

ABSTRACT
Genotype-phenotype relationships are at the heart of biology and medicine. Numerous advances in genotyping and phenotyping have accelerated the pace of disease gene and drug discovery. Though now that there are so many genes and drugs to study, it makes prioritizing them difficult. Also, disease model assays are getting more complex and this is reflected in the growing complexity of research papers and the cost of drug development. Herein we propose a way out of this arms race. We argue for synthetic interaction testing in mammalian cells using cell growth – changes in cell number that could be due to a number of factors – as a readout to judge the potential of a genetic or environmental variable of interest (e.g., a gene or drug). That is, if a gene or drug of interest is combined with a known perturbation and causes a strong cell growth phenotype relative to that caused by the known perturbation alone, this justifies proceeding with the gene/drug in more complex models like mouse models where the known perturbation is already validated. This recommendation is backed by the following: 1) Genes required for cell growth involve nearly all classifications of cellular and molecular processes; 2) Nearly all genes important in cancer – a disease defined by altered cell number – are also important in other complex diseases; 3) Patient condition and patient cell growth responses to many drugs are comparable. Taken together, these findings suggest cell growth could be broadly applicable phenotype for understanding gene and drug function. Measuring cell growth is robust, and requires little time and money. These are features that have long been capitalized on by pioneers using model organisms that we hope more mammalian biologists will recognize.
INTRODUCTION
Biological model systems have made numerous contributions to human health, which is notable because often they were used without regard to their eventual applications. For example, many transformative medicines and field-changing paradigms exist because of discoveries in organisms such as yeast and worms (1-4). Still, many argue that disease models even mice aren’t good models of disease. Some researchers are fortunately ignoring these debates and focusing on creating new ways to study human disease using model organisms (5-7). Yet, many important human genes aren’t conserved in model organisms. Moreover, even if one uses a mammalian system, it is assumed that the context being studied, e.g., tissue type, matters. There is therefore a need for models that strike a balance between the ease and robustness of model organisms while preserving the signaling networks important to human disease.
Modeling disease has gained new relevance as the number of human population genetic studies have exploded (8). Thousands of genes are being implicated in human diseases across categories from cancer to autism, Alzheimer’s, and diabetes. Among the surprises from these data are findings such as “cancer” genes being linked to diseases seemingly unrelated to cancer. For example, one of the earliest genes found to mutated in the behavioral condition, autism (9), is the well-known tumor suppressor gene, PTEN. On first pass, it might be difficult to glean what autism might have to do with the uncontrolled cell growth that PTEN inactivation causes. However, it is clearer when one considers that patients with autism often have larger brains and more cortical cells (10). Nevertheless, the extent to which the various functions of genes like PTEN are relevant to disparate diseases begs the question of how we molecularly classify and model disease.
As with studying disease genes, new treatments have often suffered from biases that prevented us from seeing their value outside of the context in which they are originally characterized. For example, going back in history, one of the largest miscalculations by the pharmaceutical industry was the cholesterol-lowering statins being held back from the clinic because they were shown to be toxic to cultured mammalian cells. Those early findings scared off drugs companies because they suggested statins might not be safe for humans (11). Merck understood these results better than their competitors and the statins went on to be one of the biggest success stories in drug development history. The successes of drug repositioning also tell us that drugs can function in multiple “unrelated” contexts. For example, malarial drugs, such as hydroxychloroquine (Plaquenil), are now used to treat rheumatoid arthritis and autoimmunity (12). There is also chemotherapeutics being considered for depression (13). Drug repositioning makes sense from a molecular perspective because many drug side effects actually can be on-target effects (14). To take it further, now with so much knowledge in the “-omic” era, it might be the expectation rather than the exception that a mechanistic target for a drug has many functions in the body. This is true even for common drugs that preceded the –omic era. For example, the serotonin transporter, SLC6A4 (a.k.a. SERT), is the target of the widely prescribed antidepressants, Prozac and Zoloft. SLC6A4 is expressed at high levels in the intestines and lungs (15), and regulates gut motility and pulmonary blood flow (16, 17) in addition to mood. This demonstrates the need to find a way to better understanding the diverse roles of drugs and their targets.
