

Abstract
The induction of antifungal proinflammatory signaling during oropharyngeal candidiasis (OPC) is crucial to limit Candida albicans proliferation and induce fungal clearance. Previously, we determined that the ephrin type-A receptor 2 (EphA2) functions as a β-glucan receptor that senses oral epithelial cell fungal burden. EphA2 plays a central role in stimulating the epithelial cell release of proinflammatory mediators that mediate resistance to OPC. Another receptor for C. albicans is the epidermal growth factor receptor (EGFR), which interacts with candidal invasins such as Als3 interact and induces epithelial cells to endocytose the fungus. Here, we investigated the interactions between EGFR and EphA2. We found that EGFR and EphA2 constitutively associated with each other as part of a physical complex. Activation of EGFR by C. albicans Als3 was required for sustained EphA2 phosphorylation and for induction of CXCL8/IL-8 and CCL20 secretion by epithelial cells. Treatment of uninfected epithelial cells with IL-17A and TNFα also induced EGFR phosphorylation, which was necessary for epithelial cells to respond to these cytokines. In mice with OPC, pharmacological inhibition of EGFR during caused a modest reduction in oral fungal burden, markedly impaired production of proinflammatory cytokines and significantly decreased accumulation of neutrophils and inflammatory monocytes. Thus, while C. albicans activation of EGFR mediates fungal invasion of the epithelium, it also sustains EphA2 signaling, inducing the epithelial cell proinflammatory response to the fungus.
Importance Host cell receptors for fungi have typically been evaluated from one of two different perspectives, their role in inducing the invasion of the organism or their role in stimulating the host inflammatory response. We had found previously that EGFR mediates the endocytosis of C. albicans by oral epithelial cells, and that epithelial cell EphA2 mediates the production of chemokines and proinflammatory cytokines in response to C. albicans. Here, we demonstrate that EGFR and EphA2 interact with each other, both physically and functionally. EGFR is required for sustained EphA2 activation and for epithelial cells to secrete proinflammatory mediators in response to both C. albicans and IL-17A. Therefore, while activation of EGFR by C. albicans enhances pathogenicity by inducing the endocytosis of the fungus, it also augments the host defense against OPC by stimulating the host inflammatory response.
Introduction
The human oral cavity hosts a multifaceted microbiome comprised of an estimated 600 bacterial and 100 fungal species (1). Among these fungal species is Candida albicans, which grows as a harmless commensal in at least 50% of healthy adults. When there is an imbalance of local or systemic immune homeostasis, C. albicans can proliferate, causing oropharyngeal candidiasis (OPC) (2). While this disease is relatively uncommon in healthy adults, it causes significant morbidity in a large, diverse population of patients, including those with HIV/AIDS, xerostomia, corticosteroid use, diabetes mellitus, and cancer of the head and neck. Each year, there are nearly 10 million cases of OPC in patients with HIV/AIDS world-wide, and nearly one fifth of these cases have esophageal involvement (3).
At least 80% of cases of OPC are caused by C. albicans. When this organism overgrows in the oropharynx, there is invasion of the superficial epithelium, leading to host cell death (4). One mechanism by which C. albicans invades oral epithelial cells is receptor mediated endocytosis. In this process, invasins, such as Als3 and Ssa1 expressed on the surface of C. albicans hyphae interact with epithelial cell receptors such as E-cadherin and a heterodimer composed of the epidermal growth factor (EGFR) and HER2. This interaction triggers rearrangement of the epithelial cell cytoskeleton, leading to the formation of pseudopods that engulf the organism and pull it into the host cell (4-7).
The epithelial cells that line the oropharynx sense the presence of C. albicans and orchestrate the host inflammatory response to fungal overgrowth. In addition to producing host defense peptides that have direct antifungal activity, oral epithelial cells secrete alarmins, proinflammatory cytokines, and chemokines that recruit phagocytes to foci of infection and enhance their candidacidal activity to limit the growth of the invading fungi (8-10). This epithelial cell response to OPC is amplified by interleukin (IL)-17, which is secreted by γδ T cells, innate TCRαβ+ cells, and type-3 innate lymphoid cells (11-14). Recently we determined that the ephrin type-A receptor 2 (EphA2) is expressed on oral epithelial cells and senses exposed β-glucan on the fungal surface. When C. albicans proliferates on the epithelial cell surface, EphA2 is activated and oral epithelial cells secrete host defense peptides and proinflammatory mediators. In mice with OPC, EphA2 also induces the production of IL-17A, and EphA2−/− mice are highly susceptible to OPC (15).
