

Abstract
Enterococcus faecalis is an opportunistic pathogen with an intrinsically high resistance to lysozyme, a key effector of the innate immune system. This high level of resistance requires several genes (oatA, pgdA, dltA and sigV) acting synergistically to inhibit both the enzymatic and cationic antimicrobial peptide activities of lysozyme. We sought to identify novel genes modulating E. faecalis resistance to lysozyme. Random transposon mutagenesis carried out in the quadruple oatA/pgdA/dltA/sigV mutant led to the identification of several independent insertions clustered on the chromosome. These mutations were located in a locus referred to as the enterococcal polysaccharide antigen (EPA) variable region located downstream of the highly conserved epaA-epaR genes proposed to encode a core synthetic machinery. The epa variable region was previously proposed to be responsible for EPA decorations, but the role of this locus remains largely unknown. Here, we show that EPA decoration contributes to resistance towards charged antimicrobials and underpins virulence in the zebrafish model of infection by conferring resistance to phagocytosis. Collectively, our results indicate that the production of the EPA rhamnopolysaccharide backbone is not sufficient to promote E. faecalis infections and reveal an essential role of the modification of this surface polymer for enterococcal pathogenesis.
Introduction
Enterococcus faecalis is a commensal bacterium found in the gastro-intestinal tract of humans and frequently isolated from the environment as a result of faecal contamination [1, 2]. Although this organism is considered as harmless in healthy carriers, E. faecalis has been proposed to contribute to the pathogenesis of inflammatory bowel disease and colorectal cancer [3, 4]. E. faecalis can also cause a wide range of hospital-acquired opportunistic infections that can be life-threatening [1]. E. faecalis infections can be difficult to treat due to the resistance of this organism to antibiotics such as cephalosporins and glycopeptides (Vancomycin Resistant Enterococci, VRE) and its capacity to form biofilms [5]. Interestingly, E. faecalis strains responsible for hospital-acquired infections are also found in healthy individuals and genes associated with virulence are not only present in clinical isolates [6]. How this organism can cause infections is therefore not entirely understood. One property of E. faecalis that contributes to the onset of infections is its resistance to the host innate immune system. Cell surface polymers including teichoic acids (TAs), a capsule and the enterococcal polysaccharide antigen (EPA) confer phagocytosis evasion and resistance to complement activation [7-9]. E. faecalis also displays an intrinsically high resistance to lysozyme, a key component of the innate immune system representing a first line of defence against pathogens. Lysozyme is found in virtually all human biological fluids including saliva, milk, serum and tears where it is found at concentrations between 1-2 mg ml−1 [10, 11]. Lysozyme has two distinct antimicrobial activities. Firstly, it hydrolyses the glycan chains of peptidoglycan, the major component of the bacterial cell wall, causing cell lysis [12]. Secondly, lysozyme displays cationic antimicrobial peptide (CAMP) activity. Lysozyme contains highly charged C-terminal sequences (RAWVAWRNR in human lysozyme) sufficient to inhibit bacterial growth [13] by causing membrane permeabilization [14].
Four genes (oatA, pgdA, dltA and sigV) contribute synergistically to lysozyme resistance in E. faecalis. Both OatA and PgdA modify peptidoglycan glycan strands, thereby inhibiting lysozyme catalytic activity. OatA is an O-acetyl transferase that transfers an acetyl group onto the C6-OH group of N-acetylmuramic acid residues [15]. PgdA is produced in response to lysozyme and is an esterase that removes the acetyl group in position 2 of N-acetylglucosamine residues [16]. DltA is a D-alanine-D-alanyl carrier ligase essential for the alanylation of TAs. It has been proposed that this modification reduces the net negative charge of TAs and inhibits the CAMP activity of lysozyme [17]. SigV is an extracytoplasmic sigma factor that controls the expression level of pgdA in response to lysozyme [18, 19]. oatA, pgdA, dltA and sigV act in concert to confer high-level resistance to lysozyme in E. faecalis. Deletions in these genes (alone or in combination) have been associated with a reduction in virulence in mice or Galleria mellonella [18, 19] and a decrease in survival within murine peritoneal macrophages [15].
In this study, we show that the quadruple mutant (oatA, pgdA, dltA and sigV; OPDV strain) still displays a relatively high resistance to lysozyme in comparison to other Firmicutes. Using transposon mutagenesis, we used this quadruple mutant to carry out transposon mutagenesis and to identify additional genes involved in lysozyme resistance. We show that several genes contributing to the decoration of the enterococcal polysaccharide antigen play an essential role in the resistance to effectors of the innate immune system and in virulence.
Results
The E. faecalis quadruple mutant harboring deletions in oatA, pgdA, dltA and sigV displays a relatively high residual resistance to lysozyme
We determined the minimal inhibitory concentrations (MIC) of lysozyme for several Gram-positive bacteria (Table 1; see S1 Fig for a representative set of MIC assays). Micrococcus luteus, used as a reference substrate to define lysozyme activity [20] had an expected very low MIC of 5 × 10−4 mg ml−1. MIC values were higher for all Firmicutes tested. Growth of several species was inhibited by lysozyme concentrations of 0.0312 mg ml−1 (Aerococcus viridans Bacillus subtilis, Bacillus megaterium and Lactobacillus cellobiosus). Lactococcus lactis growth was inhibited by concentrations of 0.125 mg ml−1. The MIC of lysozyme for all pathogens tested was relatively high: 4 mg ml−1 for Listeria monocytogenes and >16 mg ml−1 for Staphylococcus aureus, and all enterococci and streptococci tested. In L. monocytogenes, lysozyme resistance was largely due to peptidoglycan de-N-acetylation. Deletion of the gene encoding the deacetylase PgdA led to a 32-fold decrease in resistance (MIC=0.125 mg ml−1). Interestingly, abolishing peptidoglycan O-acetylation or de-N-acetylation in E. faecalis only had a minor impact on lysozyme resistance [16, 19]. The combined deletions in the four genes contributing to E. faecalis lysozyme resistance (oatA, pgdA, dltA and sigV; OPDV strain) was required for 10-fold reduction in the MIC of this antimicrobial compound (0.5 mg ml−1). However, the lysozyme MIC for the OPDV mutant was still higher than that of non-pathogenic Gram-positive bacteria.