Herein, we address these aforementioned issues. We propose that synthetic interaction testing in mammalian cells using cell growth as a readout is a relevant and scalable approach to understand surprisingly diverse types of molecular functions, diseases, and treatments. This proposal requires a change in thinking because synthetic interaction testing using human cells has historically almost always been discussed in the context of cancer (18, 19). However, we argue it has a much more general purpose. We provide evidence that cell growth is a generalizable phenotype because it is an aggregation of phenotypes. To the extent that it might be an aggregation of all possible phenotypes – an omniphenotype – suggests its potential as a pan-disease model for biological discovery and drug development.
RESULTS
Nearly all cell processes affect cell growth
Synthetic interactions are often tested between two genes. It is said there is a genetic interaction between two genes if the combined effect of mutating both on cell growth, g, is much greater than the effects of the individual mutations (Fig. 1). If there is an interaction, it can be either be a positive synergy, where the double mutant cells grow much worse, or a negative synergy, where the cells grow much better, respectively, than the individual mutant cells (Fig. 1).
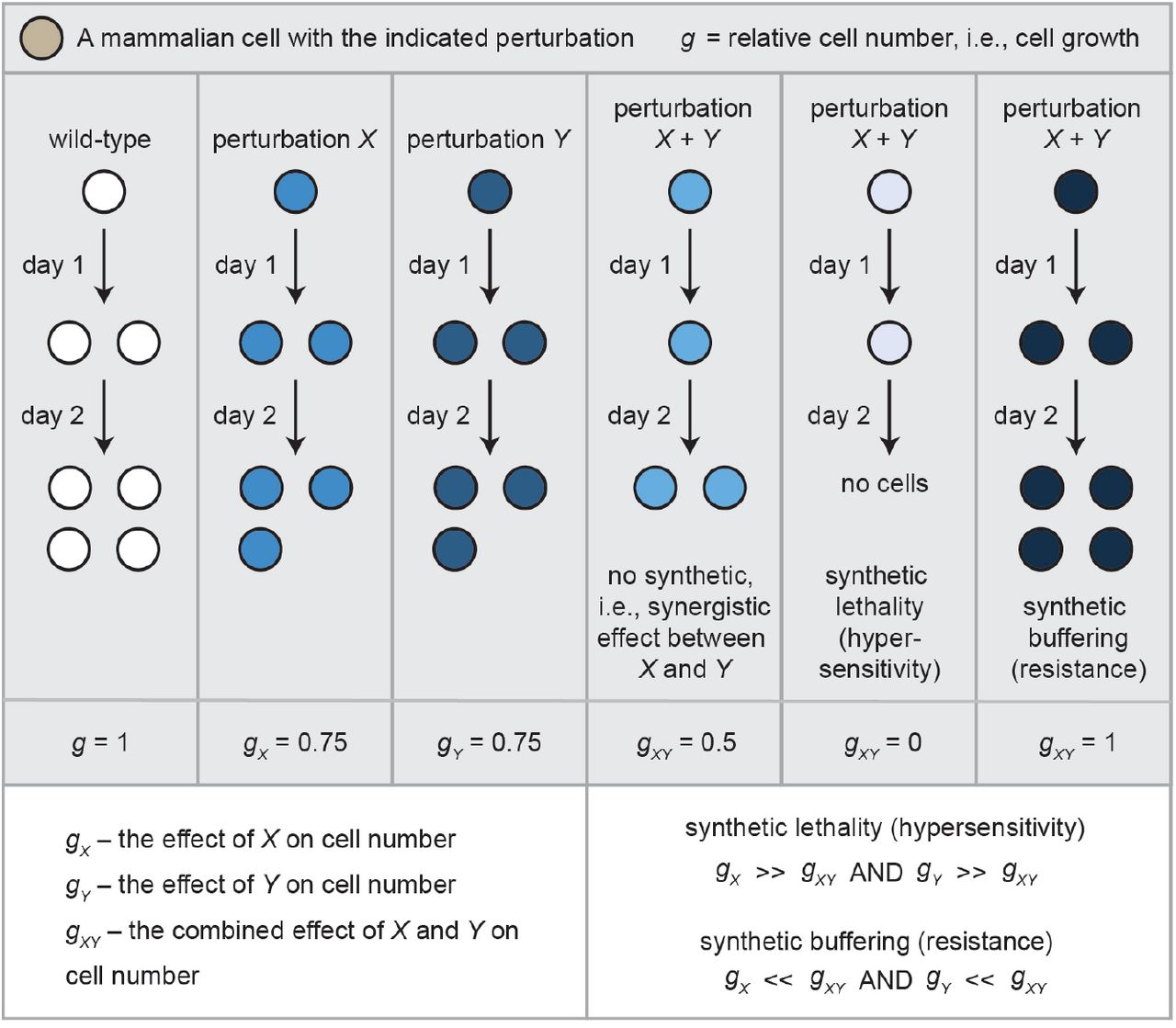
A synthetic interaction between two perturbations, X and Y, means the effect of X combined with Y on cell growth is significantly greater than the effect of either alone. Synthetic interactions can cause lethality, a.k.a., hypersensitivity, or can be buffering, a.k.a., confer resistance. Both are depicted mathematically in the figure (where g is relative cell number).
