

Abstract
The developmentally important Notch pathway is implicated in the maintenance of cancer stem/progenitor cells in tumor hypoxia. Yet, the mechanisms that lead to Notch activation in hypoxia are not clear. AMP-activated protein kinase (AMPK), a major player in energy homeostasis, is activated by hypoxia. These considerations led us to investigate whether AMPK facilitates cancer progression through the Notch pathway under hypoxia. Our data revealed that activating AMPK through pharmacological agents or genetic approaches led to an increase in the levels of cleaved Notch1 protein, and Notch signaling, in invasive breast cancer cell lines. In contrast, inhibition or depletion of AMPK reduced cleaved Notch1 levels. Significantly, we show that the hypoxia-induced increase in cleaved Notch1 protein levels requires AMPK activation. Furthermore, we show that AMPK modulates cleaved Notch1 protein levels by increasing its stability. Mechanistically, we identified a reduction in interaction between Notch1 and Itch/AIP4, a known ubiquitin ligase for Notch, upon AMPK activation, thus bringing about reduced degradation of cleaved Notch1. This interaction was also disrupted under hypoxia and was influenced by AMPK modulation. Further, we identified AMPK-mediated positive regulation of Fyn activity in turn regulates Itch phosphorylation and its interaction with Notch1. Finally, we observed that inhibition of AMPK under hypoxia affected self-renewal and drug resistance adversely, suggesting that hypoxia-induced AMPK activation might potentiate breast cancer aggressiveness through the Notch pathway. Furthermore, high grade breast cancers that commonly show hypoxic regions revealed an association between AMPK activity and Notch signalling. Altogether, our study sheds light on context-specific oncogenic role of AMPK by reinforcing Notch1 signaling under hypoxia to facilitate breast cancer progression.
Financial support This work was majorly supported by grants from the Wellcome Trust-DBT India Alliance (IA) Senior Research Fellowship (500112/Z/09/Z) to AR.
Introduction
Developmental pathways such as Notch are known to regulate self-renewal and cell fate decisions in embryonic and tissue specific stem cells by regulating cell differentiation, survival and proliferation, whereas deregulation of Notch signaling is associated with malignant transformation (1). Accumulation of intracellular domain of Notch1, and elevated expression of Jagged1 in primary breast tumors and its correlation with poor patient survival, serve as most compelling evidence towards involvement of Notch signaling in breast cancer (2–4). In addition, recent studies have shown reduction in mammosphere formation capability and tumor initiation upon Notch inhibition (5), highlighting the role of Notch signaling in breast cancer stem cell self-renewal.
Ligand binding results in proteolytic cleavages and the release of the Notch intracellular domain (NICD) from the plasma membrane and translocation to the nucleus. NICD forms a DNA-binding complex comprising of other co-activators like MAML, CSL, and p300 and activates target gene expression. Post-translational modifications including phosphorylation, ubiquitination, hydroxylation and acetylation affect the stability, transcriptional activity and localization of NICD. Hyper-phosphorylation at the PEST domain at the C-terminal end of NICD is responsible for ubiquitination by FbxWD7 and subsequent proteasomal degradation. Other ubiquitin ligases such as AIP4/Itch and c-Cbl are also known to target Notch1 for degradation (6). Together, these post translational modifications regulate the fine-tuning of half-life of Notch, and thus, its downstream signaling.
Hypoxic conditions results from an inadequate oxygen supply in solid tumor environments due to rapid cell proliferation and poor vascularisation, and is strongly associated with tumor progression, malignant spread and resistance to therapies, hindering effective cancer treatment (7). Cellular adaptation to hypoxia is mainly orchestrated by the transcription factor Hif1-α, activating pathways such as Notch and others like, TAZ (8), CD47 (9), PHGDH (10), ALKBH5 (11) and transcription factors like Oct4, c-Myc, SOX2 which are associated with stem cell self-renewal and multipotency (12). While Hif1-a has been shown to physically interact with NICD to augment Notch signaling, stabilization of NICD in hypoxia has also been demonstrated (13). In spite of the importance of hypoxia in modulating Notch-mediated stem cell self-renewal (14), the molecular mechanisms involved in hypoxia-mediated Notch1 stabilization remain poorly understood. Hypoxia is also known to stimulate 5’-AMP-activated protein kinase (AMPK) activity (15, 16). Previously considered to be a tumor suppressor, the role of AMPK in tumorigenesis is under debate in recent times (17). Reports have shown an increasing, grade-specific activation of AMPK in breast cancer patient (18). Moreover, emerging studies as well as work from our lab highlight the role of AMPK in enabling tumor cells survive the challenges faced in the tumor microenvironment such as energy stress and matrix-deprivation (19, 20).
Thus, although the levels of activated AMPK and activated Notch1 are reported to be high in breast cancer, and hypoxia stress which is prevalent in breast cancer is shown to activate both of these pathways, yet, a relationship between AMPK and Notch signaling has remained unexplored. In this study, we demonstrate that AMPK activation mediates stabilization of cleaved Notch1 levels through changes in Notch1 ubiquitination and degradation. Specifically, we detected weakened interaction of Notch1 with ubiquitin ligase Itch upon AMPK activation. Finally, we show that inhibition or depletion of AMPK lowers cleaved Notch1 levels and adversely affects cancer stem cell phenotype and drug resistance under conditions of hypoxia.
Results
1. Cleaved Notch1 levels are elevated by activated AMPK in breast cancer cell lines
Protein kinases have been implicated in promoting Notch signaling, however, a specific role for AMPK in the regulation of Notch pathway has not been explored. To investigate this, we treated invasive breast cancer cell line MDA-MB-231 with pharmacological activators of AMPK like A769662 and metformin. We used phosphorylation of downstream substrate ACC at Ser 79 as markers of AMPK activation. Immunoblotting using antibody that recognizes the C-terminal end of Notch1 showed an increase in cleaved Notch1 (~120 kDa) levels upon AMPK activation (Figure 1A and Supplementary Figure 1 B). Treatment of MDA-MB-231 cells with increasing concentrations of A769662 led to a dose-dependent increase in cleaved Notch1 levels (Figure 1B). There was no change in the full length protein level (300 kDa) (Figure 1C) or transcript level of Notch1 on A769662 treatment (Supplementary Figure 1B), thus suggesting a possible role of post-translational modification on cleaved Notch1 upon AMPK activation. Similarly, we found elevated cleaved Notch1 levels in invasive BT 474 and HCC 1806 breast cancer cells when treated with A769662 (Supplementary Figure 1C). However, in non-invasive MCF-7 and immortalized HMLE breast cells (21), cleaved Notch1 levels were not modulated by AMPK activation (Supplementary Figure 1C). This suggests that the regulation of Notch1 level by AMPK could be context-specific. Since pharmacological agents can be non-specific, we additionally used genetic approaches to activate AMPK. Over expression of constitutively activated AMPK with γ R70Q mutation and a constitutively active form of AMPK upstream kinase CaMKK β in MDA-MB-231 cells showed higher level of cleaved Notch1 (Figure 1D). Thus, increasing AMPK activity led to elevated cleaved Notch1 levels in invasive breast cancer cells.