Transposon mutagenesis of the OPDV epa variable region confers resistance to lysozyme
The relatively high lysozyme MIC of the quadruple mutant (OPDV strain) prompted us to further explore E. faecalis properties modulating lysozyme activity. We constructed a transposon mutant library in the OPDV background using the Mariner-based system previously described for E. faecium [21]. Transposon mutants were selected on agar plates containing lysozyme at a concentration of 2 mg ml−1, four times the MIC for the parental OPDV strain. Approximately 2 × 105 mutants were plated and after 24 h incubation at 37°C, 16 mutants forming colonies at this concentration were isolated and further analysed. Mapping the transposon insertion sites revealed that 9 mutants had insertions downstream of the conserved epaA-epaR region encoding the core synthetic apparatus likely required to produce the enterococcal polysaccharide antigen EPA (Fig 1A) [22]. The region containing the insertions displays genetic variability between strains and was proposed to be responsible for the decoration of the EPA polysaccharide [22-24]. Mutations were clustered around three genes encoding putative glycosyltransferases and a homolog of wcaG, an epimerase/dehydratase (Fig 1B).

A. Description of individual transposon insertions. B. Mapping of transposon insertions in the epa variable region. Insertion sites are indicated by vertical arrows. ORFs in the epa variable region are depicted in grey.
Transposon mutants were complemented to formally establish that the insertions in the epa variable region were responsible for lysozyme resistance. Four plasmids were constructed to express OG1RF_11720, OG1RF_11715 (epaOX), OG1RF_11714 (epaX-like) and OG1RF_11707 under the control of the inducible tet promoter. Following transformation into E. faecalis, gene expression was induced in the presence of anhydrotetracycline (ATc) and the production of his-tagged proteins was checked by western blot (S2 Fig). Complementation was evaluated by measuring susceptibility to lysozyme (Fig 2). Lysozyme resistance associated with transposon insertions in genes OG1RF_11720, OG1RF_11715, OG1RF_11714 and OG1RF_11707 could be complemented when the disrupted gene was expressed in trans. By contrast, the parental susceptibility to lysozyme could not be restored when complementation experiments were carried out with plasmids encoding a gene distinct form the one disrupted (Table 2). Altogether, these experiments confirmed that the resistance phenotypes of the mutants analysed were due to the disruption of the genes indicated in Fig 1.

Cell suspensions were prepared as described in S1 Fig and 1.5 µl of serial dilutions were spotted on BHI-agar plates containing 10 ng ml−1 anhydrotetracycline and various concentrations of lysozyme. Concentrations showing a clear difference in susceptibility are shown.
Mutations in the epa variable region alter the negative surface charge of E. faecalis and are associated with minor changes in sugar composition
The impact of epa transposon insertions on the production of EPA was investigated. Polysaccharides were extracted from cultures at the end of exponential growth as previously described [25]. Similar amounts of EPA were extracted as assessed by neutral sugar assays and dry weight (between 20 and 30 mg l−1). Each purified EPA sample was run on a polyacrylamide gel and stained with the cationic dye alcian blue (Fig 3A). Whilst OG1RF and OPDV polysaccharide bands previously named PS1 and PS2 were present [8], these were not detected in mutant samples, possibly because the reduced negative charge no longer allowed EPA to migrate in the gel or no longer allowed these polymers to be stained by alcian blue. As expected, complementation restored the detection of EPA after staining by alcian blue (Fig 3A).

A. Analysis of purified EPA by acrylamide gel electrophoresis. 40 µg of material was loaded on a 10 % (v/v) acrylamide-bisacrylamide (33:0.8) gel and stained with the cationic dye alcian blue. B. Electrophoretic mobility of E. faecalis OG1RF, OPDV and insertion mutants resistant to lysozyme. Representative mutants harbouring a transposon insertion in OG1RF_11720 (OPDV_11720::Tn2.5), OG1RF_11715 (OPDV_11715::Tn2.13), OG1RF_11714 (OPDV_11714::Tn2.14) or OG1RF_11707 (OPDV_11707::Tn2.8) were analysed. Wild-type OG1RF and parental OPDV strains were included as controls. C. Carbohydrate composition of purified EPA polysaccharides. The relative percentage corresponding to each monosaccharide was determined from three independent extractions.
We tested the impact of the transposon insertions on EPA charge by measuring the electrophoretic mobility of E. faecalis cells using micro-electrophoresis, which allows single particle tracking (Fig 3B) [26]. OG1RF cells displayed a negative electrophoretic mobility (migration towards the anode), even at low pH, indicating a negative surface charge. Despite the increased susceptibility of OPDV cells to lysozyme, the electrophoretic mobility measured with this strain was not significantly different from the mobility of OG1RF cells at all pHs tested (Fig 3B; S1 and S2 Table). Three of the four insertion mutants (OPDV_11720::Tn2.5, OPDV_11715::Tn2.13 and OPDV_11720::Tn2.8) displayed similar electrophoretic mobilities, very distinct from the parental OPDV strain. The negative surface charge of these mutants was significantly reduced as compared to that of the parental OPDV strain (****P<0.0001 for all pH conditions), the difference being most prominent at pH 3.0 (Fig 3B). As expected, differences between OPDV, OPDV_11720::Tn2.5, OPDV_11715::Tn2.13 and OPDV_11720::Tn2.8 cells were abolished when the mutations were complemented. Mutant OPDV_11714::Tn2.14 only showed a difference with the parental OPDV strain at pH 2.0 (****P<0.0001). This difference was no longer detected when the mutation was complemented.