Amongst the ~4 billion possible synthetic interactions between protein coding human genes, less than 0.1% have been mapped to date (20). It is challenging to scale synthetic interaction testing in human cells both because of the size of the experiments needed and also because often neither interactor has a known function in the ultimate biological context of interest (21). Therefore, we sought to develop methods that would allow us to infer synthetic interactions at scale as well as leverage interactors of known importance to explain interactors of unknown significance. We heretofore refer to the known interactor in a synthetic interaction pair as, K, and the unknown as, U. With this convention, U is what’s new and interests the researcher. Several recent works identified essential genes – genes that are required for cell viability (22-24) – and we wondered what gene ontology (GO) pathways they participate in. We applied the principal of the minimal cut set (MCS), which is an engineering term used by biologists to mean the minimal number of deficiencies that would prevent a signaling network from functioning (7,25). MCS fits well with the concept of synthetic lethality (18), and this was recognized by Francisco Planes and colleagues to identify RRM1 as an essential gene in certain multiple myeloma cell lines (7).
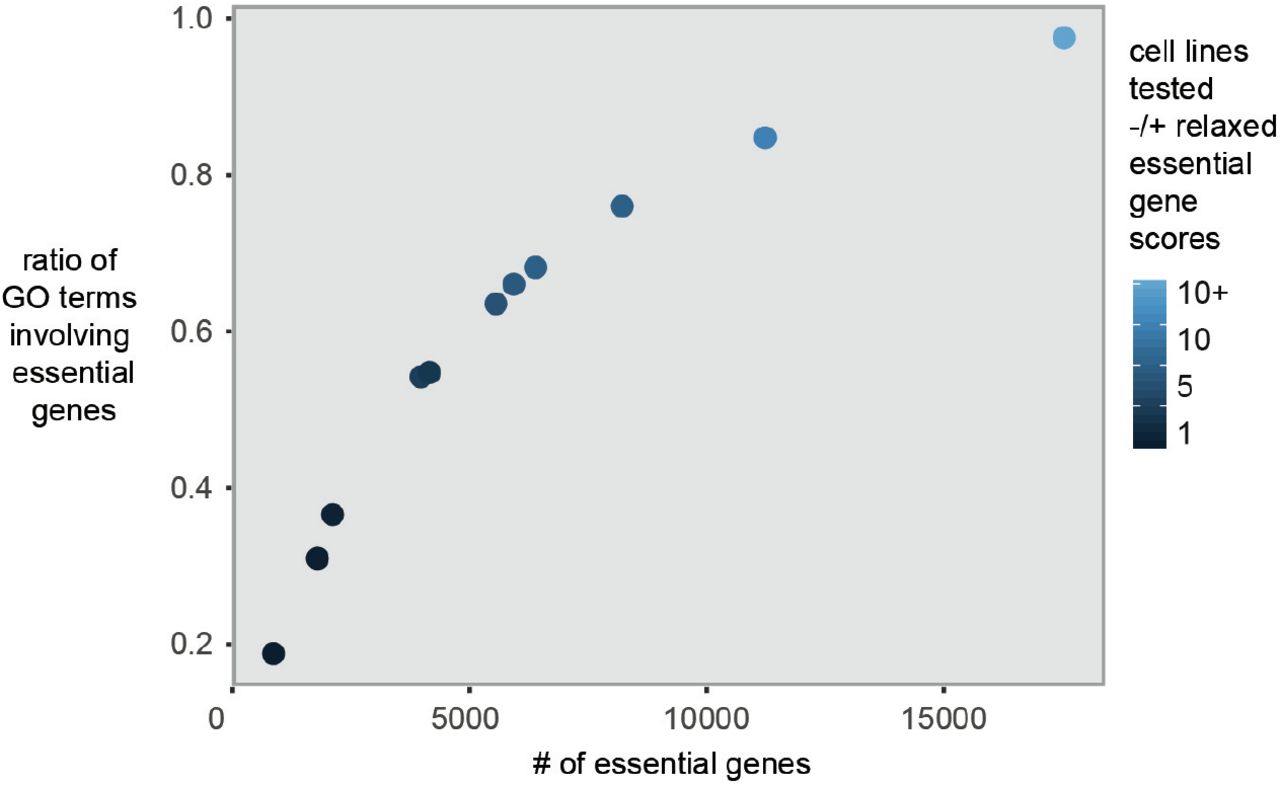
We generalized the idea of MCS to hypothesize that different cell types might be relatively defective in components of specific GO pathways such that if a gene in one of these pathways was knocked out, it would produce a synthetic lethal interaction with those components. In this case, the synthetic interaction testing is as follows: the known, K, is the gene that is essential in at least one cell type and U is the unknown, synthetically-interacting GO pathway of interest. Indeed, we found that essential genes are involved in diverse GO processes and the number of GO processes they participate in scales with the number of cell lines examined (Fig. 2, Table 1). For example, if one cell line is assessed, 2054 out of 