(A) MDA-MB-231 cells were treated with A769662 (A76; 100 μM) or metformin (Met; 500 μM) and immunoblotted for indicated proteins. α Tubulin was used as loading control. (B) MDA-MB-231 cells were treated with 100 μM, 300 μM and 500 μM concentration of A76 for 24 h and harvested for immunoblotting and probed for cleaved Notch1 levels (N=3). (C) MDA-MB-231 cells were treated with A76 (100 μM) for 24 h and subjected to immunoblotting for indicated protein (N=3). (D) MDA-MB-231 cells transfected with constitutively active gamma subunit of AMPK (γ R70Q) and immunoblotted for indicated proteins (N=3). MDA-MB-231 cells were transfected with constitutively active AMPK kinase CaMKKβ or EGFP as control and were harvested for immunoblotting and probed for cleaved Notch1. (E) MDA-MB-231 cells were treated with Compound C (Comp C; 10 μM) for 24 h and immunoblotted for indicated proteins. (N=6). (F) MDA-MB-231 cells stably expressing shAMPK α2 (#4) was induced with doxycycline (5μg/μl) for 48 h and immunoblotted for indicated proteins (N=4). (G) Immortalized wild type MEF and AMPKα1/2 DKO MEFs (DKO) were immunoblotted for indicated proteins (N=4). (H) MDA-MB-231 cells were treated with A76 (100 μM) for 24 h and subjected to immunoblotting for indicated protein (N=3) (I) MDA-MB-231 cells were transfected with 12xCSL-Luciferase (FFL) and Renilla TK (RL) and treated with A76 (100 μM) for 48 h. Luciferase activity is represented as a ratio of Firefly (FFL) to Renila (RL) luciferase (N=4). (J) MDA-MB-231 cells treated with A76 (100 μM) for 48 h and RT-PCR analysis was carried out for HES1, housekeeping gene GAPDH. Graph represents the fold change in HES1 transcript levels normalized to GAPDH. (N=4). Error bars represent ±SEM.
To further address the effects of AMPK on cleaved Notch levels, we studied the effects of AMPK inhibition and depletion. Upon treatment with Compound C, a pharmacological inhibitor of AMPK, cells showed reduced phosphorylated ACC level along with lower level of cleaved Notch1 (Figure 1E). To further confirm this, we used doxycycline-inducible knockdown of AMPK. In keeping with higher expression of AMPKα2 in breast cancer cell lines (20), we studied the effect of AMPK α2 knockdown in MDA-MB-231 cells stably expressing a doxycycline-inducible shAMPKα2. Induction of AMPK knockdown in MDA-MB-231 cells led to depletion of AMPK as well as led to lower level of cleaved Notch1 compared to un-induced (Figure 1F) or vector control cells (Supplementary Figure 1D). Knockdown of AMPK also led to reduced levels of cleaved Notch1 in HEK 293T cells overexpressing NICD (Supplementary Figure 1E), additionally suggesting that the effect of AMPK on Notch may be downstream to gamma secretase cleavage. Further, AMPK α1/α2 double knockout MEFs (DKO) showed lower cleaved Notch1 levels (Figure 1G) which was elevated upon exogenous expression of AMPKα 1 and 2 (Supplementary Figure 1F), further confirming an AMPK-dependent increase in cleaved Notch1 levels. These results, together, indicated that AMPK plays a positive role in regulating cleaved Notch1 levels.
We next investigated whether an increase in cleaved Notch1 levels upon AMPK activation led to higher Notch downstream signaling, we checked the levels of gamma secretase-specific cleaved Notch1 (Valine 1744) levels. We saw elevated NICD levels referred to as gamma secretase-specific cleaved Notch1 (Valine 1744) levels subsequently, by immunoblotting and immunostaining (Figure 1H and Supplementary Figure 1G), suggesting that the cleaved Notch1 (~120 kDa) may be active. Further, treatment with A769662 led to a 2.6-fold increase in Notch responsive 12xCSL-luciferase reporter activity (Figure 1I), indicative of increase in canonical Notch signaling. It also led to an increase in the levels of Hes1 and Hes5 (Figure 1J and Supplementary Figure 1H) – two Notch downstream effector genes. In addition, a microarray based data analysis of A769662-treated MDA-MB-231 cells further revealed upregulation of several other Notch pathway-regulated genes, such as, HES5, HEY2, SNAI2, HEYL, IFNG, CDH6 (Supplementary Figure 1I). These results show that the elevated level of cleaved Notch1 upon AMPK activation leads to higher Notch downstream signaling.
2. Reduction in cleaved Notch1 levels upon AMPK inhibition under hypoxic condition
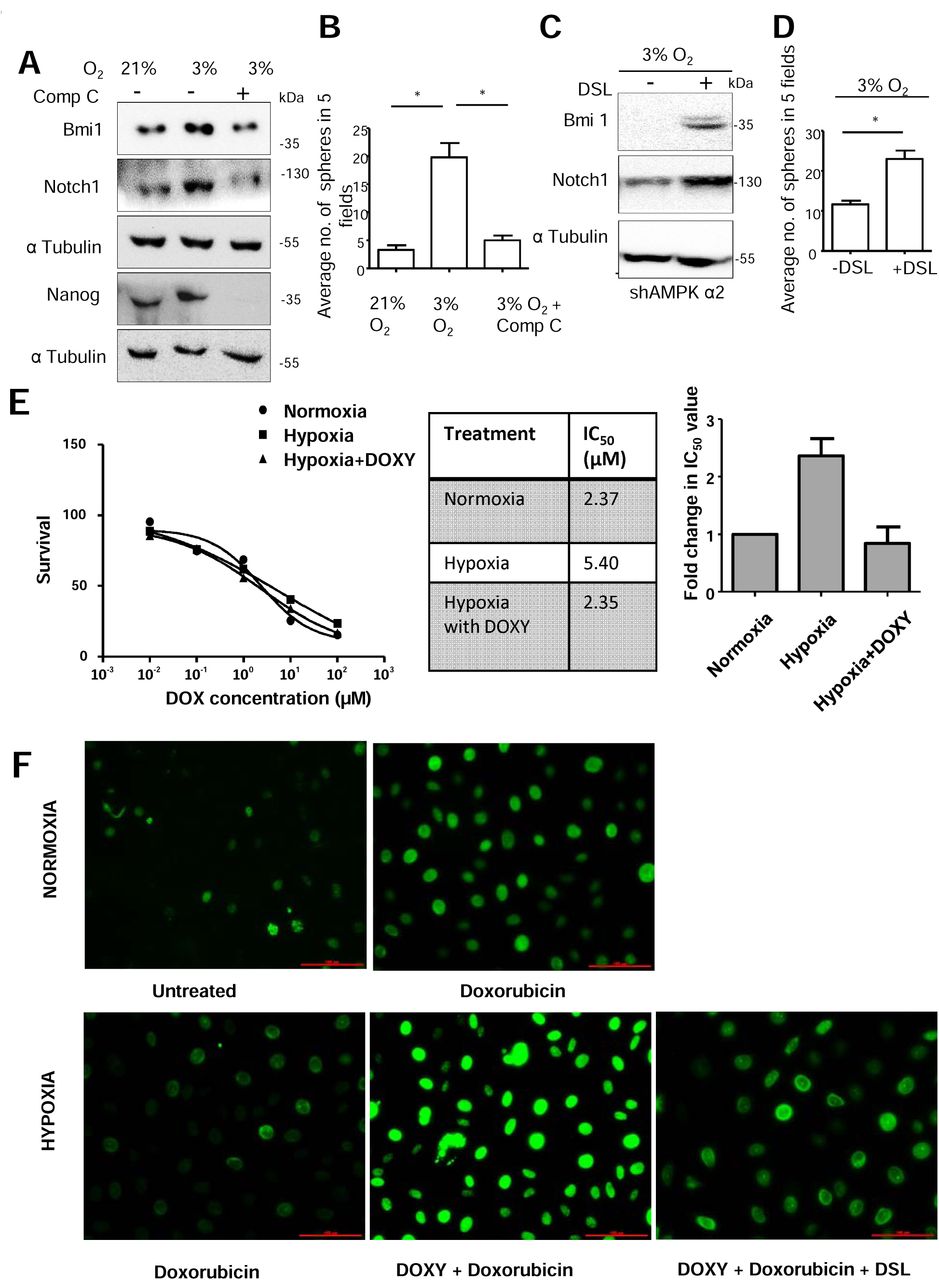
Since hypoxia is known to increase Notch1 levels in various cancer cells (22) as well as activate AMPK (15, 16), and our data revealed that AMPK activation leads to elevated cleaved Notch1 levels, we investigated into the requirement of AMPK in hypoxia-induced Notch1. We treated MDA-MB-231 cells with CoCl2, a hypoxia-mimetic, for 24 hrs. Treatment with CoCl2 led to an increase in phosphorylated AMPK levels (Supplementary Figure 2A). Further, it also led to an increase in cleaved Notch1 levels (Supplementary Figure 2A) which was inhibited in the presence of AMPK inhibitor (Figure 2A). Similar results were seen in BT-474 cells (Supplementary Figure 2B). Moreover, upon treatment with CoCl2, we did not observe changes in cleaved Notch1 level in the inducible AMPKα2 knockdown cells (sequences 1 and 4), while the control pTRIPZ expressing cells showed the expected increase in cleaved Notch1 levels (Figure 2B and Supplementary Figure 2C). Thus, these data suggested a role for AMPK in the hypoxia-induced increase in cleaved Notch1 levels. To further confirm these data, we used oxygen-regulatable trigas incubator. Cells cultured in 3% oxygen showed increased levels of Hif1α, carbonic anhydrase 9 (CA IX) and pAMPK (Supplementary Figure 2D-E), all of which are markers of hypoxia (15, 23). Further, these cells also showed increase in cleaved Notch1 and gamma secretase-specific cleaved Notch1 (Valine 1744) by immunoblotting (Supplementary Figure 2E). In addition, we observed, increased nucleus-localized gamma secretase specific cleaved Notch1 (Valine 1744) compared to cells grown in ambient oxygen (~21 %) condition (Supplementary Fig. 2F). Cells grown in 3% oxygen, when treated with inhibitor of AMPK (Figure. 2C) or upon induction of AMPK knockdown (Figure. 2D and Supplementary Fig. 2G), showed reduction in cleaved Notch1 levels. Further, hypoxia-induced increase in Notch signaling was impaired upon AMPK inhibition (Figure. 2E). These results suggest involvement of AMPK in the regulation of the Notch signaling pathway in response to hypoxic stress.