To confirm that the lack of detection of EPA on polyacrylamide gels was due to a loss of negatively charged groups rather than a lack of rhamnopolysaccharide production, we carried out carbohydrate composition analyses on purified EPA (Fig. 3C). As anticipated, EPA composition was very similar in OG1RF and OPDV strains; rhamnose, glucose, N-acetylglucosamine (GlcNac) and N-acetylgalactosamine (GalNAc) accounted for approximately 95% of the sugars identified (ca. 30%, 30%, 20% and 15%, respectively) and galactose was found in limited amounts (5%). The proportion of glucose and GlcNAc remained similar to parental OPDV levels in all epa mutants, whilst both GalNAc and galactose amounts decreased to an increase of rhamnose. Interestingly, these changes were different depending on the mutant considered. For example, EPA extracted from OPDV_11718::Tn2.8 was the only mutant EPA that still contained some galactose. The relative proportion of rhamnose increased in all mutants. GalNAc content decreased dramatically in all mutant EPA and could not be detected in OPDV_11715::Tn2.13.
NMR analyses reveal that the epa variable region contributes to minor modifications of the EPA polysaccharide
To gain further insight into the contribution of epa variable genes to the structure of EPA, we carried out NMR analyses on purified polysaccharides. The 1D proton NMR spectra of all polysaccharides were overall similar but mutations in the epa variable region were associated with modifications in the anomeric region (Fig 4A). Clear differences were also detected in the relative intensities of methyl protons in the mutant spectra. For all mutants, the intensity of N-acetyl signals (1.9-2.2 ppm) decreased whilst the intensity of methyl protons corresponding to rhamnose residues (1.2-1.6 ppm) increased, suggesting a lower content of hexosamine in mutant EPAs and a relative increase of rhamnose. This result is in agreement with the carbohydrate analyses following acid hydrolysis (Fig 3C and S3 Fig). 1H-13C HSQC spectra revealed that EPA has a very complex structure, as evidenced by the detection of over 30 signals in the anomeric region (Fig 4B). The comparison of 2D spectra corresponding to OPDV and mutant polysaccharides revealed that each epa mutation only led to a limited number of changes including changes in the signal intensity, signal shifts and disappearance (S4 Fig). The number and the nature of the signals affected in the epa mutants were different depending on the mutation considered. Altogether, NMR and sugar analyses supported the idea that the epa variable genes are involved in limited modifications of the EPA rhamnopolysaccharide previously described as “decorations”.