21246 total genes were essential and these essential genes involve 36% (6444 out of 17793) of all possible GO categories. In examining five cell lines, 3916 essential genes were identified that involve 54% of all GO categories (9577/17793). With ten cell lines, 6332 essential genes were identified that involve 68% of all GO categories (12067/17793). Therefore, essential genes involve the majority of GO processes. Because the Fig. 2 graph appears asymptotic, this suggests if enough cell types are tested, potentially all GO processes would be essential. To extrapolate beyond the ten mammalian cell lines that have been assessed so far for gene essentiality, we relaxed the significance on the cell growth scores, e.g., from p < 0.05 to p < 0.10. This relaxed list of growth-regulating genes encompasses nearly all GO processes (Fig. 2, 97.2%, 17291/17793 GO terms; 17473/21246 genes). Taken together, this suggests the majority if not all GO processes are required for cell growth.

The y-axis is the ratio is the number of distinct GO terms for the essential genes in the tested cell lines divided by the total number of distinct GO terms for all tested human genes (21,246). The x-axis is the number of essential genes. + indicates relaxed, i.e., p < 0.10, cell growth scoring.
Nearly all common disease genes are cancer genes
The omnigenic model states that all genes are important to complex disease (26). Yet, there is a scale and some genes are more important than others. These relatively more important genes are referred to as “core” (a.k.a. “hub”) genes. We reasoned that the published literature with its millions of peer reviewed experiments would be a comprehensive and unbiased way to inform us on what are a “core” genes for a given complex disease. At the same time, we were interested in leveraging the literature to identify genes that would most strongly synthetically interact with these core genes.