Although EphA2 is required for the normal host defense against OPC, exposure of oral epithelial cells to purified β-glucan induces only transient EphA2 activation and is not sufficient to initiate a significant inflammatory response. By contrast, exposure to live C. albicans induces sustained EphA2 activation and a strong inflammatory response (15). In the current study, we sought to elucidate how C. albicans infection prolongs EphA2 activation and induces a proinflammatory response during OPC. We found that activation of host EGFR by C. albicans Als3 maintained EphA2 phosphorylation and was required for the fungus to stimulate oral epithelial cells to produce the chemokines, CXCL8/IL-8 and CCL20. Treatment of uninfected epithelial cells with IL-17A and TNFα induced transient phosphorylation of EGFR, which was necessary for epithelial cells to respond to IL-17A. In mice with OPC, pharmacological inhibition of EGFR caused a modest reduction in oral fungal burden and a markedly impaired inflammatory response. Thus, while C. albicans activation of EGFR mediates fungal invasion of the epithelium, it is also plays a central role in inducing the local inflammatory response to this fungus.
Results
The cellular fate of epithelial EphA2 differs depending on the type of stimulation
When oral epithelial cells are infected with yeast-phase C. albicans, the organisms germinate and begin to form hyphae within 60 min. Previously, we found that EphA2 is autophosphorylated on serine 897 within 15 min of infection, and it remains phosphorylated for at least 90 min. By contrast, when oral epithelial cells are exposed to β-glucan in the form of zymosan or laminarin, EphA2 is phosphorylated for only the first 30 min of contact; at later time points, EphA2 phosphorylation returns to basal levels, even though β-glucan is still present (15). To investigate how oral epithelial cells respond to prolonged EphA2 stimulation, we compared the response of these cells to the native EphA2 ligand, ephrin A1 (EFNA1) and to C. albicans. After 15 min of exposure, both stimuli induced phosphorylation of EphA2 (Fig. 1A; S1). However, after 60 min, the cells exposed to EFNA1 had minimal phosphorylation of EphA2, and total EphA2 levels had declined (Fig. 1B; S1). By contrast, the cells exposed to C. albicans had sustained EphA2 phosphorylation and no decrease in total EphA2 levels. Thus, exposure to C. albicans not only induces EphA2 phosphorylation, but prevents subsequent EphA2 dephosphorylation and degradation that normally occurs with prolonged exposure to its native ligand EFNA1 (16).

Interactions between EGFR and EphA2. (A and B) Immunoblot analysis of EphA2 phosphorylation and total protein levels in oral epithelial cells after stimulation for 15 min (A) and 60 min (B) with ephrin A1-Fc (EFNA1-Fc) or yeast-phase C. albicans SC5314 (Ca). (C and D) Effects of EGFR siRNA (C) and the EGFR kinase inhibitor gefitinib (GEF) (D) on the time course of EphA2 and EGFR phosphorylation in oral epithelial cells infected with C. albicans. Phosphorylation was analyzed at 30 and 90 min post-infection (p.i.). (E) Lysates of oral epithelial cells infected with C. albicans for 30 and 90 min were immunoprecipitated (IP) with antibodies against EphA2 (left) and EGFR (right), after which EphA2, and EGFR were detected by immunoblotting (Top). Immunoblots of lysate prior to immunoprecipitation, demonstrating equal amounts of input protein (Bottom). (F) Densitometric analysis of 3 independent immunoblots such as the ones shown in (E). Results are mean ± SD. Statistical significance relative to uninfected control cells was analyzed by the Student’s t test assuming unequal variances. NS; not significant.
EGFR sustains C. albicans-induced EphA2 activation and constitutively interacts with EphA2
Previously, we determined that EphA2 and EGFR function in the same pathway to mediate the endocytosis of C. albicans by oral epithelial cells. Also, EphA2 is required for C. albicans to activate EGFR because siRNA knockdown of EphA2 in oral epithelial cells blocks phosphorylation of EGFR induced by the fungus (15). Cross talk between EphA2 and EGFR has also been observed in other cell types (17, 18). To investigate the nature of this cross-talk, we tested the effects of inhibiting EGFR on C. albicans-induced EphA2 activation. When EGFR was either knocked down with siRNA or inhibited with the specific EGFR kinase inhibitor, gefitinib (19), EphA2 phosphorylation was transient, occurring within 30 min of infection, but declining to basal levels by 90 min (Fig. 1C, D; S1). Therefore, activation of EGFR by C. albicans is required to sustain EphA2 phosphorylation.
One potential mechanism for the cross-talk between EphA2 and EGFR is a physical interaction between these two receptors. We investigated this possibility by immunoprecipitation experiments. When EphA2 was immunoprecipitated from lysates of epithelial cells, EGFR was pulled down; the amount of EGFR that was associated with EphA2 did not increase when the epithelial cells were infected with C. albicans (Fig. 1E, F). In reciprocal experiments, we determined that immunoprecipitation of EGFR from infected and uninfected epithelial cells pulled a constant amount of EphA2 (Fig. 1E, F). These results indicate that EphA2 and EGFR constitutively associate with each other, likely forming part of a complex that enables each receptor to influence the activity of the other.