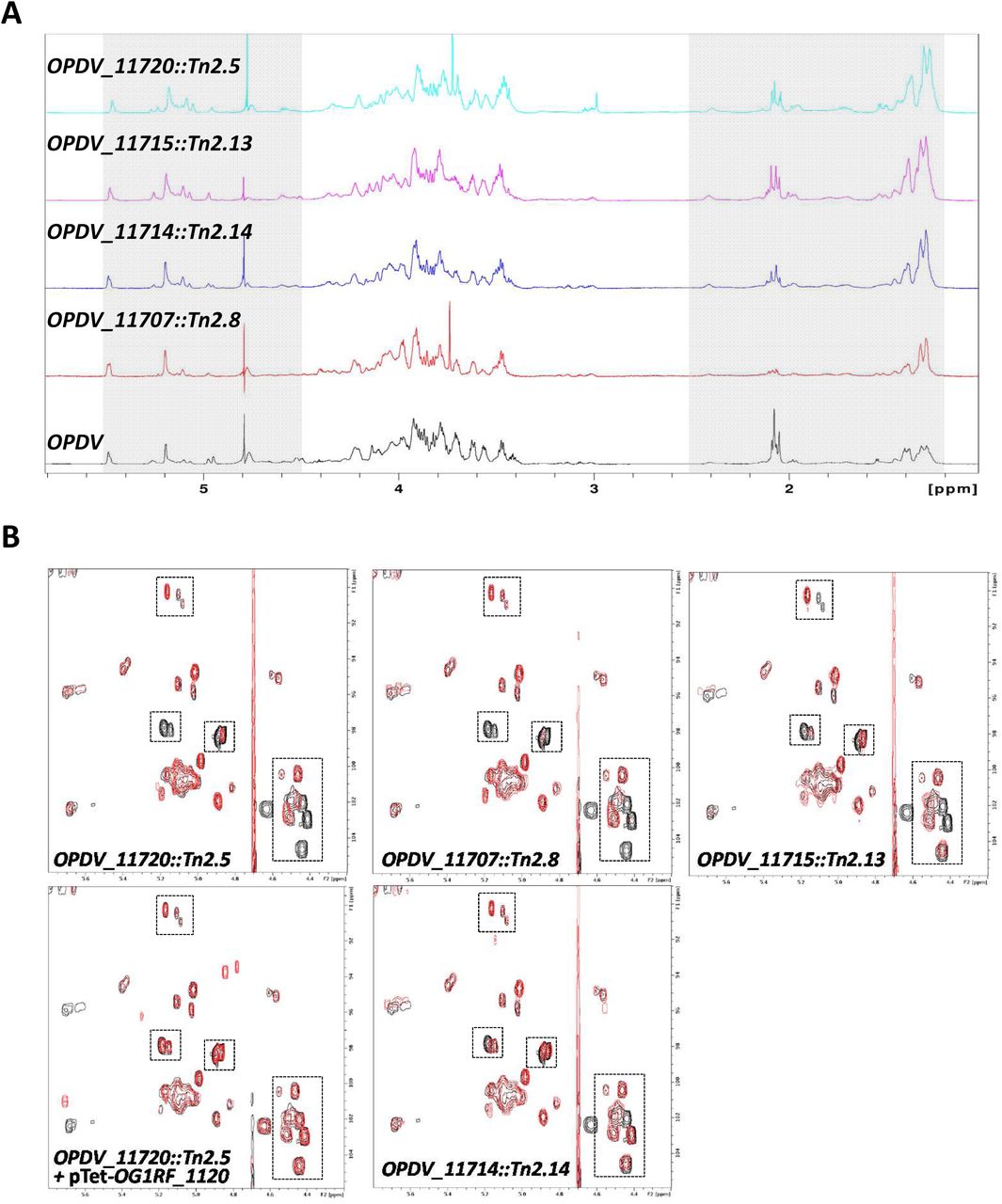
A. 1D proton spectra of the EPA polysaccharides extracted from strains OPDV, OPDV_11720::Tn2.5, OPDV_11715::Tn2.13, OPDV_11714::Tn2.14 or OPDV_11707::Tn2.8. The grey boxes indicate anomeric (4.5-5.5 ppm) and methyl protons (1.2-2.5 ppm). B. 2D 1H-13C HSQC spectra of EPA polysaccharides. The region corresponding to anomeric protons (4.2-5.5 ppm) and anomeric carbons (90-105 ppm) is shown. OPDV signals are in black, mutant signals in red. Boxes show signals with a lower intensity or a shift in the mutant EPA samples. Close-up views of the boxed regions are shown in S4 Fig.
Epa decoration determines susceptibility to antimicrobials targeting the cell envelope
All epa transposon insertions were combined with four other mutations present in the in OPDV strain (oatA, pgdA, dltA and sigV), leaving the possibility of epistatic interactions between these mutations. To avoid this potential issue, we built in-frame epa deletions in the OG1RF genetic background (S5 Fig) before testing the impact of EPA decorations on resistance to antimicrobials targeting the cell envelope (Fig 5). All epa mutants were more sensitive to SDS than OG1RF, mutants Δ11714 and Δ11707 being less sensitive than the two others. Interestingly, mutant Δ11714 (ΔepaX-like) was the only one that did not display increased susceptibility to sodium cholate, a primary bile salt. Mutant Δ11720 was the only one with an increased susceptibility to both polymyxin B and nisin, two cationic peptides targeting the cell envelope. Mutants Δ11707 and Δ11715 were more susceptible to polymyxin B than the wild-type strain, but barely more susceptible to nisin. Finally, deletion of OG1RF_11714 had no detectable impact on resistance to either of the CAMPs tested. Taken together, these results indicated that genes in the epa variable region are required for resistance to antimicrobials targeting the cell envelope.
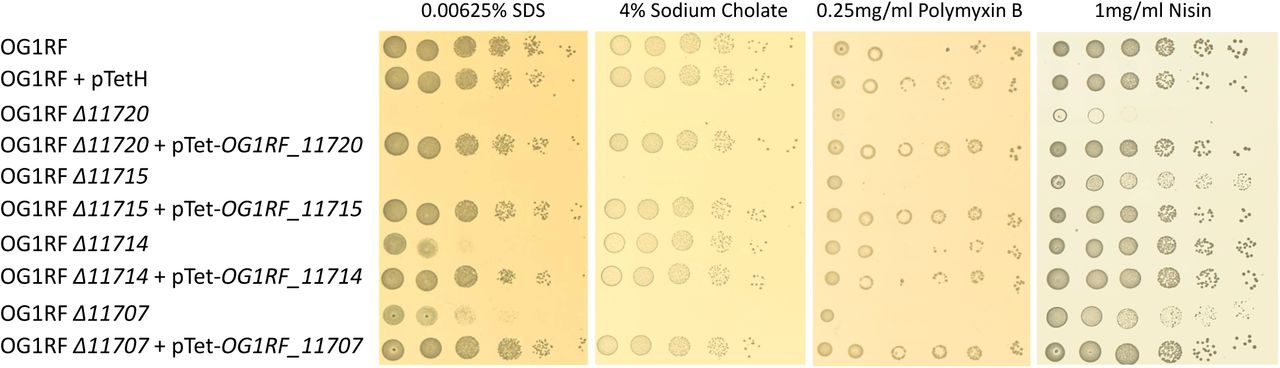
Cell suspensions were prepared as described in S1 Fig and 1.5 µl of serial dilutions were spotted on BHI-agar plates containing 10 ng ml−1 anhydrotetracycline and SDS, sodium cholate, polymyxin B or nisin. For each antimicrobial, one concentration showing a difference in susceptibility is shown.