Here we defined a synthetic interaction as follows: the gene(s) mentioned in a citation is the unknown, U. This is by convention because the gene(s) must not be known in whatever context was studied, e.g., diabetes, in order to warrant a publication. Whereas all other published genes in that context are the known, K. To measure synthetic interaction strength between U and K, we assessed their co-occurrence with the term, “cancer” as a proxy for cell growth because cancer is a complex disease defined by altered cell number. In analyzing all currently available PubMed citations (~27M), we identified a compelling relationship: most genes are cited with cancer to a similar extent as with non-cancer, complex diseases/conditions: infection, Alzheimer’s, cardiovascular disease, diabetes, obesity, depression, inflammation, osteoporosis, hypertension, stroke, and inflammation (Fig. 3). This suggests many genes synthetically interact with other genes to regulate cell growth. That some genes are cited much more than others across disease categories is consistent with the multiple functions of core/hub genes.

The co-occurrences of each human gene with cancer (x-axis) and non-cancer (y-axis) conditions in all ~27 million PubMed citations were counted. The following non-cancer conditions were considered: infection, Alzheimer’s, cardiovascular disease, diabetes, obesity, depression, inflammation, osteoporosis, hypertension, stroke, and inflammation. Line of best fit – Slope, 0.75; Correlation coefficient, R2 = 0.69.
Drug effects on patient cell growth are reflective of the patient condition response to drug
Like with mutations in genes, drugs can have different effects in different genetic backgrounds. We analyzed the published literature to identify reports where the growth of patient-derived cells in response to a drug was predictive of its effect on the same patient’s condition. In this case, U is the genetic background of the patient cells and K is the drug. What’s notable from the studies we identified is that cell growth is predictive of drug response for widely divergent medications (Fig. 4). These results might be surprising outside the context of cancer therapy, but they should not be because the growth inhibition (inhibitory concentration - IC50) for these drugs varies widely in different cell lines, which have different genetic/epigenetic backgrounds (Fig. 4). Note that this latter result is similar to that presented in Figure 2, though using chemical genetics rather than genetics.

Listed drugs met two criteria: 1) Their effect on a patient’s condition and the same patient’s cell growth were significantly correlated; 2) The drug had differing IC50 values – the concentration at which cell numbers are 50% of their untreated values – in multiple, distinct human cell types or cell lines.
DISCUSSION
We propose cell growth is a simple, yet comprehensive approach to rank less-studied genes, cell processes, diseases, and therapies in importance relative to better-studied ones (Fig. 5). Rather than needing ever more refined experiments, this work suggests the antithetical approach. That is, one can use cell growth to estimate the importance of a new factor in a biological context of interest by comparing the new factor’s individual effect on cell growth vs. its combined effect with a factor with a known role in the biological context (Fig. 5). We recently published a proof-of-concept of this idea – which can be thought of as an expanded definition of synthetic interaction testing – with the osteoporosis drugs, nitrogen-containing bisphosphonates (N-BPs) (20, 27, 28). In this work, we identified a gene network, ATRAID-SLC37A3-FDPS, using cell-growth-based, drug interaction screening with the N-BPs, and subsequently demonstrated this network controls N-BP responses in cells, in mice, and in humans (20, 27, 28). Besides this and the statins, there are numerous other examples in the literature such as with the mTOR pathway (29) where the steps from cell growth to people can be readily traced.