EGFR induces a subset of EphA2-mediated pro-inflammatory responses in oral epithelial cells
The physical association of EphA2 with EGFR suggested the possibility that EGFR may also induce the epithelial cell proinflammatory response to C. albicans infection. To test this hypothesis, we compared the effects of siRNA knockdown of EGFR and EphA2 on cytokine production in oral epithelial cells that were infected with C. albicans. We found that knockdown of EGFR inhibited the secretion of CXCL8/IL-8 and CCL20 similarly to knockdown of EphA2 (Fig. 2A). Although knockdown of EGFR had no effect on the release of IL-1α, and IL-1β, knockdown of EphA2 blocked both of these cytokines. Treatment of the epithelial cells with gefitinib had a similar effect to siRNA knockdown of EGFR, resulting in inhibition of CXCL8/IL-8 and CCL20 secretion but no reduction of IL-1α, and IL-1β release (Fig. 2B). These results suggest that EGFR governs a subset of the epithelial cell inflammatory response to C. albicans infection.

EphA2 and EGFR are required for induction of a pro-inflammatory response to C. albicans by oral epithelial cells. (A) Effects of siRNA knockdown of EGFR and EphA2 on the production of cytokines and chemokines by oral epithelial cell in response to 8 h of C. albicans infection. Box whisker plots show median, interquartile range, and range of 3 experiments, each performed in duplicate. (B) Inhibition of EGFR with gefitinib reduces oral epithelial cell pro-inflammatory response to C. albicans. Shown are results of 3 experiments, each performed in duplicate. (Ca, C. albicans; ctrl, control; GEF, Gefitinib; UNINF, uninfected). Statistical significance is indicated by *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (Mann-Whitney test with Bonferroni correction for multiple comparisons).
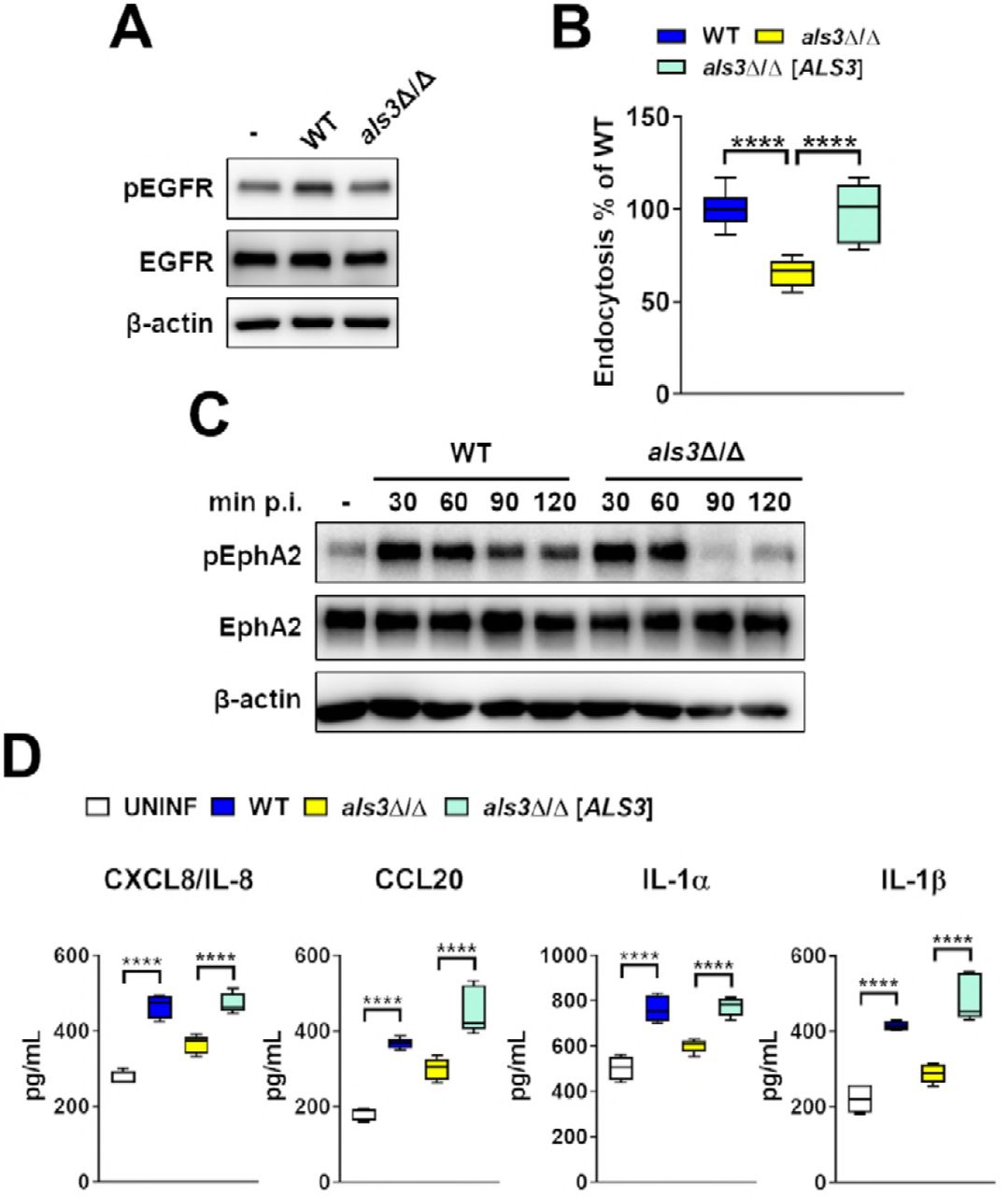
C. albicans Als3 is required for prolonged EphA2 activation and induction of epithelial cell chemokine and cytokine secretion