(A) MDA-MB-231 cells were subjected to CoCl2 (150 μM) and Comp C (10 μM) treatment for 24 h and immunoblotted for indicated protein. Graph represents the densitometric analysis for quantification of relative amount of indicated protein (N=4). (B) MDA-MB-231 cells stably expressing shAMPK α2 (#1) and vector control pTRIPZ induced with doxycycline (5μg/μl) for 24 h followed by treatment with CoCl2 (150 μM) and immunoblotted for indicated proteins (N=3). (C) MDA-MB-231 ells were grown in 3% O2 and simultaneously treated with and without Comp C (10 μM) and immunoblotted for indicated proteins (N=3). (D) MDA-MB-231 cells stably expressing shAMPK α2 (#4) induced with doxycycline (5μg/μl) for 24 h and subsequently grown in 3% O2 and immunoblotted for indicated proteins (N=3). (E) HEK-293T cells were transfected with 12xCSL-Luciferase (FFL) and Renilla TK (RL) and NICD. The cells were treated with CoCl2 for 48 h with or without Compound C. Luciferase activity is represented as a ratio of Firefly (FFL) to Renila (RL) luciferase (N=3). Error bars represent ±SEM.
3. AMPK inhibition during hypoxia lowers stability of cleaved Notch1
To investigate into the mechanisms by which AMPK activation leads to higher cleaved Notch1 levels, we first checked the levels of Notch ligands Delta-like 1, 3, 4 and Jagged 1 and 2. However, we failed to find a change in the expression of Notch-ligands under AMPK activation (Supplementary Figure 3A). Since mTOR has been associated with both positive (24) and negative (25) regulation of Notch full length receptor and transcript levels respectively, and AMPK activation leads to mTOR inhibition (20, 26), we next gauged the involvement of mTOR in the regulation of cleaved Notch1. Addition of rapamycin, an inhibitor of mTOR, failed to rescue the AMPK inhibition-mediated reduction in cleaved Notch levels (Supplementary Figure 3B), suggesting possible mTOR-independent mechanisms of regulation of cleaved Notch1 levels downstream of AMPK activation.
We investigated if AMPK affected the stability of cleaved Notch1 using cycloheximide chase assay. Addition of cycloheximide revealed that AMPK knockdown lowers the stability of cleaved Notch1 (Figure 3A). Similarly, we observed reduced cleaved Notch1 stability in AMPK DKO MEFs when treated with cycloheximide (Supplementary Figure 3C). These data suggested that the absence of AMPK enhances the rate of Notch1 degradation.

(A-B) MDA-MB-231 cells stably expressing shAMPK α2 (#4) were induced by doxycycline (5μg/μl) for 48 h and (A) treated with cycloheximide for 4 and 8 h and immunoblotted for indicated proteins and graphs represent the densitometric analysis for quantification of relative amount of indicated protein (N=3) (B) grown in 3% oxygen for another 24 h followed by cycloheximide treatment for 8 h and immunoblotted for indicated. (N=3). (C) MDA-MB-231 cells were treated with Comp C (10 μM) for 24 h and subsequently treated with or without MG-132 (5 μM) for 8 h. Cells were immunoblotted for indicated proteins (N=3). Error bars represent ±SEM.
To explore if AMPK plays any role in hypoxia-induced stabilization of Notch1 (13), we grew MDA-MB-231 cells with inducible AMPK knockdown in 3% oxygen and subsequently treated with cycloheximide. Immunoblotting revealed an aggravated reduction in cleaved Notch1 levels upon AMPK knockdown (Figure 3B). Similarly treating MDA-MB-231 cells with CoCl2 for 24 h along with Compound C revealed aggravated reduction in cleaved Notch1 levels on AMPK inhibition (Supplementary Figure 3D). Since cleaved Notch1 is reported to be targeted for degradation through 26S proteasomal machinery (27), we investigated whether AMPK inhibition enhances Notch1 degradation through 26S proteasome. We found that Compound C-mediated reduction in cleaved Notch1 levels was rescued in the presence of proteasomal complex inhibitor MG132 (Figure 3C). Thus, AMPK activation plays a critical role in cleaved Notch1 stabilization under hypoxia and AMPK inhibition brings about an increase in cleaved Notch1 proteasomal degradation.
4. AMPK inhibits Notch1 ubiquitination by modulating its interaction with Itch
To further explore the mechanism of AMPK-mediated cleaved Notch1 stability, we investigated into post-translational modifications that are known to stabilize Notch1 protein levels (6). We did not detect any alteration in the levels of serine/threonine phosphorylation or acetylation on immuno-precipitated cleaved Notch1 upon AMPK activation (Supplementary Figure 4A and Supplementary Figure 4B). Interestingly, immunoblotting with degradation-specific K48-linked polyubiquitin antibody revealed a reduction in ubiquitinated cleaved Notch1 in A769662 treated cells (Figure 4A). In contrast, when we inhibited AMPK, the ubiquitinated Notch1 levels were higher (Supplementary Figure 4C). These data suggest that AMPK activity affects Notch1 ubiquitination levels, which possibly regulates its degradation by proteasomal pathway thereby leading to its stabilization.

