

Abstract
Ortholog inference has fundamental importance across the biological sciences, underpinning phylogenetics, comparative genomics and prediction of gene function. We developed OrthoFinder (https://github.com/davidemms/OrthoFinder), which achieves higher ortholog recall than all current methods as assessed by community-standard benchmarks. Uniquely, OrthoFinder also infers orthogroups, genes trees, gene duplication events, the rooted species tree and extensive comparative genomic statistics, thus enabling the next generation of phylogenomic analyses.
Introduction
The rapid growth in biological sequence data means that many clades of species are densely sampled 1-4, and ambitious projects aim to sequence most of the tree of life. To realise the full potential of these resources requires orthology inference that is accurate, fast and scalable. However, the scale of the data, coupled with processes such as gene duplication and loss, incomplete lineage sorting, and variable sequence divergence rates make ortholog inference a challenging task.
While gene orthology is defined by phylogenetic relationship, a number of methods have been developed that approximate phylogenetic relationships between genes using ‘reciprocal best hits’ (RBH) obtained from BLAST scores. Notable methods include InParanoid 5, OrthoMCL 6 and OMA 7. However, common evolutionary events such as gene duplication can lead to misidentification of orthology relationships using these approximate methods that would otherwise be resolvable using phylogenetic trees of genes 8, 9. As orthology is defined by phylogenetic relationship, it follows that relationships between genes should be more accurately resolved using phylogenetic trees. However, methods for genome-wide ortholog inference using gene trees are unavailable.
The use of gene trees for large scale orthology inference presents numerous technical challenges. Principal among these challenges are the scalable inference of the complete set of gene trees for a set of species, the automated rooting of those gene trees (which affects orthology inference 10), and the accurate inference of orthologs in the presence of gene-tree/species-tree discordance. These challenges are all addressed by OrthoFinder (see methods) to provide accurate and scalable ortholog inference using gene trees. OrthoFinder also infers orthogroups, gene trees for all orthogroups, the rooted species tree, gene duplication events, and provides extensive comparative genomics statistics. This complete, automated analysis is performed with a single command using only protein sequences as input.
OrthoFinder is composed of three principal stages: first, orthogroup inference 11; second, inference of rooted species 12 and gene trees 13; and third, the inference of orthologs and gene duplication events from these rooted gene trees (Fig. 1A). An example of the complete set of results produced by OrthoFinder for 10 metazoan species are shown in Fig. 1B-I. The default, and fastest version, uses DIAMOND 13 for sequence similarity searches, which simultaneously provides the raw data for orthogroup inference 11 and for gene tree inference using DendroBLAST 13. Species tree inference and rooting is achieved using the STAG 12 and STRIDE 10 algorithms that were developed for OrthoFinder. To avoid low ortholog recall due to incomplete lineage sorting and gene tree error, orthologs are inferred from rooted gene trees using an enhanced, more scalable version of the powerful duplication-loss-coalescent model of DLCpar 14 (see methods).

A) OrthoFinder workflow. Step 1: Genes assigned to orthogroups. 2: Unrooted gene trees inferred for each orthogroup. 3: Rooted species tree inferred using STAG & STRIDE algorithms. 4: Gene trees resolved to find 5: Orthologs and 6: Gene duplications events 7: Summary statistics calculated. B-I) Summary of OrthoFinder analysis of a set of Chordata species: Ciona intestinalis, Danio rerio, Oryzias latipes, Xenopus tropicalis, Gallus gallus, Monodelphis domestica, Mus musculus, Rattus norvegicus, Pan troglodytes & Homo sapiens. Bar charts and heat map contain data for each species, aligned to the corresponding species in the tree in (B). B) The species tree inferred by STAG and rooted by STRIDE C) Percentage of genes from each species assigned to orthogroups. D) The number of species-specific orthogroups E) Number of genes with orthologs in any/all species. F) Heat map of the number of orthogroups containing each species-pair (top right) and orthologs between each species (bottom left) G) Ortholog multiplicities for two species, C. intestinalis and H. sapiens, with respect to all other species H) Number of gene duplications events on each terminal branch of the species tree. I) Number of duplications on each branch of the species tree and retained in all descendant species. Abbreviations: OG=orthogroup, sp.=species, spp.=species (plural), dups.=gene duplication events
The default implementation of OrthoFinder been designed to enable ortholog inference from gene trees with maximum speed and scalability. OrthoFinder has also been designed to allow the use of alternative methods for tree inference and sequence search to allow users to choose the balance between accuracy and speed suitable to their research. For example, BLAST 15 or MMseqs2 16 can be used for sequence similarity searches in place of DIAMOND, and gene trees can be automatically inferred from multiple sequence alignments using any user preferred alignment and tree inference method. Moreover, if the species tree is known prior to the analysis, this can be provided as input, rather than inferred by OrthoFinder.