The C. albicans invasin Als3 activates EGFR and induces oral epithelial cells to endocytose the fungus (Fig. S2) (5, 20). To determine if Als3 also activates EphA2, we analyzed the response of oral epithelial cells to infection with an als3Δ/Δ null mutant. As expected, this mutant induced weak EGFR autophosphorylation and was endocytosed poorly (Fig. 3A, B, S2). Infection with the als3Δ/Δ null mutant induced transient EphA2 phosphorylation that returned to basal level by 90 min of infection (Fig.3C; S3). Infection with the als3Δ/Δ null mutant also caused significantly less secretion of CXCL8/IL-8, CCL20, IL-1α, and IL-1β (Fig. 3D). Collectively, these results indicate that C. albicans Als3 activates EGFR, which prolongs EphA2 phosphorylation and stimulates oral epithelial cells to secrete chemokines and pro-inflammatory cytokines.

Infection of oral epithelial cells with the C. albicans als3Δ/Δ null mutant induces only transient phosphorylation of EphA2 and a diminished proinflammatory response. (A). Immunoblot analysis showing the effects of the indicated C. albicans strains on the phosphorylation of EGFR after 90 min of infection (WT; wild-type strains DAY185). (B) Epithelial cell endocytosis of the indicated C. albicans strains after 120 min of infection. Data are the combined results of three experiments, each performed in triplicate. (C) Immunoblot analysis showing the time course of EphA2 phosphorylation in oral epithelial cells that had been infected with wild-type and als3A/A mutant strains of C. albicans for the indicated times. (D) Oral epithelial cell production of chemokines and cytokines after 8 h of infection with the indicated C. albicans strains. Data are the results of 3 experiments, each performed in duplicate. Statistical significance is indicated by ****P < 0.0001 (Mann-Whitney test with Bonferroni correction for multiple comparisons).
Inhibition of EGFR impairs the host inflammatory response during OPC
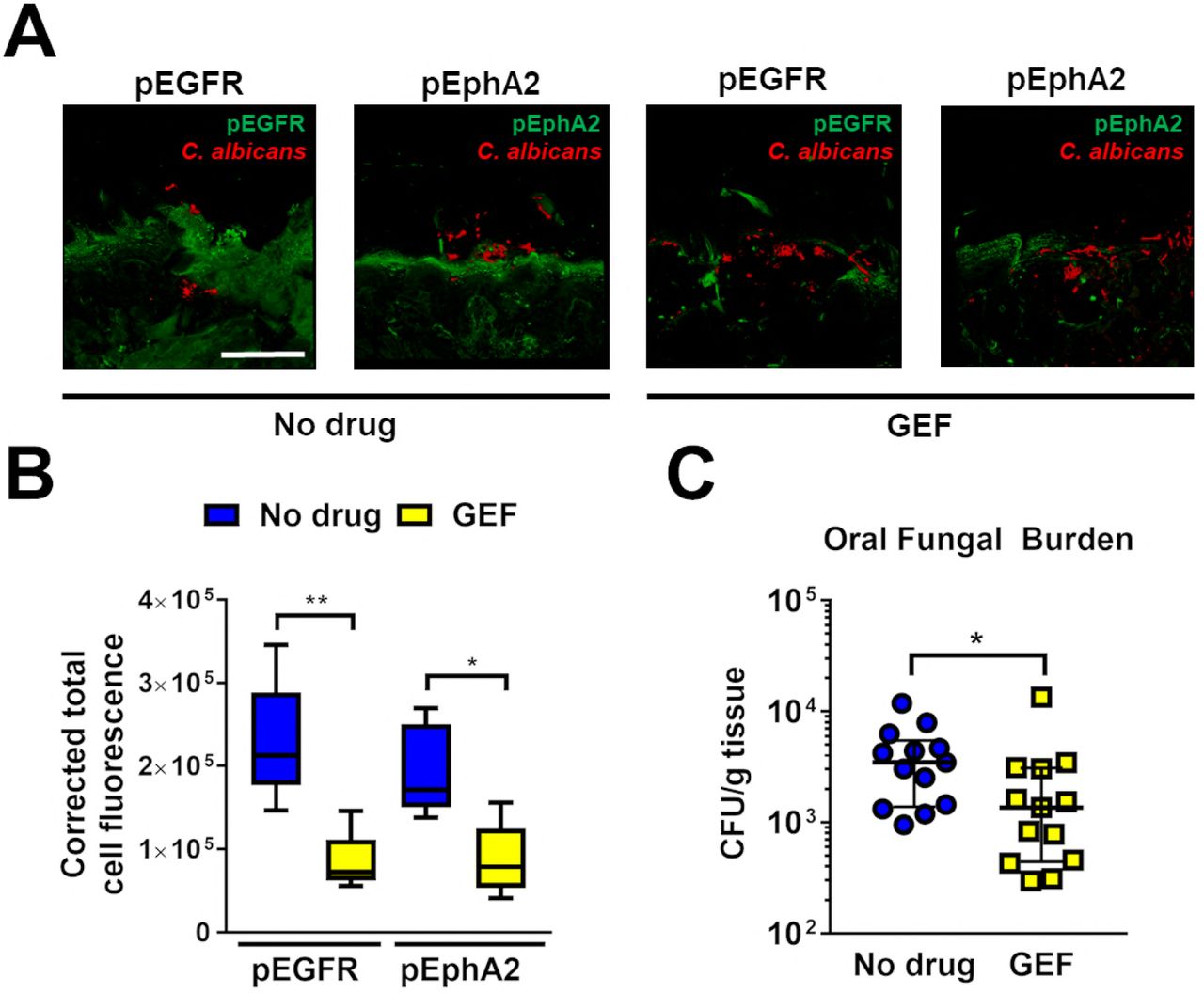
To further assess the role of EGFR in mediating the host inflammatory response to C. albicans during OPC, we treated immunocompetent mice with gefitinib (20, 21) and then orally inoculated them with C. albicans. Using immunostaining with phosphospecific antibodies, we assessed the effects of gefitinib on EGFR and EphA2 phosphorylation in the oral epithelium. Similar to its effects in vitro, treatment with gefitinib reduced the phosphorylation of both EGFR and EphA2 after 1 d of infection (Fig. 4A, B). At this time point, the gefitinib-treated mice had a 3-fold reduction in