(A-D) MDA-MB-231 cells were subjected to A76 (100 μM) for 24 h and (A) subsequently treated with MG132 (5 μM) for 2 h prior to Notch1 was immuno-precipitation followed by immunoblotting for indicated proteins (N=3) (B) immunoblotted for Itch protein (N=3) (C) Notch1 was immuno-precipitated and immunoblotted for Itch interaction (N=3) (D) Itch was immuno-precipitated and immunoblotted for pan-ubiquitin. (E) MDA-MB-231 cells were transfected with Myc tagged Itch plasmid and immunoblotted for indicated proteins. Graphs represent the densitometric analysis for quantification of relative amount of indicated protein (N=3). (F-G) MDA-MB-231 cells stably expressing shAMPK α2 (#1) were grown in 3% O2 for 24 h with and without induction by doxycycline (5μg/μl) and Itch was immuno-precipitated and (F) immunoblotted for Notch1 interaction. Same lysates were used for both the panels. Immunoprecipitation was carried out separately for the 2 panels. (G) immunoblotted for phospho serine/threonine and phospho tyrosine levels. (H) MDA-MB-231 cells stably expressing shAMPK α2 (#1) were grown in 3% O2 for 24 h with and without induction by doxycycline (5μg/μl) and were immunoblotted for phospho Itch Y420 levels and other indicated proteins (I) MDA-MB-231 cells transfected with or without dominant negative Fyn (DN Fyn) were grown in 3% O2 for 24 h and harvested for Itch immuno-precipitation followed by immunoblotting for phospho tyrosine levels.
Notch1 is known to be a substrate for several ubiquitin ligases such as FBXW-7, Itch/AIP4, NEDD 4, Deltex 1, c-Cbl (28). Noticeably, a recent study (29) has shown that AMPK activation disrupts the interaction of Itch/AIP4 with its substrate p73. We therefore examined a possible role for Itch in AMPK-mediated Notch1 stability. Although there was no change in Itch protein levels (Figure 4B), interestingly, we found that Itch interaction with Notch1 reduced on AMPK activation (Figure 4C). Auto-ubiquitination at K-63 linked polyubiquitin enhances the activity of Itch (30).We detected less ubiquitin smear on immuno-precipitated Itch (Figure 4D) indicating reduced activity of Itch under AMPK activation. Furthermore, we tested the effect of AMPK on Itch activity towards its substrates. We found that while the levels of cleaved Notch1 reduced on Itch overexpression, AMPK activation inhibited the Itch-mediated down-modulation of cleaved Notch1 (Figure 4E). Similar effects were observed for c-Jun, another substrate of Itch (Figure 4E). Overexpression of Itch in BT-474 yielded similar down-modulation of cleaved Notch1 and the same could be rescued on AMPK activation (Supplementary Figure 4D). These results suggest a protective role for AMPK in Itch-mediated Notch degradation.
We next asked whether Itch-mediated ubiquitination is involved in cleaved Notch1 stabilization under hypoxia. Although Itch levels remained unchanged in hypoxic condition (Supplementary Figure 2F), we observed reduced interaction of Itch with Notch1 in hypoxia, which was negated by AMPK knockdown (Figure 4F). Similar to our observations with AMPK perturbation, we found Notch1 K-48 linked ubiquitination reduced in hypoxia and increased upon AMPK inhibition in hypoxic conditions (Supplementary Figure 4E). Thus, these data uncover a novel AMPK-mediated regulation of Notch1 stability in hypoxia through modulating its interaction with ubiquitin ligase Itch.
To study if changes in Itch phosphorylation are involved in Notch regulation, we immuno-precipitated Itch and observed an increase in tyrosine phosphorylation of Itch in hypoxia which decreased markedly upon AMPK knockdown (Figure 4G). Subsequently, we probed for the levels of phospho Itch at Y420 residue and observed an increase in its levels in hypoxia; AMPK knockdown however reduced its levels (Figure 4H).
Phosphorylation of Itch has been shown to regulate its interaction with substrates (31). We investigated the role of Fyn which mediates inhibitory tyrosine phosphorylation of Itch leading to decrease in ubiquitination of Notch1 (31). We inhibited Fyn using Src family kinase inhibitor PP2 under condition of hypoxia and observed lower levels of cleaved Notch1 (Supplementary Figure 4F). Further, we transfected cells with dominant negative Fyn, and found lower levels of cleaved Notch1 in hypoxia (Figure 4I), suggesting a possible role for Fyn in Notch1 stabilization in hypoxia. Moreover, dominant negative Fyn transfected cells had lower tyrosine phosphorylation on Itch, suggesting that Fyn-mediated regulation of Itch phosphorylation might regulate Notch1 levels in hypoxia (Figure 4I).
To check a possible role for AMPK in the Fyn-mediated Itch regulation, we investigated the effects of AMPK activity on Fyn. The activity of Src-family kinases (SFK), of which Fyn is a member (32), is regulated by phosphorylation at specific tyrosine residues. Since phospho-Fyn specific antibodies are not available, we used Src-family (Tyr 416) specific antibodies which detect Tyrosine 416 specific phosphorylation on Src-family members that leads to their activation (33). AMPK activation led to an increase in the levels of phosphorylated Src-family tyrosine 416 (Supplementary Figure 4G). In contrast, AMPK knockdown led to a reduction in the levels of hypoxia-mediated activated SFK (Supplementary Figure 4H). These data suggested a possible role for AMPK in positively regulating the activity of Fyn.
To further unravel the mechanisms by which AMPK augments Fyn activation, we carried out LC-MS to detect interacting proteins of Fyn in cells treated with AMPK activator A769662 and vehicle control DMSO. FLAG-tagged Fyn expressing HEK293T stable cells were immunoprecipitated with FLAG specific antibodies and protein complexes were isolated and subjected to LC-MS. PTPRF (34), a phosphatase known to dephosphorylate the inhibitory phosphorylation at Y528, was associated with Fyn only in AMPK activated condition (Table 1). PTPN5 (35) and PTPRC (34) are phosphatases known to dephosphorylate the activating phosphorylation of Fyn at Y416, thus decreasing its activity. These were detected in DMSO condition only (Table 2). Our proteomic analysis further substantiated the role of AMPK in positively modulating Fyn activity, by facilitating differential association of phosphatases with Fyn. These results together indicate an important role for Fyn kinase in AMPK-dependent Itch regulation in hypoxia. Together, these data identify a novel AMPK-dependent regulation of tyrosine phosphorylation of Itch by Fyn kinase in the stabilization of cleaved Notch1.
5. AMPK activation-mediated Notch1 stabilization leads to an increase in stem-like properties
Since Notch signaling is implicated in cancer stem cell (CSC) maintenance (36), we asked if AMPK-mediated enhanced Notch1 stabilization is involved in this process. AMPK activation with A769662 led to an increase in the stemness-associated factors Nanog and Bmi1 which was attenuated in the presence of DAPT, a pharmacological inhibitor of Notch signaling (37), in both MDA-MB-231 (Figure 5A) and BT-474 cells (Supplementary Figure 5A). To further confirm this, we abrogated Notch signaling by knocking down Notch1 in MDA-MB-231 cells using two different shRNA targeting sequences. Treatment with A769662 led to a moderate increase in Bmi1 and Nanog protein levels in control scramble cells, but not in Notch1 knockdown cells (Figure 5B and Supplementary Figure 5B). Moreover, the transcript levels of stemness-associated genes like Bmi1 and Sox2 increased with AMPK activation in control cells but not in Notch1 knockdown cells (Figure 5C). In addition, a microarray-based data analysis of A769662-treated MDA-MB-231 cells further revealed upregulation of several other stemness-related genes such as, NKAP, DLL1, NOTCH2, ERBB2, FOXC1 (data not shown). This suggests the likely involvement of Notch pathway in AMPK-mediated increase in stemness in these cells. Serial sphere formation in non-adherent, serum-deprived condition serves as an in vitro mimic of stemness activity. We observed that AMPK activation with A769662 increased primary and secondary sphere formation in control cells, whereas it failed to do so in Notch 1 inhibited or depleted-cells (Figure 5D). We next measured the CD44high/24low stem-like sub-population in AMPK activated cells. While treatment with A769662 led to a 2-fold increase in the CD44high/24low population in control cells, it failed to do so in Notch1 inhibited or depleted-cells (Figure 5E and Supplementary Figure 5C). These results together suggest that AMPK activation leads to elevated cancer stem cell-like properties in breast cancer cells through Notch signaling.