The decoration of the EPA rhamnopolysaccharide is essential for virulence and underpins phagocyte evasion
The impact of epa mutations on E. faecalis virulence was tested in the zebrafish experimental model of infection (Fig 6). Cell suspensions corresponding to approximately 1,200 CFUs were injected in the bloodstream of LWT embryos 30 h post fertilization (hpf) and the survival of larvae was monitored for 90 h post infection (hpi). As a preliminary experiment, we analysed the virulence of the OPDV transposon mutants (S6 Fig). Each epa transposon mutant had a significantly reduced virulence as compared to the wild-type OG1RF strain. Even though the combined deletions in oatA, pgdA, dltA and sigV did not impair the virulence of E. faecalis in the zebrafish model of infection (S7 Fig), we could not exclude the possibility of an epistatic relation between the OPDV and epa mutations. We therefore repeated the zebrafish infections using the in-frame epa deletion mutants in the wild-type OG1RF genetic background (S5 Fig). All epa mutants showed a significant decrease in virulence as compared to the wild-type OG1RF (Fig 6), killing only between 0-10% of the larvae as opposed to the 40 to 55% of killing following injection of the wild-type strain; Fig 6A-D). As expected, the complementation of each epa deletion fully restored the virulence. Although the epa deletion mutants (except the ΔepaX-like strain) present a slight defect in their growth rate, it is unlikely that this accounts for the lack of virulence; all complemented strains (and the wild-type OG1RF harbouring an empty complementation vector) also present a growth defect (S8 Fig) and yet kill zebrafish larvae as well as the wild-type strain (Fig 6).

Survival of zebrafish larvae (n>20) following infection with E. faecalis OG1RF (WT) and epa deletion mutants was monitored over 90 h post infection. A. Mutant Δ11720. B. Mutant Δ11715. C. Mutant Δ11714. D. Mutant Δ11707. E. Statistical significance determined by Log-rank test; ns, P>0.05; **P<0.01; *** P<0.001; **** P<0.0001. All injections presented in Fig 4A and 4D were carried out on the same day. The same data corresponding to the OG1RF strain are therefore shown for the 2 experiments.
The production of EPA has been associated with an increased resistance to phagocytosis, which represents a key step during pathogenesis [8, 27]. We therefore quantified phagocytosis in zebrafish larvae infected with the wild-type OG1RF and one representative epa mutant (OG1RF_Δ11714, epaX-like) expressing the red fluorescent protein mCherry (Fig 7). Confocal microscopy images were used to measure bacterial uptake by phagocytes labelled with anti L-plastin antibodies coupled to Alexa-488, a green fluorophore, as previously described [28]. The ratio between red fluorescence inside to red fluorescence outside phagocytes was significantly higher for the epaX-like mutant (OG1RF_Δ11714) than for the wild-type strain (***P=0.0006) or the complemented strain (OG1RF Δ11714 + pTetH-OG1RF_11714; **P = 0.0049). As expected, no difference in phagocytosis was observed between the wild-type and complemented strains (ns, P>0.05) (Fig 7A). Representative pictures shown in Fig 7B-D clearly indicate that unlike the wild-type strain, the epaX-like mutant was no longer able to evade phagocytes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A. Quantification of E. faecalis uptake by zebrafish phagocytes. Embryos were infected with 1,600 CFUs of E. faecalis cells constitutively producing mCherry and fixed in 4 % paraformaldehyde 1.5 h post infection. Phagocytes were immunolabelled using rabbit anti L-plastin antibodies and detected with goat anti-rabbit antibodies conjugated to Alexafluor 488. The infected and immunolabelled embryos were imaged using scanning confocal microscope. The ratio of mCherry fluorescence signal area associated with phagocytosed and free bacteria was measured using the Fish Analysis Fiji plugin. The uptake of mutant OG1RF Δ11714 (ΔepaX) was significantly higher when compared to the wild-type (OG1RF; ***P = 0.0006) and complemented strain (OG1RF Δ11714 + pTetH-OG1RF_11714; **P = 0.0049). No difference in phagocytosis was observed between the wild-type and complemented strains (ns, P>0.05). Representative images showing E. faecalis uptake in zebrafish embryos are shown. Each picture corresponds to the quantification result indicated with a red dot in A, following infection with OG1RF (B), OG1RF Δ11714 (ΔepaX) (C) and the complemented strain (D). Phagocytes labeled with L-plastin appear in green, mCherry labelled bacteria in red. Scale bar is 25 μm.
Altogether, our data therefore indicate that decoration of the EPA polysaccharide is essential for E. faecalis pathogenesis.
Discussion
Previous studies revealed that E. faecalis resistance to lysozyme is unusually complex and results from several mechanisms acting synergistically. These include peptidoglycan O-acetylation and de-N-acetylation, D-alanylation of teichoic acids and transcriptional control by the extracytoplasmic sigma factor SigV. Despite a 100-fold decrease as compared to the wild-type strain, the residual resistance of the quadruple OPDV mutant is still relatively high (MIC=0.5 mg ml−1) as compared to other Firmicutes. This result contrasts with other bacteria in which a limited number of genes play a key role in resistance. For example, the combined deletions of oatA and dltA in S. aureus led to a decrease of at least 2,000-fold in resistance as compared to parental strain [13]. Deletion of pgdA alone in L. monocytogenes is associated with a 32-fold decrease resistance.