oral fungal burden compared to untreated mice (Fig. 4C). The oral tissues of gefitinib-treated mice also contained significantly less IL-1α, IL-1β, CXCL1/KC, IL-17A, and the p19 subunit of IL-23 than the control mice (Fig. 5A). By contrast, the levels of CCL2, CCL3, CCL4, and the host defense peptide S100A8 in the gefitinib treated mice were not significantly different from the control mice, indicating that gefitinib selectively inhibited a subset of the host inflammatory response. Treatment with gefitinib also caused a dramatic reduction in the number of neutrophils and inflammatory monocytes in the oral tissues relative to the mice (Fig. 5B, C, S4). Thus, in immunocompetent mice, gefitinib inhibits C. albicans-induced phosphorylation of EGFR and EphA2, thereby reducing both oral fungal burden and the host inflammatory response.

Pharmacological inhibition of EGFR reduces EphA2 phosphorylation and oral fungal burden during OPC. (A) Confocal images of thin sections of the tongues of immunocompetent mice that were administered gefitinib and then infected with C. albicans for 1 d. The thin sections were stained for pEGFR, or pEphA2 (green) and C. albicans (red). Scale bar, 100 μm. (B) Quantification of corrected total cell fluorescence of pEGFR and pEphA2 in the oral mucosa of mice with OPC. Results are the combined analysis of a total of 6 mice per condition from 2 separate experiments. (C) Oral fungal burden of control and gefitinib-treated mice after 1 d of infection. Results are median ± interquartile range of at total of 14 mice per group from two independent experiments. The y-axis is set at the limit of detection (100 CFU/g tissue). Statistical significance is indicated by *P < 0.05, **P < 0.01 (Mann-Whitney test).

Pharmacological inhibition of EGFR inhibits innate antifungal responses during OPC. (A-C) Levels of chemokines and cytokines (A), neutrophils (B), and inflammatory monocytes (C) in the tongues of control and gefitinib-treated mice treated with OPC after 1 d of infection. Scatter plots show median and interquartile range of a total of seven mice in each group from two independent experiments. Statistical significance is indicated by *P < 0.05, **P < 0.01 (Mann-Whitney test with Bonferroni correction for multiple comparisons).