The accuracy of key component algorithms of the OrthoFinder method have been independently assessed 11-13, 17, 18 To demonstrate the accuracy of overall ortholog inference, OrthoFinder (in multiple standard configurations) was submitted to the Quest for Orthologs (QfO) benchmarking server 19 (Fig. 2A-H, Supplementary Fig. 1, Supplementary Table 1, https://questfororthologs.org). All versions of OrthoFinder inferred more orthologs (higher recall/recovered ortholog sets—see methods) than all other tested methods (Figure 2A-H). Across the four tests, the default and fastest version, OrthoFinder (DIAMOND) achieved between 17% (Fig. 2A) and 29% (Fig. 2C) higher ortholog recovery. OrthoFinder with BLAST+MSA achieved between 21% (Fig. 2B) and 39% (Fig. 2C) higher ortholog recovery. No other method was consistently second best to OrthoFinder in terms of ortholog recall/recovered ortholog sets. Precision/Robinson-Foulds distance was between 0.3% better and 6.9% better for default OrthoFinder (Fig. 2B,D). OrthoFinder with BLAST+MSA was between 1.3% worse and 1.7% better than the average for the other methods (Fig. 2C,A). Similarly, OrthoFinder infers more orthologs than most static, online databases at comparable levels of precision/Robinson-Foulds distance (Supplementary Figure 2). For each inferred orthology relationship, OrthoFinder provides the gene tree that supports the inference. A further analysis of the QfO benchmarks by taxonomic grouping is provided in Supplementary Figure 1.

A-H) Quest for Orthologs benchmarks and Pareto frontier for methods (see 19) A-B) Agreement of orthologs with SwissTree/FreeFam-A trees, better scoring methods are at top right C-D) X-axis: percentage of randomly selected genes with orthologs in required species. Y-axis: Robinson-Foulds (RF) distance between selected set of putative orthologs and the known species tree, better scoring methods are at bottom right. E-H) Zoomin of plots A-D. See methods for full description Quest for Orthologs benchmarks. I) Runtime for each method with 4-256 input Fungi proteomes. J) Results returned by methods.
OrthoFinder uses a novel, fast and scalable duplication-loss-coalescent resolution algorithm to identify gene duplication events and map them to the species tree (see methods). The accuracy of these gene duplication detection was demonstrated on two simulation datasets 14, 20 and compared to four popular methods: Notung 21, GSDI Forester 22, DLCpar (exact and search)14 and the overlap method23. The OrthoFinder algorithm out-performed all methods other than DLCpar (exact) (Supplementary Fig. 2A, Supplementary Table 2). On real-world data, it analysed all gene trees from a 128 Fungi species dataset in 141 seconds, whereas DLCpar (exact) was unable to analyse the smallest, 4 species dataset within the 120 hour upper time limit imposed (Supplementary Fig. 2B). Thus, OrthoFinder is the most accurate method that is scalable to realistic datasets.
To demonstrate the scalability of the complete OrthoFinder program, it was run on sets of between 4 and 256 fungal species with 16 parallel processes (Figure 2I). All other software tools available on the Quest for Orthologs benchmarks were similarly tested. There was a large range of runtimes across the methods. Many methods timed out at larger species sets, with 64 species being the largest set on which all were runnable. At this point of comparison, the slowest method took 200 times longer to run than the fastest. The default version of OrthoFinder ran in 192s on the 4 species and 1.8 days on the 256 species datasets. In this time, it inferred orthogroups, all gene trees, the rooted species tree, orthologs and gene duplication events (Fig. 2J). Overall, it was second only to the RBH-based method, SonicParanoid, which took 1.2 days on the 256 species set, but does not provide gene trees, the rooted species tree or gene duplication events.
In summary, OrthoFinder resolves several key technical challenges in the accuracy and scalability of orthology analysis. Its substantial gains in speed and ortholog recall are mirrored by its substantial advance in data provision. For example, no other orthology inference method provides gene trees, rooted species tree and gene duplication events. Thus OrthoFinder should facilitate rapid novel analyses of large datasets that were previously beyond the reach of most research groups. The OrthoFinder source code and executables are available at https://github.com/davidemms/OrthoFinder. A compressed archive of all data is available at the Zenodo research data archive at https://doi.org/10.5281/zenodo.1481147.
Methods
OrthoFinder workflow
A gene tree is the canonical representation of the evolutionary relationships between the genes in a gene family. Thus, ortholog inference from gene trees is an important goal. However, no automated software tools are available that provide accurate, genome-wide ortholog inference from gene trees. A number of challenges had to be addressed to enable this. These included: the efficient partitioning of genes into small, non-overlapping sets such that all orthologs of a gene are nevertheless contained in the same set as the original gene; automatic rooting of gene trees without a user-provided species tree; and robust ortholog inference in the presence of imperfect gene-tree inference. The OrthoFinder workflow was designed to address these challenges and is described in detail below.
OrthoFinder infers orthologs from gene trees using the steps shown in Figure 1A. Input proteomes are provided by the user using one FASTA file per species. Each file contains the amino acid sequences for the proteins in that species. Orthogroups are inferred using the original OrthoFinder algorithm 11; a gene tree is inferred for each orthogroup; the species tree is inferred from the unrooted gene trees using the STAG algorithm 12; the species tree is rooted using the STRIDE algorithm 10; the rooted species tree is used to root the gene trees; speciation and duplication events are inferred from the rooted gene trees by a restriction of the Duplication-Loss-Coalescent model 14 to apply to the problem of ortholog inference using the ‘overlap’ method 23; orthologs and gene duplication events are thus inferred; summary statistics are calculated. Only orthogroup inference was provided in the original implementation of OrthoFinder 11; the subsequent steps are new, and described below.
Use of orthogroups for gene tree inference
For ortholog inference, orthogroups are the optimum partitioning of genes for gene tree inference. An orthogroup is the natural extension of orthology to multiple species. Orthologs are the set of genes in a species-pair descended from a single gene in the last common ancestor of those two species. An orthogroup is the set of genes from multiple species descended from a single gene in the last common ancestor of a set of species. An orthogroup is thus the smallest set of genes such that, for all genes it contains, the orthologs of these genes are also in the same set. Since gene tree inference scales more slowly than linearly in the number of taxa, partitioning genes into the smallest possible sets is the most efficient way of constructing a set of gene trees that encompass all orthology relationships. The original OrthoFinder orthogroup inference method is still the most accurate method on the independent Orthobench test set 11 and thus is used for this step.