(A) MDA-MB-231 cells were treated with A76 (100 μM) with and without DAPT (100 nM) for 48 h and immunoblotted for indicated proteins (B) MDA-MB-231 cells stables with scrambled and shNotch1 (#E7) were treated with A76 (100 μM) for 48 h and immunoblotted for indicated proteins (C) MDA-MB-231 cells with scrambled and shNotch1 (#E7) were treated with A76 (100 μM) for 48 h and Bmi1, Sox2, CD44, Oct4 and KLF4 transcript levels were analysed (D) MDA-MB-231 cells treated with A76, DAPT and A76 together with DAPT and Notch1 knockdown cells treated with A76 were subjected to primary and secondary sphere formation assay in methyl cellulose; the plot represents the average number of spheres in independent experiments and (E) dual staining with anti CD44-PE and anti CD24-FITC and FACs analysis was performed to measure the CD44 high /24 low population. The plot represents CD44high/24low populations from each treatment (N=3). Error bars represent ±SEM.
6. Hypoxia-induced AMPK activation promotes stem-like properties and drug-resistance in breast cancer cells through Notch signaling
Hypoxic condition is known to facilitate cancer stem-like properties and promote epithelial to mesenchymal transition through involvement of Notch signaling (22, 38). Since we uncovered an important role of AMPK in modulating Notch1 levels, and thus maintenance of breast cancer stem cell population, we investigated the requirement of AMPK activation to maintain CSCs under hypoxic condition. Compared to MDA-MB-231 cells grown in 21% oxygen, cells grown in 3% oxygen showed elevated levels of Bmi1 and Nanog which was impaired on AMPK inhibition (Figure 6A). To further confirm this, cells were grown with hypoxia-mimetic CoCl2 and simultaneous inhibition of AMPK brought about reduction in CoCl2-mediated increase in Bmi1 level (Supplementary Figure 6A). Bmi1 and Nanog levels were also high in BT-474 cells when treated with CoCl2 and disrupting AMPK activity brought about similar effects in presence of hypoxia (Supplementary Figure 6B). Further, hypoxia-induced increase in sphere formation was also abrogated by AMPK inhibition (Figure 6B). We obtained similar results with CoCl2 treatment in the presence of AMPK inhibition (Supplementary Figure 6C), clearly indicating the requirement of AMPK in hypoxia-induced sphere forming potential. Next, to gauge if hypoxia-induced AMPK mediates its effects via Notch, we treated AMPK knockdown cells with synthetic Notch ligand DSL under hypoxia. We observed that the ligand-treated AMPK-knockdown cells had higher levels of cleaved Notch1, showed elevated levels of Bmi1, and also formed higher number of spheres (Figure 6C and D). These data suggest that hypoxia-induced AMPK activation promotes stem-like properties in breast cancer cells through Notch signaling.
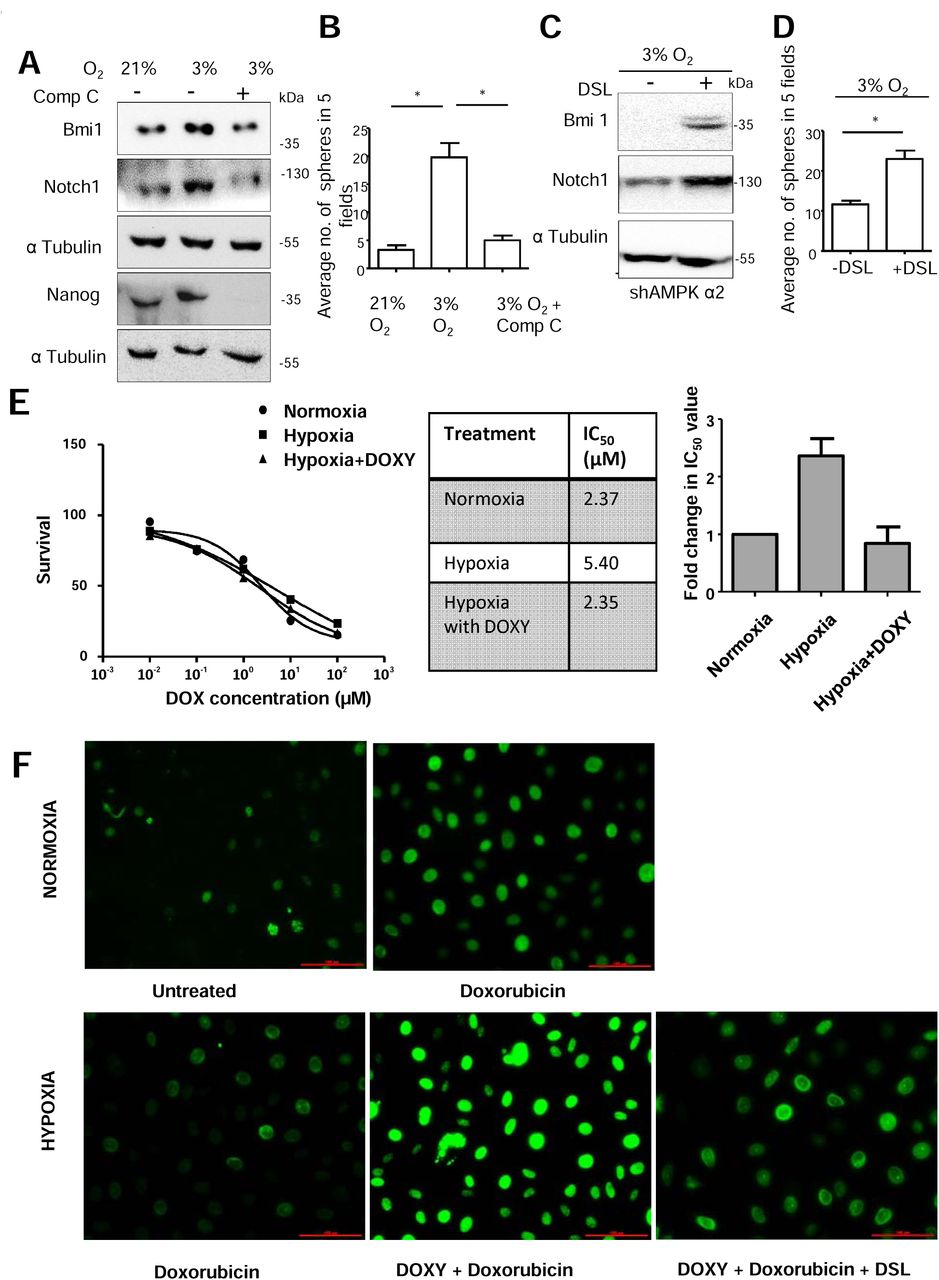
(A-B) MDA-MB-231 cells grown in 3% O2 were treated with and without Comp C (10 μM) for 48 h and (A) immunoblotted for indicated proteins and (B) subjected to sphere formation assay. The plot represents the average number of 7 day spheres in 5 fields in two replicates of 3 independent experiments. (C-D) MDA-MB-231 cells stably expressing shAMPK α2 (#1) were grown in 3% O2 for 24 h with doxycycline induction and subsequently treated with and without DSL ligand (100 nM) and (C) immunoblotted for indicated proteins and (D) were subjected to sphere formation assay. The plot represents the average number of 7 day spheres of 3 independent experiments. (E) MDA-MB-231 cells stably expressing inducible shAMPK α2 were treated with indicated concentrations of doxorubicin for 48 h under normoxic and hypoxic conditions with and without with doxycycline induced AMPK α2 knockdown (N=3). Representative survival curve, the IC50 values and fold change in IC50 values have been indicated. (F) MDA-MB-231 cells stably expressing inducible shAMPK α2 were treated with 1μM of doxorubicin for 24 h under normoxic and hypoxic conditions with and without with doxycycline induced AMPK α2 knockdown and additional Notch activation by DSL ligand and DNA damage was assessed by pH2A.X staining.