Using random transposon mutagenesis, we showed that several genes located downstream the conserved epaA-epaR region modulate susceptibility to lysozyme. It was proposed that epaA-epaR encode a core synthetic machinery whilst downstream genes contribute to the decoration of EPA rhamnopolysaccharide [22, 24]. In agreement with previous studies, three distinct polysaccharide bands (named PS1, PS2 and PS3) were detected on polyacrylamide gels ([8] and Fig 3A). The two upper bands simultaneously disappeared in all the transposon mutants, suggesting that both are structurally related to EPA. The nature of the third band is unknown and could be either a metabolic intermediate of EPA or an unrelated polymer. The lack of detection of PS1 and PS2 in EPA polysaccharides from mutants suggested that either their charge did not allow them to enter the gel and/or that they were no longer stained by the cationic dye alcian blue. A similar result was previously described for mutants harbouring deletions in OG1RF_11715 (epaOX) [23] and the homolog of OG1RF_11714 in V583 (epaX) [24]. By comparing the electrophoretic mobility of the transposon mutants to that of the parental OPDV strain, we confirmed that the epa genes downstream of the epaA-epaR locus contribute to the negative charge of the EPA polysaccharide. This negative charge could at least in part be due to the presence of phosphate in the polymer. Interestingly, the OG1RF and OPDV strains displayed similar electrophoretic mobilities. Previous studies also showed that alanylation of teichoic and/or lipoteichoic acids in L. lactis had no detectable impact on the electrophoretic mobility of this organism [29]. The dlt operon has been shown to modify lipoteichoic acids [17]. Since these polymers are embedded inside the cell wall, it is likely that their modification does not lead to a change in the bacterial surface charge. Further experiments are required to test whether alanylation of cell wall polymers only has a moderate impact on the charge of the cell wall or if such modifications can simply not be detected by measuring electrophoretic mobility. Three of the mutants identified in this work carry a transposon in genes encoding putative glycosyl transferases. Despite the low amino acid identity between the glycosyl transferase sequences (19-27% depending on the comparison), these proteins have very similar predicted secondary structures, with two transmembrane domains and both the N- and C-termini exposed at the cell surface. Tertiary structure predictions suggest that all 3 proteins have a very similar fold and are GalNAc transferases. These predictions are in agreement with our NMR and sugar analyses indicating that EPA polysaccharides from all glycosyl transferase mutants (OPDV_11720, OPDV_11715 and OPDV_11714) contain less GalNAc and less intense N-acetyl proton signals. In addition to a reduced amount of GalNAc, EPA polymers from the glycosyl transferase mutants also contained a reduced amount of galactose. Further analyses are required to explore the catalytic activity of these glycosyl hydrolases. It remains unclear whether they can use distinct sugars as a substrate or if the addition of GalNAc is required for the activity of other glycosyl transferases adding Gal residues. Since none of the heterologous complementations of the transposon mutants were able to restore the parental phenotype, we anticipate that the 3 glycosyl transferases identified play distinct roles. This idea is supported by several independent observations: (i) mutants OPDV_11720::Tn2.5, OPDV_11715::Tn2.13 and OPDV_11714::Tn2.14 present distinct alterations in their EPA carbohydrate compositions and the deletion mutants present differences in their antimicrobial susceptibility profiles; (ii) 2D-NMR spectra indicate that each mutation is associated with distinct modifications of the signals in the anomeric region; (iii) the OPDV_11714::Tn2.14 (epaX-like) mutant behaved differently from the other epa mutants studied since it displayed a less pronounced defect in surface charge. Altogether, our results suggest that glycosyltransferases in the epa variable region fulfil distinct roles. The complexity of EPA structure precludes any conclusion about the specific role of individual epa genes. However, based on the 2D NMR spectra, it is tempting to assume that OG1RF_11720, the first gene of the epa variable region encodes a glycosyl transferase that is adding the entire EPA decorations, whilst EpaOX and EpaX contribute to the transfer of smaller decorations.
epa deletion mutants displayed distinct resistance to antimicrobials targeting the cell envelope. Deletion of OG1RF_11720, which had the most pronounced impact on EPA structure led to the most pronounced increased susceptibility to antimicrobials. Deletion of OG1RF_11714 (epaX), which had the least pronounced impact on EPA structure only led to an increased susceptibility to SDS. Since epa mutations could confer resistance to both negatively charged antimicrobials and CAMPs, these results suggested that the charge of EPA does not entirely account for the resistance to these compounds. We therefore speculate that EPA decorations are critical to maintain cell integrity, as previously suggested [23, 24]. A contribution of epa decoration genes in biofilm formation [30], resistance to antimicrobials [23] and colonisation [24] was previously suggested, but no information was available on a potential role in the context of pathogeny. E. faecalis pathogenesis in the zebrafish model of infection involves two critical steps: phagocyte evasion and tissue damage caused by the metalloprotease GelE [27]. Since oatA, pgdA, dltA and sigV are unlikely to contribute to these processes, it was expected that their simultaneous deletion would have a very limited impact on virulence. By contrast, EPA has been reported to mediate resistance to phagocytic killing [8] and plays a critical role in virulence both in experimental mouse and zebrafish infections [27, 31]. We therefore hypothesized that epa mutations altering the decoration of EPA would impair virulence. In agreement with this hypothesis, all epa transposon mutants (in the OPDV background) and in-frame deletions in the wild-type OG1RF background were avirulent in the zebrafish model of infection. Further investigations revealed that the epaX mutation leads to a significant increase in E. faecalis uptake by phagocytes, suggesting that the decoration of EPA mediates immune evasion and underpins virulence.
Collectively, the results provide a paradigm shift in our understanding of E. faecalis pathogenesis, revealing that the modifications of EPA, rather than EPA backbone itself, underpin phagocyte evasion, an essential step during host infection. Whether epa is directly recognized by the host immune system or is shielding other surface components remains an open question.
Materials and methods
Ethics statement
Animal work was carried out according to guidelines and legislation set out in UK law in the Animals (Scientific Procedures) Act 1986 under Project License P1A417A5E. Ethical approval was granted by the University of Sheffield Local Ethical Review Panel.