Gene tree inference
OrthoFinder provides two methods for gene tree inference. The default method uses DendroBLAST13 trees, inferred using sequence similarity scores already calculated in the first stage of the OrthoFinder algorithm. This is the recommended method since it is fast while achieving high accuracy on the Quest for Orthologs benchmarks 19 (Figure 2A-D). The alternative method infers multiple sequence alignments (MSA) and infers trees from these MSA. OrthoFinder provides the option to restart an existing analysis at the tree inference stage (Figure 1A, Stage 2). This skips the computationally costly all-versus-all sequence search from Stage 1. Thus, if a user wishes to use the more traditional MSA tree inference, it is recommended to first complete a faster OrthoFinder analysis using the default options and to perform a check of the results for quality control. The user can then proceed to an updated analysis using gene trees inferred using MSA.
User-specified sequence search and tree inference programs
OrthoFinder allows the user to specify a program of their choice for the initial sequence search in Step 1 and for the MSA and tree inference from MSA in Step 2 (if MSA trees are used). The default sequence search method is DIAMOND 17, the default MSA method in MAFFT 24 (mafft-linsi is used for trees with fewer than 500 taxa, otherwise default mafft is used) and the default tree inference from MSA method is FastTree 25. A simple configuration file is provided in JSON format. This allows a user to specify the command line format of any alternative program they would like to use for any of these three steps. Any configured program can then be used by selecting it using OrthoFinder command line arguments. In the main text of this paper, three variants of OrthoFinder were tested to place the accuracy of the default options in context: The default version uses DIAMOND search and DendroBLAST gene trees. The first alternative uses BLAST in place of DIAMOND, the second alternative additionally uses tree inference from MSA.
Species tree inference
The rooted species tree is required by OrthoFinder in order to identify the correct out-group in each gene tree, as correct out-group rooting influences the orthology assignments from that tree. If the user knows the rooted species tree for the set of species being analysed then it is recommended to specify this tree manually at the command line. Such a tree can be provided as a Newick format text file. In the event that a species tree is not provided (or not known), then OrthoFinder must infer it.
Sets of one-to-one orthologs are often used for species tree inference but, especially for large scale analyses, these can be rare12. A new algorithm, STAG (Species Tree from All Genes), was developed to allow species tree inference even for species sets with few or no complete sets of one-to-one orthologs present12. Without this algorithm, species tree inference could fail if no one-to-one orthologs were present. STAG infers the species tree using the most closely related genes within single-copy or multi-copy orthogroups. In benchmark tests, STAG13 had higher accuracy than other leading methods for species tree inference; including maximum likelihood species tree inference from concatenated alignments of protein sequences, ASTRAL26 & NJst27.
If the MSA method for tree inference is selected instead then these MSA are also used for species tree inference. Since one-to-one orthologs can be rare, orthogroups are analysed to identify those that are single-copy in as high a fraction of the species as possible. This is balanced with the need for a sufficiently large number of orthogroups that meet this threshold fraction of species which have single-copy genes. OrthoFinder aims for a minimum of 100 orthogroups meeting this criterion using the method described in STAG12. A concatenated MSA is constructed from these orthogroups, with gap characters for species with multiple genes or no genes in a particular orthogroup. Columns with more than 50% gap characters are removed. The species tree is inferred from this concatenated MSA using the user-selected tree inference method.
Species tree rooting
For orthogroups containing gene duplication events, the choice of root for the gene tree can affect ortholog inference10. Since orthogroups can potentially contain any subset of the species in the analysis, it is not sufficient to simply know the out-group for the complete species set. Instead, the complete rooted species tree is required so that the out-group is known for any gene tree containing any subset of the species. The STRIDE algorithm (Specie Tree Root Inference from Duplication Events)10 is used to root the species tree in OrthoFinder. It is particularly suited to species tree rooting for ortholog inference. This is because, the correct root only affects ortholog inference if gene duplication events have occurred. In such cases, these gene duplication events can be identified by STRIDE and used to identify the root of the species tree. Out-group rooting (i.e. knowledge in advance as to the out-group species) can be used by providing the rooted species tree in advance (if the complete species tree is known) or by allowing OrthoFinder to infer the species tree and then the user re-rooting it on the known out-group, before continuing the analysis using the rooted species tree.
Gene tree rooting
For the majority of gene trees, the correct root is unambiguous given the rooted species tree. However, factors such multiple species in the out-group, gene duplication events and gene-tree/species-tree discordance can all frustrate gene tree rooting. A robust algorithm was developed to root any gene tree regardless of the severity of the above mentioned factors.
The correct root for a gene tree is on the out-group species clade, unless the tree contains a gene duplication event prior to the first speciation event. This could occur if two orthogroups derived from a duplication in same gene family have not been correctly separated. If such a duplication event exists in the tree, there will be no unique out-group clade and the correct location for the root is the bipartition corresponding the ancient duplication event. In the rare cases that there are multiple duplication events in the tree prior to the divergence of any of the species then one of these ancient duplications will be the oldest and hence the correct root. However, the topology of the gene tree cannot be used to determine which duplication is the oldest. Fortunately, rooting on any of the ancient duplications correctly preserves all orthology and paralogy relationships. Thus, for orthology inference it is necessary to decide if the gene tree can be best rooted on a clade separating the out-group species from the in-group species or on any bipartition corresponding to an ancient duplication.