Notch signalling is known to bring about increased drug resistance in hypoxic condition in various tumours such as osteosarcoma, ovarian cancer and breast cancer (39–41). Various mechanisms have been elucidated, such as increase in Sox2 levels or MRP-1 downstream of Notch1 activation upon hypoxia which helps mediate higher resistance to chemotherapeutic drugs. As we have elucidated a novel role for AMPK in hypoxia-induced increase in Notch1 signalling, therefore, we wanted to investigate whether the AMPK-mediated increase in Notch1 signalling affected drug sensitivity of breast cancer cells. We observed a 2-fold increase in the IC50 value of doxorubicin when exposed to hypoxia and knockdown of AMPK in cells in hypoxia restored IC50 value (Figure 6 E). DNA damage accumulated by the cells in hypoxic condition upon doxorubicin treatment was assessed by pH2A.X staining. We observed an increase in pH2A.X staining intensity upon treatment with doxorubicin (Figure 6F). In hypoxic condition, the cells did not accumulate DNA damage upon doxorubicin treatment, but knockdown of AMPK signalling in hypoxia led to excessive accumulation of DNA damage (Figure 6F) which suggests that hypoxia-induced AMPK enhances drug resistance. To gauge if hypoxia-induced AMPK indeed mediates its effects via Notch, we treated AMPK knockdown cells with synthetic Notch ligand DSL to activate Notch signalling under hypoxia and assessed the DNA damage. We observed a reduction in pH2A.X staining compared to cells without Notch activation, indicating restoration of drug resistance phenotype (Figure 6F). Thus, AMPK-Notch axis plays a role in mediating drug resistance in hypoxic condition.
7. Hypoxia-induced AMPK-Notch1 signalling in vivo
Since hypoxia conditions are prevalent in tumors (42), we validated the AMPK-Notch1 signaling axis in tumor xenografts. BT 474 cells stably expressing shRNA against AMPK α2 were injected sub-cutaneously in immunocompromised mice; tumor formation by these cells were impaired compared to scramble control cells (20). We found that cleaved Notch1 (Valine 1744) levels were highly diminished in tumor sections derived from BT-474 cells with knockdown of AMPK α2 whereas the control cells expressed cleaved Notch1 (Valine 1744) in majority of the cells (Figure. 7A and Figure 7B).

(A) Representative images from immunohistochemical analysis performed on tumors from BT-474 cells stably expressing scrambled or shAMPK to assess cleaved Notch1 (Valine 1744) levels (Scale bar: 100 μm). (B) Quantification of staining intensity in (A). (C) Representative images from immunohistochemical analysis of pACC and cleaved Notch1 (Valine 1744) in breast cancer patient samples (N=19) (D) Patient derived breast cancer epithelial cells grown in 3% O2 were treated with and without Comp C (10 μM) for 24 h and immunoblotted for indicated proteins (N=3).
To address the relevance of AMPK-Notch signaling in breast cancer, we undertook immunohistochemical analysis for AMPK activation (by pACC staining) and Notch1 (Valine 1744 staining) on breast cancer patients’ samples (N=19), and found a correlation (p= 0.0422, Fischer exact test) between activation of AMPK and cleaved Notch1 in these samples (Figure. 7C). Furthermore, we investigated into the response of patient-derived breast cancer cells to hypoxia. The primary tissue-derived cells exposed to hypoxic condition showed upregulation of cleaved Notch1 and phospho Itch levels, whereas pharmacological inhibition of AMPK in these cells under hypoxic condition reduced the cleaved Notch1 and phospho Itch levels (Figure. 7D). We further investigated the gene signature of hypoxia, AMPK and Notch pathways in breast cancer patient samples. Microarray dataset from GEO repository was analysed and hierarchical clustering of gene expression profiles revealed co-expression of hypoxia, AMPK and Notch-responsive genes in majority of cases (Figure 8), thus underscoring the relevance of the hypoxia-AMPK-Notch axis in breast cancers.

Heat map depicting clustering of genes involved in Hypoxia, Notch and AMPK signalling across breast invasive carcinoma patients’ data available in NCBI GEO data set. Genes of interest are color-coded; red in the heat map represents highly expressed genes, whereas green represents downregulated genes.
Discussion
The Notch pathway, because of its importance in numerous cancers, has been a major candidate for developing therapeutic agents. While gamma secretase inhibitors and antibodies against receptor and ligands have reached clinical trials, gastrointestinal toxicity and other side effects have led to shifting of focus on therapeutic targeting of other pathways impinging on regulation of Notch signaling (43, 44). One such pathway that we have examined is the metabolic regulator AMPK. We found AMPK to be a positive modulator of cleaved Notch1 contributing to elevated stemness and drug resistance in breast cancer cells, suggesting that inhibition of AMPK can alleviate stem-like properties, thus rendering cancer cells susceptible to existing anti-cancer agents.
Hypoxia in solid tumors is considered a malice aiding cancer progression hindering successful therapy. Both AMPK and Notch1 are activated under hypoxia, a condition prevalent during cancer progression (15, 38, 45). Effects of hypoxia-induced Notch1 signaling have been found in, melanoma (46), adenocarcinoma of lungs (47), glioma (48), as well as breast carcinomas (22). Mechanistically, it has been shown that Hif1α and NICD directly interact and augment transcription of Notch responsive genes (13). Another mechanism implicated in this crosstalk involves FIH (factor inhibiting Hif1-α) which is critical in blocking differentiation in myogenic and neural progenitor cells (49). Moreover Hif-1a binding to NICD also enhances proliferation and decreases differentiation in GSC (50). Hypoxia also influences breast cancer cell EMT and migration through Notch signaling, as Hif-α synergises with Notch co-activator MAML to increase expression of Snail and Slug and reduce E-cadherin expression (51). Interestingly, Gustaffson et al., reported the stabilization and increased half-life of NICD in hypoxia, although the molecular mechanism remained unknown. Our results indicate elevated breast cancer stemness in hypoxia and further show that this is dependent on AMPK-mediated stabilization of Notch1.
Notch signalling along with a central role in self-renewal of cancer stem cells, has also been implicated in imparting resistance to therapy cancer (39–41). Recent studies have also implicated AMPK in mediating hypoxia-induced drug resistance to doxorubicin in osteosarcoma (52). Another report also implicates AMPK in mediating drug resistance to cisplatin (53). Thus, from these reports it becomes clear that inhibition of AMPK along with chemotherapy can sensitize cancer cells to chemotherapeutic agent. In this study we show that hypoxia-induced drug resistance properties are supported by AMPK activation, and this effect can be abrogated by Notch inhibition/depletion.