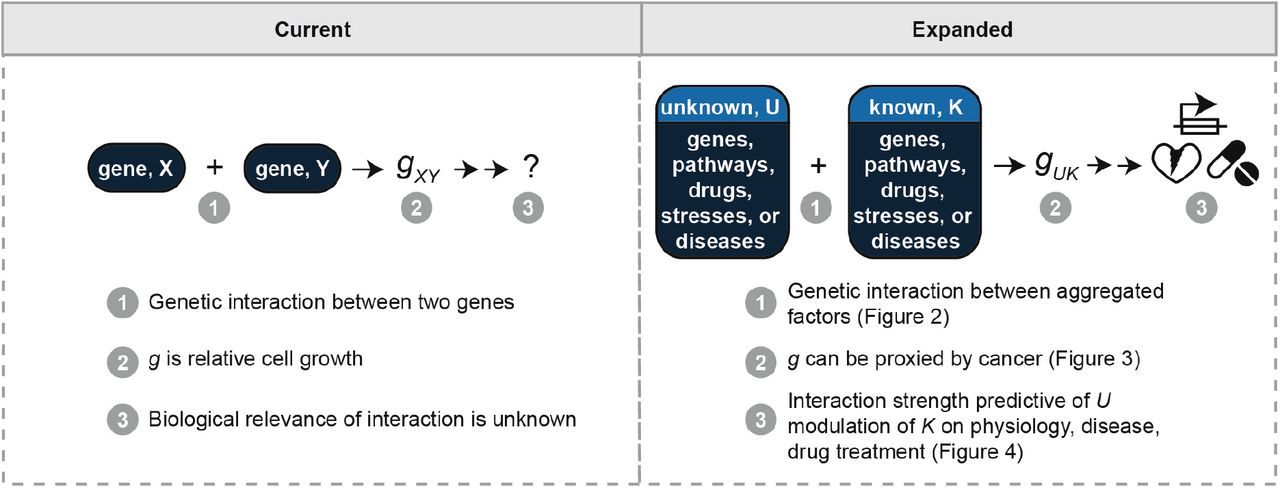
A currently, commonly used framework for synthetic interaction testing is as follows: 1) Genetic interactions are tested between two genes, e.g., X and Y; 2) g, the phenotype being measured, is relative cell growth; 3) It isn’t known how the interaction strength between X and Y might be relevant to physiology or disease. The expanded framework described herein is as follows: 1) Genetic interactions can be tested between aggregated factors such as multiple genes and/or pathways (related to Figure 2); 2) g can be proxied by the relative involvement of the interactors in cancer (related to Figure 3); 3) One interactor, K, has a known effect on a physiology or disease of interest. The effect of the other, U, in that context can therefore be measured relative to K (related to Figure 4).
Below we address the relevance of cell growth as a phenotype to the different levels at which biomedical scientists tackle their work: at the level of cell and molecular processes, at the level of disease, and at the level of environmental factors such as drugs.
Relevance to cell and molecular processes (Fig. 2)
Our Figure 2 results suggest one can study the majority of cell and molecular processes using cell growth-based, synthetic interaction testing. This is useful for molecular biologists because we often spend considerable time developing experimental models. For example, large efforts are often spent upfront (before getting to the question of interest) developing cell types to model various tissues. This is done because of the often, unquestioned assumption that the cell type being studied matters. While this can be true, it is worth considering whether it’s the rule or the exception. In support of the latter, in single cell RNAseq analysis, 60% of all genes are expressed in single cells and that the percentage jumps to 90% when considering 50 cells of the same type (30). This argues that the majority of the time, there should be a generic cell context to do at least initial “litmus” synthetic interaction testing of a hypothesis about a new genetic or environmental factor of interest before proceeding with a more complex set-up. Like with cellular phenotypes, many molecular phenotypes such as reporter assays require domain expertise, can be hard to elicit, and might not be easily reproducible. Whereas cell growth is robust and readily approximated by inexpensive assays such as those to measure ATP levels.
Relevance to diseases (Fig. 3)
Our PubMed analysis lends growing support to the use of the published literature as a data source (31). The costs to perform the wet-lab experiments required to publish in the “-omic” era aren’t small and favor well-resourced teams. Gleaning insights from the existing literature, which is large and growing rapidly, but at the same time knowable especially when approached computationally, is an underappreciated way to level the playing field.
One counterintuitive insight from Figure 3 is that core genes on the diagonal might not be as good drug targets as genes off the diagonal because the core genes are involved in more diseases. This might mean that because of their importance, hub genes might be too networked with other genes to be targeted by therapies without the therapies causing off-target effects. Fortunately, this might then also mean those off the diagonal might make more appealing drug targets because they are less networked. Thus, in this case we used cell growth as a filter to prioritize genes for further study. This is important because certain genes get cited over and over (21), and approaches to aggregate data are known to favor the “discovery” of already well-studied genes over less studied genes (like with income inequality where the “rich get richer”) (32). When applied in unbiased ways such as with genome-wide screening (20), we expect our approach to be particularly impactful in addressing this issue of identifying important genes that historically are ignored.