For each bipartition in the gene tree two comparable quantities are calculated, SIO and SD, which quantify how well a bipartition separates the genes into in-group/out-group (SIO) and how well the bipartition represents an ancient duplication separating it into two clades, both clades containing the in-group and the out-group (SAD):
 where I/O are the sets of species in the out-group/in-group respectively and A/B are the sets of species in the two clades either side of the bipartition. SIO and SAD range between 0 and 1. A bipartition with a value of 1 for SIO implies that it perfectly divides the tree into an in-group and out-group, and implies a value of 0 for SAD for all bipartitions in the tree. Conversely, a bipartition with a value of 1 for SAD implies that it is a duplication event before the divergence of any of the species, with all species present for both duplicates. It implies a value of 0 for SIO for all bipartitions in the tree. A high value for either SIO or SAD shows that the bipartition is close to one of these perfect cases for a root for the tree. The bipartition with the highest value of SIO or SD is used as the best root for the gene tree.
where I/O are the sets of species in the out-group/in-group respectively and A/B are the sets of species in the two clades either side of the bipartition. SIO and SAD range between 0 and 1. A bipartition with a value of 1 for SIO implies that it perfectly divides the tree into an in-group and out-group, and implies a value of 0 for SAD for all bipartitions in the tree. Conversely, a bipartition with a value of 1 for SAD implies that it is a duplication event before the divergence of any of the species, with all species present for both duplicates. It implies a value of 0 for SIO for all bipartitions in the tree. A high value for either SIO or SAD shows that the bipartition is close to one of these perfect cases for a root for the tree. The bipartition with the highest value of SIO or SD is used as the best root for the gene tree.
Ortholog inference and identification of gene duplication events from gene trees
Ortholog inference from gene trees has been performed using a number of methods. The ‘overlap’ method is employed in a number of ortholog databases 23, 28 and was originally described in a method for determining orthologs of human genes 23. Nodes in the gene tree are identified as duplication nodes if the sets of species below its child nodes overlap, otherwise the node is a speciation node. Genes that diverged at a speciation node are orthologs, those that diverged at a duplication node are paralogues. DLCpar 14 uses a parsimony model to carry out gene tree/species tree reconciliation and identify each node as a duplication or speciation node. It searches for the most parsimonious reconciliation of the gene and species tree under a duplication-loss-(deep) coalescent (DLC) model that addresses incongruence between the gene and species trees. PHYLDOG 20 jointly infers the gene tree and species tree under a maximum likelihood model, thereby implying a reconciliation between a gene tree and the species tree. These methods were tested for ortholog prediction and runtime (in parallel, using 16 cores) on the fungi orthogroups used previously and described in detail below.
DLCpar is available in different versions. There is an exact version, referred to in this paper as DLCpar (exact), which exhaustively explores all possible reconciliations. This is unable to analyse gene families large gene families (>200 genes) and its computational complexity is too high 14 to analyse the gene trees considered by OrthoFinder. Nevertheless, OrthoFinder was compared to it in terms of accuracy at recovering gene duplication events for smaller, simulated gene trees. An approximate version, referred to here as DLCpar (search), uses a hill-climbing algorithm to search for a locally optimum reconciliation. With its default parameters this was found to stop before it has converged to an optimal solution and so it was also tested with parameters that test for convergence. These parameters were: “-i 100 --nprescreen 100 --nconverge 100” for gene trees with fewer than 25 taxa; “-i 25000 --nprescreen 100 --nconverge 1000” for fewer than 200 taxa greater than or equal to 25; and “-i 50000 --nprescreen 100 --nconverge 2000” for greater than or equal to 200 taxa. This variation is referred to here as DLCpar (search, converged). PHYLDOG was used with the LG08 model of sequence evolution (model=LG08), the bionj method for estimating the initial gene tree (init.gene.tree=bionj) and a SPR limit of 5 (SPR.limit.gene.tree=5). The complete option files are given in Supplementary Text 1. Jointly inferring the species tree and all gene trees with PHYLDOG consistently resulted in an error (“optimizeModel.c:2752: modOpt: Assertion ‘tr->likelihood >= currentLikelihood’ failed”) and no results. This was circumvented by inferring the gene trees individually with a pre-calculated species tree inferred using STAG. This species tree was also used to root the gene trees (as described above) for DLCpar and the overlap method. It was also provided as the input species tree to DLCpar.
Ortholog inference using the overlap method, DLCpar (search), DLCpar (search, converged) and PHYLDOG was tested on the OrthoFinder orthogroups 11 of varying size. These were the complete sets of orthogroups for sets of fungi species ranging from 4 species to 256 species. For the overlap method, DLCpar (search) and DLCpar (search converged) the default DendroBLAST gene tree for an orthogroup was provided as input. PHYLDOG requires a multiple sequences alignment (MSA) as input rather than a gene tree as input. The MSA were inferred using MAFFT, as described above. All these options are made available for testing in OrthoFinder using the options: “-R only_overlap”, “-R dlcpar”, “-R -dlcpar_convergedsearch”, “-M phyldog”.