Cleaved Notch is targeted for proteasomal degradation after ubiquitination. Notch and its interaction with ubiquitin ligases have been often shown to be influenced by regulators like Numb, which suppresses Notch signaling by recruiting Itch and facilitating degradation of Notch (54). Our data identified AMPK activity as a regulator of Itch-mediated Notch degradation. Several studies have identified phosphorylation as a major regulator of Itch activity, where kinases like ATM, JNK activate Itch by increasing its enzymatic activity for a specific set of targets like c-Jun or c-FLIP, other kinases like Fyn reduce interaction of Itch with substrates including Jun-B as well as Notch leading to lesser ubiquitination (55). A previous study showed an inhibitory effect of AMPK activator on interaction of Itch to its substrate p73 leading to lesser degradation (29). We found that interaction of cleaved Notch1 with Itch is disrupted upon AMPK activation by the regulation of Itch tyrosine phosphorylation by Fyn (Figure 9). We also observed that AMPK could facilitate Fyn activation by modulating the protein tyrosine phosphatases associated with Fyn. Changes in protein phosphatase activity by LKB1 and AMPK has been previously reported in lung and cervical cancer cell lines (56) and similar changes may take place in breast cancer cells as well.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In conditions of normoxia, basal activity of AMK does not interfere with interaction of E3-ubiquitin ligase Itch/AIP4 with its substrate cleaved Notch1, which is ubiquitinated and targeted for proteasomal degradation. In conditions of hypoxia, activated AMPK levels, enhance Fyn activation which phosphorylates and hinders binding of ligase Itch/AIP4 with its substrate cleaved Notch1, thereby resulting in cleaved Notch1 stabilization. This results in elevated stemness and drug resistance of the tumours.
AMPK is traditionally viewed as a tumour suppressor as mTOR is negatively regulated by AMPK bringing about slower growth, and AMPK is also known to stabilize p53 bringing about cell cycle arrest (57). We did not observe transcriptional changes of Notch1 through mTOR that has been previously described (25). While p53 is known to positively regulate Notch1 in epithelial cells (58), we have tested our signalling axis in p53 mutant cell line (MDA-MB-231) indicating that p53 does not play a role in mediating the effects of AMPK on cleaved Notch1. We on the other hand show a pro-tumorigenic role of AMPK through positive regulation of Notch1 through post-translational regulation.
Interestingly, our study shows a high correlation between AMPK and Notch activation in grade 3 primary breast cancer samples that are known to contain hypoxic regions (11). In support of our observations, abundant expression of activated AMPK has also been found in human gliomas (59) and breast cancer (60). Contextual oncogenic properties of AMPK, has been reported, especially in bioenergetic stress conditions such as nutrient deprivation and hypoxia, which are often faced by cells in a growing tumour (61, 62). While we have shown the effects of AMPK in increasing breast cancer stemness in hypoxia, previously, a protective role for AMPK has been demonstrated in androgen dependent prostate cancer cells in hypoxia (63) and in solid tumours with low oxygen condition (45). Further, studies from our lab (20) and others (19) show in-vivo evidence of tumour promoting role of AMPK. Another study showed that this role of AMPK is evident only in a metabolically stressed tumour microenvironment context (64). Thus, our report compliments such studies on tumour promoting role of AMPK in the context of tumour hypoxia.
Inhibitory effect of metformin, which also activates AMPK, on breast cancer stem cells has been earlier reported (65). Although multiple mechanisms have been shown in this regard, these effects of metformin on cancer stem cells have been shown to be independent of AMPK (66–68). Our study revealed a novel role for AMPK in regulating hypoxia-mediated Notch1 stabilization leading to elevated stemness in breast cancer, and hence we propose that inhibition of AMPK can be a potential therapeutic strategy, alleviating self-renewal of breast cancer cells.
Materials and Methods
Cell Culture
Cell lines MDA-MB-231, BT-474, HCC-1806, MCF-7 (procured from ATCC, VA, USA) were cultured in DMEM (Sigma, MO, USA) with 10% fetal bovine serum (Invitrogen, CA, USA), penicillin and streptomycin. HMLE (kind gift from Dr. Robert Weinberg, MIT, USA) were cultured in DMEM-F12 with growth factors. All cell lines were maintained in standard 5% CO2 incubator at 37 °C. AMPKα1/2 DKO MEFs were a kind gift from Dr. Benoit Viollet (INSERM, France). For generation of hypoxic conditions cells were kept in 3% O2 in a hypoxia incubator (Eppendorf, Germany).
Plasmids, transfection and stable cell line generation
Transfections were carried out using Lipofectamine 2000 (Invitrogen) or Polyethyleneimine (Sigma). EGFP CA CaMKK and pMT2 HA AMPK γ1 R70Q were a kind gift from Dr. Grahame Hardie (University of Dundee, Scotland) and Dr. Lee Witters (Dartmouth College, USA), respectively. pCMV Tag3B AMPK α1 and α2 were a kind gift from Dr. Ronald Evans (Salk institute, USA). pTRIPZ with shRNA against AMPK α2 (2 sequences) were procured from Dharmacon. 12XCSL-Luc was obtained as a kind gift from Dr. Rajan Dighe (IISc, Bangalore). pRK5 Myc-Itch was received as a kind gift from Gerry Melino (University of Leicester, UK), pTriEx-4 Neo DN Fyn K299M was a kind gift from Dr. Akash Gulyani (InStem, Bangalore). The shNotch1 (2 sequences) and scrambled constructs were purchased from TransOMIC technology (AL, USA). Stable cells were generated using puromycin (Sigma) selection.
Pharmacological compounds
Pharmacological compounds used were: AMPK activators A-769662 (100 μM) (Abcam, Cambridge, UK), Metformin (500 μM) (Sigma Aldrich), cobalt chloride hexahydrate (150 μM) (Sigma Aldrich), cycloheximide (100μg/mL) (Sigma Aldrich), DAPT (N-[N-(3,5-difluorophenacetyl-L-alanyl)]-S-phenylglycine t-butyl ester) (5 μM) (Sigma Aldrich), AMPK inhibitor compound C (10μM) (Calbiochem, Merck Biosciences, CA, USA), Nicotinamide 0.5 mM (Calbiochem, Merck Biosciences), doxycycline (5 μg/mL), MG-132 (5μM), PP2 (10μM) (Calbiochem, Merck Biosciences).
Immunoblotting
Whole cell lysates were prepared with lysis buffer (50 mM Tris pH 7.4, 5 mM EDTA, 250 mM NaCl, 0.5% TritonX, 50 mM NaF, 0.5 mM sodium orthovanadate, 100mM sodium pyrophosphate (Sigma Aldrich), and protease inhibitor (Roche, Switzerland)). 8% SDS PAGE was used to resolve the proteins. α tubulin was used as loading control. Densitometric analysis of the blots was done using Multi Gauge V2.3 (Fujifilm). Intensity of bands were normalised to loading control and represented as fold change over control.
Immunoprecipitation
Cells grown in 90 mm dish were lysed with cold lysis buffer (25mm Tris pH7.4, 1mM EDTA, 150 mM NaCl, 5% glycerol, 0.5 mM sodium orthovanadate, 100mM sodium pyrophosphate, protease inhibitor). 1mg protein from each lysate was estimated and incubated with 1 μg of antibody and 10 μL sepharose G beads (Invitrogen) at 4° C overnight. After incubation the beads were washed with lysis buffer and boiled with Laemmli’s buffer and loaded on SDS-PAGE.