Relevance to environmental factors, e.g., drugs (Fig. 4)
With the exception of oncology, it could be argued pharmacogenomics is only slowly going mainstream in many fields. One reason for this is the poor feasibility and high cost-to-benefit ratio of current genetic testing strategies to predict drug response (33). Though more work needs to be done to test the generalizability and “real-world” application of the studies we highlight in Figure 4, they do provide important initial evidence for using cell growth as a diagnostic assay for precision medicine. In addition to drug diagnostics, these studies have implications for drug discovery. Both target-based and phenotypic-based drug discovery have drawbacks (34). Target-based approaches don’t necessarily tell you about the phenotype and phenotypic screens don’t tell you about the target. On the other hand, cell growth-based, drug response genetic screening (35) can potentially be the best of both worlds because it can provide a phenotype and the gene target in the same screen.
Still, we acknowledge limitations in our findings. Our work focuses on genes and drugs as the genetic and environmental variables of interest. Future work must determine to what extent our work would apply to other variables, such as epigenetic signatures. Also, many genes aren’t published on due to human factors, which bias our PubMed-based analyses. As more methods get developed and more knowledge accumulates, a scientist’s job of figuring out where to focus their attention gets harder. Our view is that cell growth could be used more often to help scientists triage their opportunities.
METHODS
Gene ontology and essential gene analysis
Gene ontology (GO) information for each human gene was obtained from BioMart, https://useast.ensembl.org/info/data/index.html. Essential gene data was obtained from Blomen et al., Wang et al., and Hart et al. (22-24). There were ten cell lines tested: K562, KBM7, Jiyoye CS, Raji CS, HAP1, HCT116, DLD1, HeLa, GBM, RPE1. The code used to intersect the GO terms for each essential gene is available at: https://github.com/tim-peterson/omniphenotype.
For the relaxed filter data point, we used growth scores at p < 0.1 for both the Wang and Hart studies. For the Blomen study, p-values > 0.05 weren’t given, therefore we took all genes with growth scores less than 0.5 standard deviations above the mean.
PubMed analysis
In both Figure 3 and 4, we analyzed PubMed using the E-utilities (https://www.ncbi.nlm.nih.gov/books/NBK25497/) API endpoint: https://eutils.ncbi.nlm.nih.gov/entrez/eutils/esearch.fcgi. We either returned PubMed IDs (PMID), which were counted (Fig. 3), or abstracts, which were manually analyzed for relevant information (Fig. 4). The code for both analyses is available at https://github.com/tim-peterson/omniphenotype.
Cancer vs. other complex disease analysis
A list of all human genes was obtained from NCBI, ftp://ftp.ncbi.nih.gov/gene/DATA/GENE_INFO/Mammalia/. A list of top conditions was curated from several sources including the World Health Organization list of top ten causes of death: http://www.who.int/en/news-room/fact-sheets/detail/the-top-10-causes-of-death. The generic structure for the E-utilities queries was the combination of each human gene with each top condition: { gene } AND { condition }. The output of these queries was lists of PubMed IDs that were counted. Correlation co-efficient was calculated using R using the lm() function on log-log data. Code is available at: https://github.com/tim-peterson/omniphenotype.
Drug response in patients vs. patient cells analysis
To identify studies that reported findings on patient cell drug responses that correlated with the patient, the phrase “human lymphoblastoid cell lines proliferation” was queried and 804 abstracts were returned. “PMBC” as in peripheral mononuclear blood cells was substituted for lymphoblastoid cell lines (LCLs) to identify the simvastatin study. The LCL and PMBC citations were manually curated to identify relevant citations that mentioned drug responses on cell proliferation. The IC50 studies were obtained by querying { drug } AND “cell viability” OR “IC50”.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}