All methods identified similar numbers of orthologs for the orthogroups for small species sets but differences emerged for orthogroups for the larger species sets (Supplementary Fig. 3A). For larger species sets, the gene trees are larger and the complexity of gene tree inference and ortholog inference from gene trees becomes more challenging. DLCpar (search) and PHYLDOG ortholog prediction fell as the number of species increased (by 45% and 22% respectively). DLCpar (search, converged) was consistent as the number of species increased, demonstrating that the DLC model was able to address gene tree/species tree discordance as the complexity of the problem increased. The overlap method showed similar consistency to DLCpar (search, converged) but identified fewer orthologs. However, DLCpar (converged) was over 500 times slower than the overlap method (Supplementary Fig. 3B). It took 230 hours to analyse the set of gene trees for the 64 fungi species. To put this in context, the overlap method required 27 minutes and a complete OrthoFinder run on the 64 fungi species, including the final ortholog inference method and all other steps, required 2.6 hours. For this reason the DLCpar (search, converged), although accurate, was judged to be too slow for genome-wide ortholog inference.
With the aim of achieving similar accuracy to the best performing method, DLCpar (search, converged), but with better scalability, a new ortholog inference algorithm was designed based on the DLC model and overlap method. For ortholog inference using the overlap method, only a restricted version of DLC gene tree reconciliation is necessary since it is only necessary to distinguish duplication nodes from speciation nodes. Subtrees with only one gene per species do not need to be reconciled with the species tree, since they do not imply a gene duplication event. Such discrepancies are costly to analyse under the standard DLC model and yet are very common. For example, in an analysis of 1030 gene trees of one-to-one orthologs from 23 fungi species all 1030 gene trees were topologically distinct from each other and from the species tree 29.
The restricted version of the DLC model that has been implemented in OrthoFinder performs a post-order traversal of the gene tree (a node is not visited until all its descendant nodes have been visited), analysing each node of the gene tree in turn. A given node is analysed to identify if the species sets below its child nodes overlap. If there is an overlap, the smallest sub-clade below each child nodes that contains the complete set of overlapping species is identified up to a maximum total topological depth of two below the node (clades O., Supplementary Fig. 5A). Note, there will always be such a sub-clade, even if the sub-clade corresponds to the child node itself. The possible sub-cases for overlaps between these clades have been enumerated (Supplementary Fig. 5B). The node is assigned to the corresponding sub-case. If a more parsimonious interpretation of the sub-case is available under the DLC model then the sub-tree below the node is rearranged to match this interpretation (Supplementary Fig. 5C). After the node has been analysed, the next node in the post-order traversal is analysed. Note, the post-order traversal can be continued unimpeded despite any such rearrangements below the node being analysed. The resulting gene trees are referred to as ‘resolved’ gene trees and correspond to the locus tree under the DLCpar model 14.
Although only a single traversal of the tree is employed, rather than the iterative search and rearrangement employed by DLCpar, the post-order traversal enables more parsimonious interpretations of child clades below a node to be identified prior to the analysis of the parent node. Thus, analysis of sub-trees below a node inform the subsequent analysis of the node itself. In theory, nodes could be categorised to sub-cases based on overlaps of clades at a greater topological depth than that employed here, however, the number of such sub-cases would increase rapidly. The limited depth proved sufficient to equal the accuracy of DLCpar (search, converged) for ortholog inference. The pre-calculated solutions for each sub-case removed the need for iterative search using random (i.e. unguided) tree rearrangement operations.
The pre-calculation of more parsimonious solutions for a given node is only made possible by the restriction of the DLC model to duplication and loss events implied by overlapping species sets. For full gene tree/species tree reconciliation problem considered by DLCpar, this is not possible. The full reconciliation problem is considerably more complex, but does not need to be solved by OrthoFinder in order to infer orthologs. Note that the species tree is not required for the restricted DLC model used by OrthoFinder. Thus OrthoFinder ortholog inference is unaffected by inaccuracies in species tree inference. The only use of the species tree is in determining the root for each gene tree. OrthoFinder reads orthologs and gene duplication events directly from the resolved gene trees using the overlap method23. For each node in the tree, its two child clades are examined to see if the species present in the clades overlap. If they do then the node is identified as a gene duplication event. If they do not, then all the genes in one of the clades are orthologs of all the genes in the other clade.
This method identified similar numbers of orthologs to DLCpar (search, converged) but was over 500 times faster, taking 27 minutes to analyse the complete set of gene trees for the 64 Fungi datastet (Supplementary Figure 3).
Ortholog Benchmarking
Orthogroup inference accuracy of OrthoFinder has already been tested using the independent Orthobench dataset 30. This showed it to be the most accurate method tested 11. The community developed ‘Quest for Orthologs’ benchmarks 19 were used to assess the accuracy of the newly developed OrthoFinder ortholog inference. OrthoFinder was tested using the default method (DIAMOND sequence search and DendroBLAST trees, no additional options). It was also tested with the BLAST replacing DIAMOND (options: “-S blast”), and with both BLAST search and multiple sequence alignment and maximum likelihood tree inference (options: “-S blast -M msa”). In the latter case the default options of MAFFT 24 and FastTree 25 were used for multiple sequence alignment and tree inference. For each of these three cases, OrthoFinder was run on the 64 reference proteomes of the Quest for Orthologs test set with a single command (“-f Proteomes/” + options) and the inferred orthologs were submitted to the Quest for Orthologs webserver for benchmarking.