Because gefitinib impaired the host inflammatory response to C. albicans, we investigated its effects on the candidacidal activity of neutrophils and macrophages. We found that gefitinib did not decrease the capacity of human neutrophils or mouse bone marrow derived macrophages to kill C. albicans in vitro (Fig. S5). Taken together, these results indicate that while EGFR signaling is required for epithelial cells to mount a pro-inflammatory response to C. albicans, it is dispensable for governing phagocyte killing of this organism.
EGFR activity is required for the epithelial cell response to IL-17A
Many of the chemokines that were suppressed by gefitinib treatment are known to be induced by IL-17A, a cytokine that plays a central role in the host defense against OPC (22-24). We therefore investigated whether EGFR is required for oral epithelial cells to respond to IL-17A. When oral epithelial cells were stimulated with IL-17A and TNFα in the absence of C. albicans, EGFR was autophosphorylated for 60 min, after which phosphorylation returned to basal levels (Fig. 6A, Fig. S6). Stimulation of uninfected epithelial cells with IL-17A and TNFα also induced the production of CXCL8/IL-8, CCL20, and GM-CSF (Fig. 6B). This stimulation was almost completely blocked when the epithelial cells were treated with gefitinb (Fig. 6C). Thus, activation of EGFR is required for IL-17A to induce a proinflammatory response in oral epithelial cells.

Interaction between EGFR and IL-17 in the epithelial cell response to C. albicans. (A) Immunoblot showing the time course or EGFR phosphorylation in oral epithelial cells that were exposed to IL-i7A and TNFα for the indicated times. (B) Effects of stimulating oral epithelial cells with IL-i7A alone or in combination with TNFα for 8 h on secretion of CXCL8/IL-8, CCL20, and GM-CSF. (C) Inhibitory effects of gefitinib on oral epithelial cells secretion of the indicated inflammatory mediators after 8 h of stimulation with IL-i7A and TNFα. Results in (B and C) are the combined data from 3 independent experiments, each performed in triplicate. Statistical significance is indicated by *P < 0.05, ***P < 0.001, ****P < 0.0001 (Mann-Whitney test with Bonferroni correction for multiple comparisons).
Discussion
Oral epithelial cells play a central role in orchestrating the host defense against C. albicans during OPC (2, 4, 25). Here, we found that EphA2 and EGFR form a complex in oral epithelial cells. When these cells are infected with C. albicans, EphA2 and EGFR functionally interact to induce a pro-inflammatory response. Previously, we found that EphA2 is required for C. albicans to stimulate EGFR (15). Here, we determined that EGFR is in turn necessary for C. albicans to induce prolonged activation of EphA2. This finding suggests that the initiation of an antifungal proinflammatory response in the oral cavity requires two signals, one induced by EphA2 binding to fungal β-glucans and the other induced by EGFR interacting with fungal invasins.
EphA2 and EGFR are known to interact in epithelial cell cancers, especially those that have become resistant to EGFR inhibitors (26, 27). In these cells, siRNA knockdown of EphA2 restores sensitivity to EGFR inhibition. Interestingly, treatment of these malignant cell lines with soluble EFNA1 has the same effect as EphA2 siRNA, presumably because EFNA1 induces EphA2 endocytosis and subsequent degradation. Our data indicate that C. albicans activates EphA2 differently than EFNA1 because binding of C. albicans stabilizes EphA2 and prevents its degradation. The sustained EphA2 protein levels likely contribute to the prolonged EphA2 signaling induced by C. albicans infection.
Functioning as a receptor for fungal β glucans, EphA2 initiates the epithelial cell production of chemokines and pro-inflammatory cytokines in response to C. albicans overgrowth (15). The finding that EphA2 and EGFR interact prompted us to investigate whether EGFR also mediates the production of pro-inflammatory mediators. Indeed, we found that knockdown of EGFR with siRNA or inhibition of EGFR with gefitinib significantly reduced C. albicans-induced production of CXCL8/IL-8 and CCL20 in vitro. Infection with the als3Δ/Δ mutant also induced less chemokine secretion by oral epithelial cells, suggesting that activation of EGFR is necessary for secretion of CXCL8/IL-8 and CCL20. However, gefitinib treatment had no effect on epithelial cell release of IL-1α and IL-1β, whereas infection with the als3Δ/Δ mutant decreased the release of these cytokines, an effect that was similar to siRNA knockdown of EphA2 (15). A potential explanation for this result is that treatment with gefitinib caused a 20%±8% increase in epithelial cell damage due to the wild-type strain (n=9, p < 0.0001 by the Student’s t-test), whereas infection with the als3Δ/Δ null mutant caused significantly less damage to oral epithelial cells relative to the wild-type strain (6). Similarly, siRNA knockdown of EphA2 reduced the extent of C. albicans induced epithelial cell damage(15). Thus, we speculate that while the secretion of CXCL8/IL-8 and CCL20 in response to C. albicans is induced by activation of EGFR, the release of IL-1α and IL-1β is stimulated by epithelial cell damage.