Bacterial strains, plasmids and growth conditions
Bacterial strains, plasmids and oligonucleotides used in this study are described in S3 Table. All strains were routinely grown at 37°C in Brain Heart Infusion (BHI) broth or BHI-agar 1.5 % (w/v) plates unless otherwise stated. For E. coli, erythromycin was added at a final concentration of 200 µg ml−1 to select pTetH derivatives. When necessary, E. faecalis was grown in the presence of 10 µg ml−1 chloramphenicol, 128 µg ml−1 gentamicin or 30 µg ml−1 erythromycin. For complementation experiments, anhydrotetracycline was used at a final concentration of 10 ng ml−1 to induce gene expression.
Construction of the OPDV strain
The work describing the contribution of oatA, pgdA, dltA and sigV to lysozyme resistance was carried out using E. faecalis JH2-2 as a genetic background [16, 19]. Since this laboratory strain is avirulent in the zebrafish model of infection, we decided to use OG1RF as a parental strain [32]. The quadruple OG1RF mutant harbouring deletions in oatA, pgdA, dltA and sigV was built using existing plasmids (S3 Table) to create in-frame deletions in the following order: oatA, pgdA, dltA and sigV.
Transposon mutagenesis
A Mariner-based transposon mutagenesis system previously described was used [21]. Plasmid pZXL5 was introduced in E. faecalis OPDV by electroporation and transformants were selected at 28°C on plates containing chloramphenicol and gentamicin. Cells harbouring pZXL5 were grown to mid-exponential phase at 28°C and transposition was induced by addition of nisin (25 ng ml−1). The culture was then transferred to 42°C overnight to counter-select the replication of the plasmid. The library was then amplified by growing the cells at 42°C in the presence of gentamicin.
Transposon mutagenesis
A Mariner-based transposon mutagenesis system previously described was used [21]. Plasmid pZXL5 was introduced in E. faecalis OPDV by electroporation and transformants were selected at 28°C on plates containing chloramphenicol and gentamicin. Cells harbouring pZXL5 were grown to mid-exponential phase at 28°C and transposition was induced by addition of nisin (25 ng ml−1). The culture was then transferred to 42°C overnight to counter-select the replication of the plasmid. The library was then amplified by growing the cells at 42°C in the presence of gentamicin.
Isolation of transposon mutants resistant to lysozyme
Serial dilutions of the transposon library were plated on BHI agar plates containing 1, 2 or 4 times the lysozyme MIC for the OPDV strain (0.5 mg ml−1) and gentamicin. After 24 to 48 h at 42°C, individual colonies growing at the highest concentration (2 mg ml−1) were chosen for further characterisation.
Mapping transposition sites
Transposon insertion sites were mapped by reverse PCR using two divergent primers (Mar_up and Mar_dn) on the transposon (S9A Fig). Chromosomal DNA was extracted using the Promega Wizard kit and digested by SspI in a final volume of 30 µl at a concentration of 4 ng µl−1 (S9B Fig). Digestion products were further diluted to 1 ng µl−1 and self-ligated at 16°C for 16 h after addition of 100 U of T4 DNA ligase (NEB) (S9C Fig). Three microliters of the ligation product were used as a template for PCR amplification using oligonucleotides Mar_up and Mar_dn (S9D Fig). PCR products were gel extracted and sequenced using oligonucleotide T7 (S9E Fig). The insertion site was defined as the first nucleotide of the E. faecalis OG1RF genome immediately downstream of the inverted repeat sequence flanking the transposon.
Construction of complementation plasmids
DNA fragments encoding OG1RF_11720, OG1RF_11715, OG1RF_11714 and OG1RF_11707 were amplified by PCR using the oligonucleotides described in S3 Table. PCR products were digested by NcoI and BamHI and cloned into pTetH, a pAT18 derivative allowing anhydrotetracycline-inducible expression (S. Mesnage, unpublished). Each open reading frame was fused to a C-terminal 6-Histidine tag.
Antimicrobial assays
Colonies from a BHI agar plate were resuspended in PBS and diluted to an OD of 1 at 600 nm. Ten-fold dilutions were prepared in PBS and 1.5 µl of each cell suspension were spotted on BHI agar containing antimicrobials at various concentrations. MICs of lysozyme were defined as the concentration of antimicrobial inhibiting the growth of 1.5 µl of a cell suspension corresponding to a 1000-fold dilution of the cell suspension at OD of 1 at 600 nm. For complementation experiments, anhydrotetracycline was added at a final concentration of 10 ng ml−1. At least two biological replicates were carried out for each susceptibility assay.
Measurement of electrophoretic mobility
An overnight culture was diluted 1000-fold in 25 ml of BHI broth and grown for 17 h at 37°C in static conditions. Anhydrotetracycline (100 ng ml−1) was added to all cultures to induce gene expression for complementation and exclude the possibility that this chemical could account for differences between strains. Cells were harvested by centrifugation (5 min, 8,000 x g at room temperature), washed twice in 25 ml of 1.5 mM NaCl and resuspended at a concentration of 3×107 CFU ml−1 in 1.5 mM NaCl at various pHs. The electrophoretic mobility was measured in an electric field of 8 V cm−1 using a laser zetaphoremeter (CAD Instrumentation, Les Essarts le Roy, France). For each measurement, results were based on the analysis of 200 individual particles. The results presented in Fig 3, S1 and S2 Tables are the combined results of three independent experiments (biological replicates).
NMR
NMR experiments were conducted on a Bruker DRX-600 (plus cryoprobe) spectrometer at 25°C. EPA polysaccharides were freeze-dried and resuspended in D2O. Spectra were processed and analysed using TOPSPIN (version 2.1). Trimethylsilylpropanoic acid was used as a reference.