The Quest for Orthologs benchmarks are described in detail in 19. The species tree discordance test and the generalised version both consider a set of species partitioned into clades with a known species tree topology connecting the clades. The benchmarking consists of a repeated test. For one of the clades of species a gene is selected at random for each instance of the test. If the orthology inference method under scrutiny predicts an ortholog for that gene for at least one species from each of the remaining clades then the test is recorded as a ‘successful ortholog set’. For each successful ortholog set an MSA is constructed and a gene tree inferred using RAxML31. The normalised Robinson-Foulds (RF) distance is calculated between this tree and the known species tree. The result of the benchmark is the percentage of successful ortholog sets and the average RF distance for the successful sets. A higher percentage success and a lower average RF distance indicates a better ortholog inference method under this test. The SwissTree32 and TreeFam-A33 tests compare the ortholog precision and recall compared with the orthologs inferred from a set of the curated SwissTree and TreeFam-A gene trees. Note that OrthoFinder can be penalized by an increased RF distance in these test in cases were OrthoFinder judges it more parsimonious that genes are orthologs under the DLC model even if the gene tree conflicts with the species tree. The benchmarks treat all such differences as an error, rather than considering what is the most parsimonious explanation for the conflict. This is an important point, as discussed above it is expected that gene trees will contain tree error (else species trees could be accurately inferred from a gene tree of any single copy gene). In general genes trees do not match the species tree and in an exemplar analysis of 1030 gene trees of one-to-one orthologs from 23 fungi species all 1030 gene trees were topologically distinct from each other and from the species tree 29...
The full set of benchmarks, the input files and the ortholog inference results can be seen online at http://orthology.benchmarkservice.org/ and are shown in Fig. 2A-H of the main paper. The complete datasets are available to download from Zenodo research archive at https://doi.org/10.5281/zenodo.1481147.
Performance Testing
We constructed sets of fungal proteomes of increasing size for performance testing. Ensembl Genomes was interrogated on 6th November 2017 using its REST API 34 to identify all available fungal genomes. To achieve an even sampling of species we selected one species per genera and excluded genomes from candidate phyla or phyla with fewer than 3 sequenced genomes. This gave a set of 272 species which were downloaded from the Ensembl FTP site 35. We created datasets of increasing size by randomly selecting 4, 8, 16, 32, 64, 128 and 256 species such that the last common ancestor was the same for each dataset. Each dataset was analysed using a single Intel E5-2640v3 Haswell node (16 cores) on the Oxford University ARCUS-B server using 16 parallel threads for OrthoFinder with DIAMOND (arguments: “-S diamond -t 16 -a 16”). The complete datasets for all analysed species subsets are available for download from Zenodo at https://doi.org/10.5281/zenodo.1481147. All methods submitted to Quest for Orthologs that provided a user-runnable implementation of the method were tested on the same Fungi datasets and same ARCUS-B server nodes and run in parallel using 16 threads (when supported by the method).
Evaluation of gene tree inference
OrthoFinder makes it possible to use any software for tree inference from MSA. Additionally, it allows gene tree inference using DendroBLAST13, which is a fast, distance-based method for gene tree inference. This saves considerable time by reusing the results from the original all-versus-all sequence search performed as the first step of the OrthoFinder workflow. To investigate the options available to the user in terms of accuracy and speed, a number of alternative gene tree inference methods were tested for performance and accuracy. These were: DendroBLAST, which infers a tree from a matrix of similarity scores derived from a local alignment tool such as BLAST 13; QuickTree, a neighbour-joining algorithm using distances inferred from a global MSA 36; FastME, which uses infers a tree from a distance matrix inferred from a multiple sequence alignment using the minimum evolution principle 37; FastTree, an approximate maximum likelihood (ML) method 25; and IQTree, a fast ML method that compares favourably to RAxML and PhyML in terms of likelihood 38. Multiple sequence alignments were inferred using MAFFT 24. The methods were tested on previously calculated orthogroups for 12 different species sets sampled from across the eukaryotic domain, with between 5 and 47 species 10. Gene trees were inferred using each method for all orthogroups from the 12 species sets containing four or more genes. For each method, the tasks were parallelised over 16 cores.
The ordering of the runtimes for the methods were as expected, with DendroBLAST the fastest method (3.8 hours for all 206575 trees) and IQ-TREE the slowest (6561 hours) (Supplementary Fig 5A). The second fastest method was QuickTree (19.0 hours).
Two methods were used to assess the accuracy of the resulting trees, which both agreed on the ranking of the methods. These two methods were used since, in general, the gene trees were multi-copy and so could not be directly compared with a high accuracy species tree (for example) to assess their accuracy. In the first test, Notung 21 was used to reconcile each gene tree against a high-confidence species tree from literature 10 using the duplication and loss model. The cost from this is the number of gene duplication and loss events that must be inferred in order to explain the gene tree topology given the species tree. This will count both true duplication and loss events but also all excess events that must be postulated to explain any topological discrepancies between the gene tree and the species tree. Using this method, gene trees for multi-copy gene families in which the branching order of species within clades matches the branching order of the species tree received a lower cost than those where the branching order differed. The assumption is made that a more accurate tree inference method will show more agreement with the species tree in terms of species branching order and thus Notung reconciliation cost will be lower. For each tree, the cost was normalised by the number of nodes in the tree. The accuracy of the tree inference methods using the normalised Notung reconciliation cost exactly mirrored the ordering of the methods in terms of runtime (Supplementary Fig. 6B).
In the second test, the IQ-TREE gene tree for each orthogroup was used as the best available estimate of the true gene tree (it was confirmed as the most accurate method in the first test). The normalised Robinson-Foulds distance between this IQ-TREE gene tree and the gene tree from each of the remaining tree inference methods was calculated. This method confirmed the ordering of the remaining methods in terms of accuracy: FastTree, FastME, QuickTree and DendroBLAST (Supplementary Fig. 6B).