The central role of EGFR in the host inflammatory response was also demonstrated in the mouse model of OPC, where treatment with gefitinib inhibited phosphorylation of both EphA2 and EGFR, and reduced the tissue levels of CXCL1/KC, IL-1α, IL-1β, IL-17A, and IL-23p19. As a result, the accumulation of neutrophils and inflammatory monocytes in the oral tissues was dramatically decreased. It was notable that treatment with gefitinib only inhibited a subset of the inflammatory mediators induced by C. albicans infection; the tissue levels of CCL2, CCL3, CCL4 and S100A8 in the gefitinib treated mice were not significantly different from control mice. This results suggests that EGFR activation is require for the production of a subset of inflammatory mediators in vivo and that other inflammatory mediators are induced by an EGFR-independent pathway. EGFR has previously been found to be important for the production of CXCL8/IL-8 and CXCL10 by pulmonary epithelial cells infected with influenza and rhinovirus. In contrast to C. albicans, which appears to activate EGFR directly (5), these viruses activate EGFR indirectly by stimulating a metalloproteinase that cleaves an EGFR proligand that in turn binds to EGFR (28, 29).
We observed that stimulation of uninfected epithelial cells with IL-17A and TNFα induced the phosphorylation of EGFR. Moreover, gefitinib blocked the release of CXCL8/IL-8, CCL20, and GM-CSF by uninfected cells that had been stimulated with IL-17A and TNFα. These data indicate that EGFR phosphorylation is required for at least some of the epithelial cell responses to IL-17A stimulation. Lee et al. (30) found similarly that in a colonic epithelial cell line, the combination of IL-17A and TNFα induces EGFR phosphorylation and that treatment with an EGFR kinase inhibitor decrease IL-17A-induced release of CXCL8/IL-8 and CXCL10. These authors suggested that EGFR potentiates and prolongs ERK signaling induced by IL-17A, leading to chemokine secretion.
Our current results combined with our previous data (15) suggest a more nuanced model for how oral epithelial cells respond to C. albicans overgrowth during OPC (Fig 7). In this model, C. albicans β-glucans initially activate EphA2. Subsequently, when C. albicans forms hyphae, it expresses invasins such as Als3 that activate EGFR and sustain EphA2 activation, leading to activation of the MEK1/2, c-Fos, and STAT3 signaling pathways, ultimately resulting in the release of proinflammatory mediators by the infected epithelial cells. EGFR is also activated by the IL-17A that is produced by intraepithelial lymphocytes, leading to further amplification of the epithelial cell proinflammatory response. The overall result is the secretion of chemokines, proinflammatory cytokines, and host defense peptides as well as recruitment of phagocytes to the focus of infection, leading to inhibition and eventual killing of the fungus.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model of the interactions of C. albicans with EphA2 and EGFR during OPC. When C. albicans interacts with EphA2 and EGFR on oral epithelial cells, there is prolonged phosphorylation of EphA2, which activates the STAT3 and mitogen-activated protein kinase (MAPK) pathways, leading to the production of chemokines, proinflammatory cytokines, and host defense peptides. The chemokines recruit proinflammatory monocytes and neutrophils to the focus of infection. Activation of EphA2 and EGFR also causes the epithelial cells to endocytose C. albicans, which subsequently causes epithelial cell damage and leads to the release of IL-1α and IL-1β. In response to C. albicans infection, leukocytes such as y§ T cells, innate TCRap+ cells, and type-3 innate lymphoid cells secrete IL-17A, which requires EGFR activation to amplify the epithelial inflammatory response.
While receptor-mediated induction of the proinflammatory response is likely beneficial to the host, activation of EphA2 and EGFR may also be beneficial to the fungus. The interaction of EphA2 and EGFR with C. albicans activates the clathrin-dependent endocytosis pathway in the epithelial cells, leading to rearrangement of the actin cytoskeleton and formation of pseudopods that engulf C. albicans and pull it into the epithelial cell (31). This process contributes to the pathogenicity of the fungus because the internalized organism is hidden from phagocytes and can utilize the epithelial cell as a source of nutrients (32).
This model explains the modest effect of gefitinib on oral fungal burden during OPC. Although EGFR inhibition impaired the host inflammatory response to C. albicans, this negative effect was counteracted by reduced fungal invasion of the epithelium. Based on this model, therapeutic strategies to block the interaction of fungi with host receptors should be evaluated for their effects on both the host inflammatory response and fungal invasion.
Materials and Methods
Ethics statement
All animal work was approved by the Institutional Animal Care and Use Committee (IACUC) of the Los Angeles Biomedical Research Institute. The collection of blood from human volunteers for neutrophil isolation was also approved by the Institutional Review Board of the Los Angeles Biomedical Research Institute.
