↵
- 4.
- 5.
- 6.
- 7.
- 8.↵
- 9.↵
- 10.
- 11.
- 12.↵
- 13.↵
- 14.
- 15.
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.
- 83.
- 84.↵