Carbohydrate extraction and analyses
EPA was extracted as previously described from standing cultures in BHI at the end of exponential growth (OD600nm=0.8) [25]. The method previously described to analyse pneumococcal polysaccharides and conjugates was followed, with the exception of the first acid hydrolysis step [33] Briefly, purified EPA polysaccharides were hydrolyzed in 4 N trifluoroacetic acid for 4 h at 100°C. Hydrolysis products were analysed by high-performance anion-exchange chromatography (HPAEC) coupled to pulsed-amperometric detection (PAD) using a Dionex DX 500 BioLC system (ThermoFisher). Monosaccharides were separated on a Carbopac PA10 (4 mm × 250 mm) analytical column (Thermofisher Scientific) at a flow rate of 1 ml min−1. Solvent A was 18 mM NaOH, solvent B was 100 mM NaOH, and solvent C was 100 mM NaOH containing 1 M sodium acetate. NaOH and NaAc gradients were used simultaneously to elute the carbohydrates by mixing the three eluents. The gradients used were as follows: after 15 min of isocratic elution in buffer A, a 3 min gradient to 100 % of buffer B was applied. A second gradient was applied between 18 and 35 min using buffer C to reach 300 mM sodium acetate. The column was re-equilibrated in 18 mM NaOH for 20 min after every run. The following pulse potentials and durations were used: E1 = 0.1V, t1 = 400 ms; E2 = −2V, t2 = 20 ms; E3 = 0.6V, t3 = 10 ms; E4 = −0.1V, t4 = 70 ms. Data were collected and analysed on computers equipped with the Dionex PeakNet software. Carbohydrate analyses were made in triplicate using three independent EPA extractions from 3 distinct colonies.
Construction of pGhost derivatives for allele replacement
All plasmids for allele replacement were constructed with the same strategy. Two homology regions were amplified: the 5’ homology region (referred to as H1) was amplified with oligonucleotides H11 (sense) and H12 (antisense). The 3’ homology region (referred to as H2) was amplified with oligonucleotides H21 (sense) and H22 (antisense). Both PCR products were purified, mixed in an equimolar ratio and fused by overlap extension using oligonucleotides H11 and H22 [34]. The assembled PCR fragment flanked by two restriction sites was digested and cloned into pGhost9 [35] similarly digested. Oligonucleotide sequences and restriction sites used for cloning are described in S3 Table.
Construction of E. faecalis OG1RF in-frame epa mutants
Isogenic derivatives of E. faecalis OG1RF were constructed by allele exchange using the procedure previously described [36]. Briefly, pGhost9 derivatives were electroporated into OG1RF and transformants were selected at a permissive temperature (28°C) on BHI plates with erythromycin. To induce single crossover recombination, transformants were grown at a non-permissive temperature (42°C) in the presence of erythromycin. The second recombination event leading to plasmid excision was obtained after 5 serial subcultures at 28°C without erythromycin. The last overnight subculture was plated at 42°C without erythromycin. A clone harboring a double crossover mutation was identified by PCR (S5 Fig) and further confirmed by sequencing of the recombined region.
Zebrafish strains and maintenance
London wild type (LWT) zebrafish were provided by the aquarium facility at the University of Sheffield. Embryos were maintained in E3 medium at 28°C according to standard procedures previously described [37].
Microinjections of E. faecalis in zebrafish embryos
Cells were grown to mid-exponential phase (OD600nm∼0.3) and harvested by centrifugation (5,000 × g for 10 min at room temperature). Bacteria were resuspended in filtered phosphate buffer saline (150 mM Na2HPO4, 20 mM KH2PO4, 150 mM NaCl [pH 7.5], PBS) and transferred to microcapillary pipettes. Embryos at 30 h post fertilization (hpf) were anaesthetized, dechorionated, embedded in 3 % (w/v) methylcellulose and injected individually with 2 nl of a cell suspension corresponding to ca. 1,000 cells as previously described [27]. The number of cells injected was checked before and after each series of injections with a given strain. Zebrafish embryos were monitored at regular intervals until 90 h post infection (hpi). At least 20 embryos per group were used.
Imaging of infected larvae by confocal microscopy and quantification of uptake by phagocytes
Immuno-labelled embryos were immersed in 0.8 % (w/v) low melting point agarose in E3 medium and mounted flat on FluoroDish™ (World Precision Instruments Inc.). Images were collected using a DMi8 confocal microscope (Leica). Image acquisition was performed with the Volocity software and the images were processed with ImageJ 1.49v software. Bacterial phagocytosis was quantified using an ImageJ custom script called Fish Analysis, which can be obtained from http://sites.imagej.net/Willemsejj/ or via ImageJ updater. All bacteria were identified based on their fluorescence (mCherry, Channel 2). Subsequently, the fluorescence intensity of the phagocytes (Alexa 488, Channel 1) surrounding the phagocytosed bacteria was measured. The phagocytosed bacteria had high fluorescence intensity of Channel 2 and low fluorescence intensity of Channel 1. The area of phagocytosed bacteria was compared with the area of non-phagocytosed bacteria and their ratio was calculated.
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 7.03. Comparisons between survival curves were made using the log rank (Mantel-Cox) test. Electrophoretic mobilities were compared using two-way ANOVA Comparison of uptake by zebrafish macrophages was carried out using an unpaired non-parametric Dunn’s multiple comparison test.
Acknowledgments
The authors would like to thank Abdellah Benachour (University of Caen) for providing the plasmids used to construct the OPDV strain and the Bateson Centre aquaria staff for their assistance with zebrafish husbandry. The authors thank Dr Philip Elks for access to the confocal microscope and Paul Martin (University of Bristol) for the kind gift of anti L-plastin antibodies.
References