Primary antibodiess
Antibodies against C-terminal end of Notch1 that recognizes full length (300 kDa) as well as cleaved Notch1 (120 kDa) were purchased from Santa Cruz Biotechnology (Texas, USA), Itch from DSHB, c-Jun antibody and phospho Itch Y420 from Abcam and α tubulin from Calbiochem, Merck. phospho Thr 172 AMPK, phospho Ser 79 ACC, total AMPKα1/2, total AMPKα2, NICD or cleaved Notch1 valine 1744 (gamma-secretase cleavage specific), cyclin D3, Bmi1, Nanog, phospho threonine, phospho serine, acetyl lysine, pan-ubiquitin, K48-linked ubiquitin and phospho Tyr 416 Src family and CA9 were purchased from Cell signalling Technology (MA, USA).
Immunocytochemistry
Cells grown in 35 mm dish were fixed with methanol: acetone, permeabilized using 0.2% Triton X 100, blocked with 0.2% fish skin gelatin and incubated with primary antibodies against NICD, Hes5, Dll1, Dll3, Dll4 and Jagged 1and 2 (Santa Cruz Biotechnology), cy3 conjugated secondary antibodies (Jackson ImmunoResearch, PA, USA) were used and Hoechst 33342 (1 μg/mL) was used to counterstain and imaged at 20X using Olympus IX71 (Japan). Images were processed using Image Pro Plus (Media Cybernetics).
Luciferase assay
MDA-MB-231 cells were seeded in a 60 mm dish were transfected with pGL3-12xCSL luciferase and pRLTK using Lipofectamine 2000 (Invitrogen). The transfected cells were seeded in 12 well plate (1x105 cell per well), and treatment was carried out for 48 h. Cells were harvested for luciferase activity using Dual luciferase assay kit (Promega, WI, USA) as per manufacturer’s instructions and analysed for luciferase activity using TEC AN infinite M200 pro plate reader (Switzerland). Firefly luciferase activity was normalised to Renila luciferase activity. Data is represented as fold change in relative light units.
RNA isolation and q-PCR
Total RNA was isolated from 60 mm dish using TRI reagent (Sigma). cDNA synthesis kit (Applied Biosystems, CA,USA) was used to convert 2 μg RNA to cDNA. q-PCR for Hes1 and Notch1, Nanog, Bmi1, Sox2, CD44, Oct4 and Klf4 was performed using SYBR green (Kappa Biosystems) in Eppendorf Mastercycler.
LCMS/MS
Liquid Chromatography Mass Spectrometry (LCMS/MS) was carried out to analyse the interactome of Fyn in HEK-293T cells. Protein complexes were isolated from FLAG-tagged expressing HEK-293T stable cells treated with DMSO or A769662 by FLAG immunoprecipitation. 100 μg of the samples was taken for digestion. The sample was diluted with 50 mM NH4HCO3. The sample was treated with 100 mM DTT at 95°C for 1 h followed by 250 mM IDA at room temperature in dark for 45 min. The sample is then digested with Trypsin and incubated overnight at 37°C. The resulting sample was vacuum dried and dissolved in 20 μl of 0.1% formic acid in water. After centrifugation at 10000 g, the supernatant was collected into a separate tube.10 μL injection volume was used on BEH C18 UPLC column for separation of peptides. The peptides separated on the column were directed to Waters Synapt G2 Q-TOF instrument for MS and MSMS analysis. The raw data was processed by MassLynx 4.1 WATERS. The individual peptides MSMS spectra were matched to the database sequence for protein identification on PLGS software, WATERS. Fyn interacting partners were identified by processing the raw data through PLGS search engine for protein identification and expression.
Sphere formation assay
1×105 cells per 35 mm dish coated with 0.6% agar were seeded in serum-free DMEM-F12 medium with growth factors containing methylcellulose. Sphere forming efficiency was assessed by culturing these cells for 7 days with or without treatment. The primary mammospheres were counted and trypsinised and were allowed to form secondary spheres for another 7 days.
FACS analysis of surface markers
MDA-MB-231 cells and shNotch1 stables with indicated treatments, were trypsinised and 50,000 cells/tube were kept at 37° C for 1 h for surface marker recovery post trypsinisation, followed by double immunostaining with primary antibodies conjugated with FITC or PE for 45 minutes on ice. Cells were washed and resuspended in serum free medium, and analysed using BD FACS Verse. The plots were obtained using BD FACSDIVA software.
DNA damage assay
MDA-MB-231 cells were incubated with 1 μM doxorubicin at 37 □C for 24 h. After quick PBS wash, cells were fixed using methanol: acetone for 5 min in -20°C. Cells were incubated with 1:200 anti-pH2A.X antibody at 4 □C, overnight and subsequently immunofluorescence was carried out. The cells were imaged at 20X and 60X using Olympus IX71. Image processing was done using Image Pro Plus and intensity of puncta was measured.
MTT assay
MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) is a colorimetric assay that measures cell viability as a function of the redox potential of cells. Actively respiring cells can convert the water soluble MTT to insoluble purple formazan crystals. MTT (1 mg/mL from a 5mg/mL stock) was added to the cells (in 100 μL media) at the end of the experiment and 4 h later the formazan crystals were dissolved in 100 μl of DMSO and the absorbance was measured at 570 nm in a spectrophotometer.
Immunohistochemistry
A total of 19 breast cancer tissue samples were obtained from previously untreated grade III invasive ductal carcinoma cases. The paraffin blocks were collected from Kidwai Memorial Institute of Oncology (KMIO; Bangalore, KA, India). Medical Ethics Committee (Institutional Review Board of KMIO) and Institutional Human Ethics Committee (IISc; Bangalore) approved the study. Cleaved Notch1 (Valine 1744) and pACC antibodies were used at 1/100 dilution. IHC was performed as described previously (71). Fischer’s exact test was used to test the correlation between NICD and pACC.
In-vivo tumor formation
BT-474 cells (1x106/100 μL) were subcutaneously injected into each flank of the 10 female athymic nude mice. The mice were allowed to form tumors upto 100 mm3. Then the mice were randomly divided into 4 groups with 5 mice in each. DMSO was injected as a vehicle control intra-peritoneally and served as the control group. Compound C (2 mg/kg) was intraperitoneally injected into second group for every 4 days. After 24th day the tumors were excised and fixed in 10% formalin for immunohistochemistry for NICD.
Signature gene expression
List of signature genes was taken from Molecular Signatures Database GSEA (http://software.broadinstitute.org/gsea/msigdb), AmiGO Gene Ontology Consortium (http://amigo.geneontology.org/amigo), Profiler PCR Array list Qiagen. Expression of genes across breast invasive carcinoma patients in NCBI GEO https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE40206. Heat maps of combined datasets were generated using GENEPATTERN software.broadinstitute.org/cancer/software/genepattern. We performed Hierarchical cluster analysis and data are categorized into different clusters showing similar expression profiles.
Statistical analysis
Statistical analysis was done using GraphPad Prism 5. All data are presented as mean ± SEM. p values < 0.05 were considered as statistically significant. Student’s t-test was used; *** represents p<0.001, **represents p<0.01 and * represents p<0.05. Fischer’s exact test was used for correlation studies.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgement
The authors thank Dr. Benoit Viollet for DKO MEFs, and Mr. Saurav Kumar and Prof. Kondaiah, for help with microarray data analyses and Sai Balaji for help with xenograft assay. This work was majorly supported by grants from the Wellcome Trust-DBT India Alliance (IA) Senior Research Fellowship (500112/Z/09/Z) to AR. ML acknowledges Council for Scientific and Industrial Research for CSIR fellowship (19-12/2010(i) EU-IV). The authors acknowledge grants from DBT-IISc partnership programme, support from DST-FIST and UGC, Govt. of India, to the Department of MRDG and flow-cytometry facility at IISc.
References