These tests showed that DendroBLAST is approximately five times faster than the next fastest method, but also the least accurate of the methods considered (Supplementary Fig. 6). However, the Quest for Orthologs benchmarks (Figure 2A-D) tests showed that gene trees from DendroBLAST provide more accurate orthologs using the OrthoFinder method than using any of the heuristic RBH-based methods of inferring orthologs. FastTree+MAFFT is slower than FASTME+MAFFT or QuickTree+MAFFT by a factor of two to three times but achieves accuracy close to that of IQ-TREE. While IQ-TREE is the most accurate method, most users will find it too slow for genome-wide gene tree inference (average runtime 547 hours for the sets of orthogroups measured; median runtime 72 hours, which was for primate dataset consisting of 11 species and 19096 orthogroups). For the faster methods, the majority of time was taken by MAFFT for MSA inference (18.7 hours of the 19.0 hours for QuickTree+MAFFT and of the 27.1 hours for FastME+MAFFT), thus the development of faster MSA inference methods could affect the relative trade-offs for choice of gene tree inference method. Such a MSA inference method could be utilised by a user of OrthoFinder simply by adding it to the JSON format configuration file. To place these times in context of the full OrthoFinder workflow, 128 fungal species took 12.9 hours from start to finish using the default DendroBLAST gene trees. DendroBLAST tree inference was 18 minutes of this time whereas MAFFT+FastTree tree inference would require 5.2 hours.
Simulation Tests of OrthoFinder Gene Duplication Event Inference Accuracy
To test the ability of OrthoFinder’s tree resolution method to identify gene duplication events, it was tested on the simulated ‘flies’ and ‘primates’ dataset from 14 and the simulated ‘metazoa’ dataset from 20. To model real data, the flies and primate datasets used known species trees and parameters for divergence times, duplication and loss rates, population sizes and generation times. Trees were simulated with varying effective population sizes and duplication rates so as to model incomplete lineage sorting. The flies dataset consisted of 12000 trees with 12 species and 12032 gene duplication events. The primates dataset consisted of 7500 trees with 17 species and 16066 gene duplication events. The metazoa dataset intended to emulate the complexity of real data by using heterogeneity in rates of duplication and loss, a complex model of sequence evolution and then inferring trees with a homogenous, simple model 20. It consisted of 2000 gene trees with 40 species and 4967 gene duplication events.
OrthoFinder’s ortholog inference algorithm, which identifies a node as a speciation or duplication, was tested on the simulated gene trees to test its ability to accurately identify gene duplication events. For comparison, Forester 22, Notung 21, DLCpar (exact), DLCpar (search) 14 and the overlap algorithm (i.e. without OrthoFinder’s tree resolution) were also tested. PHYLDOG was the source of the primates dataset, and provided gene trees generated under different modelling assumptions from those used in DLCpar. However, it was not included in the tests presented here since it considers the problem of generating reconciled gene trees from multiple sequence alignments (MSA) and is not applicable to the inference of gene duplication events from gene trees. The flies and primates datasets consist of gene trees generated directly from a multispecies coalescent process. Since they do not include multiple sequence alignments there would be no way of testing PHYLDOG on these two datasets. To measure performance, all methods were run in parallel using 16 cores on the gene trees for the 4 to 128 species fungi orthogroups sets.
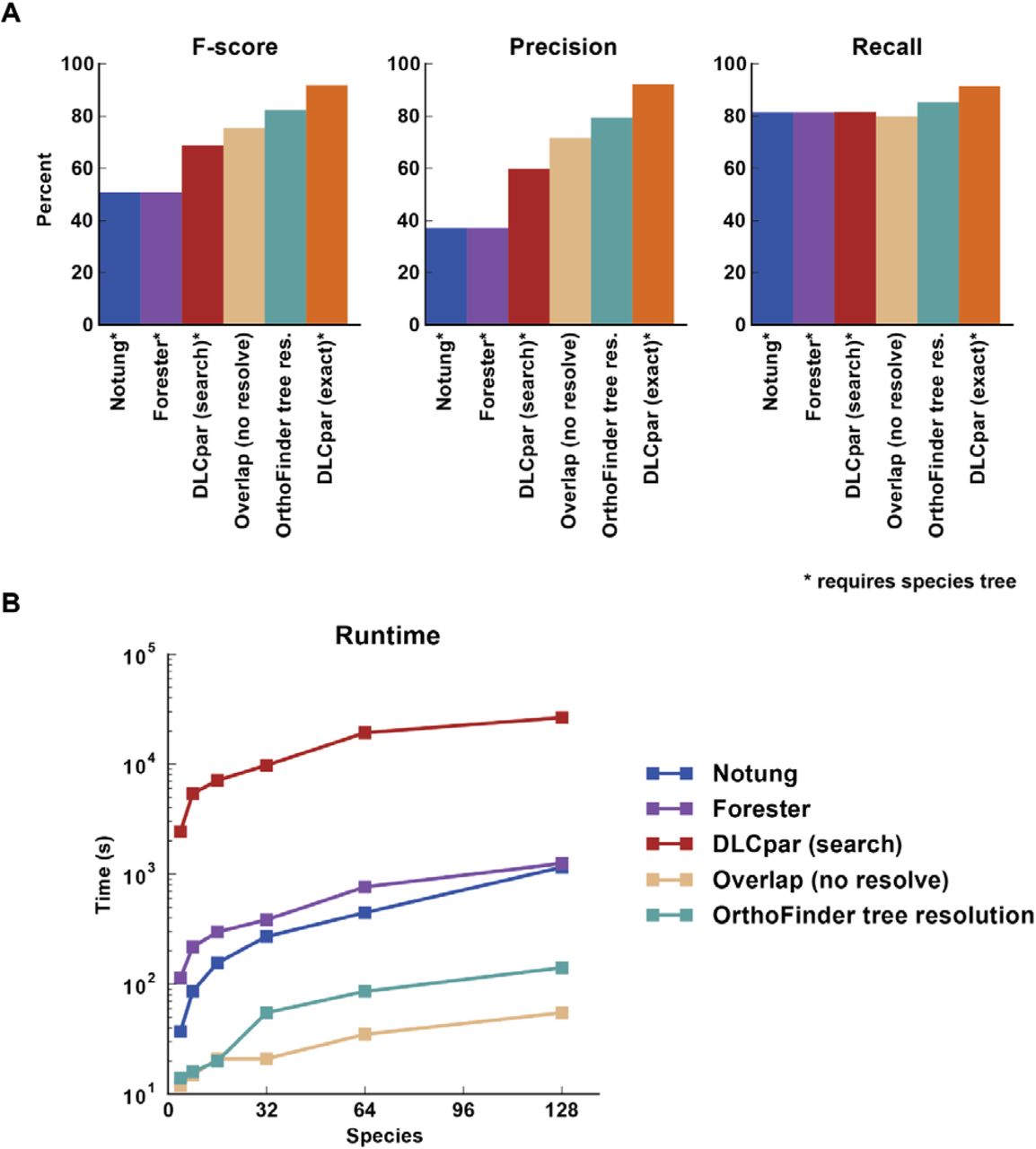
The results showed that the OrthoFinder was second only to DLCpar (exact) in terms of accuracy (Supplementary Fig. 2A). However, the DLCpar (exact) was unable to analyse the trees from the smallest fungi species dataset in 120 hours so no run times were recorded for it on the real world datasets. For comparison, the next slowest method DLCpar (search) was able to analyse this dataset in 40 minutes and the largest dataset (gene trees for the 128 fungi species) in 7.3 hours (Supplementary Fig. 2B). The OrthoFinder method achieved an F-score of 82.8% versus 91.8% for DLCpar (exact). It analysed all gene trees for the 128 fungi species in 2 minutes 21 seconds. Thus the OrthoFinder method had higher accuracy than any of the methods capable of analysing real world data in a reasonable runtime. Additionally, the use of the OrthoFinder tree resolution plus overlap algorithm increased both precision and recall over using the overlap algorithm on its own (Supplementary Fig. 5A).
Chordata Dataset
The data for the OrthoFinder analysis of the ten chordata species for the illustration of the results of an OrthoFinder analysis (Fig. 1A-I) are provided in the Zenodo archive https://doi.org/10.5281/zenodo.1481147. This includes the input proteomes, the OrthoFinder results and the script used to generate the figures from the results. OrthoFinder was run with default settings (DIAMOND sequence search and DendroBLAST gene trees).
Supplementary Figure Legends

Results of the Generalised Species Tree Discordance tests from the Quest for Orthologs benchmarks for four clades of species. A) Vertebrata B) Fungi C) Eukaryota D) LUCA (Last Universal Common Ancestor, a species set covering bacteria, archaea and eukaryotes). See methods for description of Quest for Orthologs benchmarks.

Quest for Orthologs benchmarks and Pareto frontier for OrthoFinder and online databases (see 19) A-B) Agreement of orthologs with SwissTree/FreeFam-A trees, better scoring methods are at top right C-D) X-axis: percentage of randomly selected genes with orthologs in required species. Y-axis: Robinson-Foulds (RF) distance between selected set of putative orthologs and the known species tree, better scoring methods are at bottom right.

A) Duplication inference accuracy on simulated gene trees. B) Runtime to analyse all trees from the 4 to 128 species Fungi datasets (see methods), a maximum time of 120 hours (4.3×105 seconds) was allowed. DLCpar (exact) did not complete the smallest dataset in this time limit and so no time points are shown.

Comparison of methods for ortholog inference from gene trees as a function of increasing number of species in orthogroups. A) Average orthologs per gene as percentage of the maximum possible number if every gene had an ortholog in every species. B) The runtime for each of the methods.

Deterministic tree reconciliation applied to nodes within a gene tree. If the sets of species below a node, n, overlap the node is analysed to find if there is a more parsimonious interpretation of the subtree under the duplication, loss, deep-coalescence (DLC) model. The analysis is done in the context of the sister clade, S, and the descendant clades, U, V, W, X, Y, Z and O. These clades may contain single or multiple genes. The overlapping clades under the two descendants of n are identified, down to a total combined depth of 2 (so that the problem remains tractable). Each case has a number of possible sub-cases according to whether the species sets in the clades overlap. The notation X&Y means that the species set for the genes in clade X overlap with the species sets in the clade Y. In order for a node to fit an archetype (the overlap in n’s descendants is in the clades ‘O’), constraints arise on some of the sub-overlaps. T=True, F=False.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comparison of gene tree inference methods. A) Runtime using 16 parallel processes for tree inference for all orthogroups from 12 species sets from 10, containing a total 206575 gene trees. B) Violin plot of the normalised Notung reconciliation cost between each gene tree and the corresponding species tree. C) Violin plot of the normalised Robins-Foulds (RF) distance between the IQTREE inferred gene tree and the gene tree inferred by each of the remaining methods.
Acknowledgements
The authors would like to acknowledge the use of the University of Oxford Advanced Research Computing (ARC) facility in carrying out this work.
Bibliography