Fungal strains and epithelial cells
The C. albicans wild type strain SC5314 (33), the als3Δ/Δ mutant, and the als3Δ/Δ+pALS3 complemented strain (6) were used. For the experiments, the C. albicans cells were grown for 18 h in yeast extract-peptone dextrose (YPD) in a shaking incubator at 30°C. The fungal cells were harvested by centrifugation, washed twice with phosphate-buffered saline (PBS), and counted using a hemacytometer.
The OKF6/TERT-2 immortalized human oral epithelial cell line was kindly provided by J. Rheinwald (Harvard University, Cambridge, MA) (34) and was cultured as previously described (20). OKF6/TERT-2 cells were authenticated by RNA-Seq (35) and tested for mycoplasma contamination.
Inhibitor and agonists
The EGFR kinase inhibitor gefitinib (Selleckchem) was dissolved in DMSO and used at a final concentration of 1 μm. It was added to the host cells 60 min prior to infection or stimulation and remained in the medium for the entire incubation period. Control cells were incubated with a similar concentration of DMSO at a final concentration of 0.1 %. EFNA1-Fc (Acro Biosystems) was used at a final concentration of 1 μg/ml. IL-17A and TNFα (PeproTech)) were used at final concentrations of 50 ng/ml and 0.5 ng/ml, respectively.
siRNA
To knockdown EGFR and IL-17RA, OKF6/TERT-2 cells were transfected with siRNA as described previously (20). Briefly, the cells were grown in 6-well tissue culture plates and transfected with 80 pmol EGFR siRNA (sc-29301), or a similar amount of random control siRNA (sc-37007; both from Santa Cruz Biotechnology) using Lipofectamine 2000 (Thermo Fisher Scientific) following the manufacturer’s instructions. The extent of EGFR knockdown was verified 72 h later by immunoblotting with specific antibodies against EGFR. Knockdown of EGFR was > 80% (Fig. S1)
Immunoblotting
OKF6/TERT-2 cells in 24-well tissue culture plates were switched to KSF medium without supplements for 1 h and then infected with 1 × 106 C. albicans yeast or incubated with IL-17A and TNFα for various times as described previously (15). Next, the cells were rinsed with cold HBSS containing protease and phosphatase inhibitors and detached from the plate with a cell scraper. After the cells were collected by centrifugation, they were boiled in sample buffer. The lysates were separated by SDS-PAGE, and phosphorylation was detected by immunoblotting with specific antibodies against pEphA2 (#6347, Cell Signaling) and pEGFR (#2234, Cell Signaling). Next, the blot was stripped, and the total amount of each protein was detected by immunoblotting with antibodies against EphA2 (D4A2, Cell Signaling) and EGFR (#4267, Cell Signaling). Each experiment was performed at least 3 times.
Co-immunoprecipitation
OKF6/TERT-2 cells were grown in 75 cm2 flasks to confluency and then switched to KSF medium without supplements for 3 h and then infected with 1×108 C. albicans yeast. After 30 or 90 min. OKF6/TERT-2 were washed with ice-cold cold PBS (with Mg2+, and Ca2+), scraped, and lysed with 100 μl ice-cold 5.8% octyl β-D-glucopyranoside (0479-5g; VWR) in the present of protease/phosphatase inhibitors. Whole cells lysates were precleared with 20μl of protein A/G plus (sc-2003; Santa Cruz Biotechnology) at 4°C for 30minutes. Bead-protein mix was centrifuged at 3000rpm for 30 sec at 4°C and supernatants were collected. 2 μg of anti-EGFR antibody (sc-101; Santa Cruz Biotechnology), or anti-EphA2 antibody (#6347, Cell Signaling) respectively, was added to 500 μg of proteins, and incubated on a rotator at 4°C for 2 hours. 25μl of protein AlG plus was added to each immunoprecipitation sample and incubated for an additional hour at 4°C. Samples were pelleted at 3000 rpm for 30 sec, and washed 3 times in 500 μl of ice-cold 1.5% octyl β-D-glucopyranoside. Proteins were eluted with 30 μl of 2X SDS buffer, and heated at 90°C for 5 minutes. Samples were centrifuged at 3000 rpm for 30 sec, and supernatants were collected, and separated by SDS-PAGE, and analyzed as described above.
Measurement of epithelial cell endocytosis
The endocytosis of C. albicans by oral epithelial cells was quantified as described previously (36). OKF6/TERT-2 oral epithelial cells were grown to confluency on fibronectin-coated circular glass coverslips in 24-well tissue culture plates and then infected for 120 min with 2 × 105 yeast-phase C. albicans cells per well, after which they were fixed, stained, and mounted inverted on microscope slides. The coverslips were viewed with an epifluorescence microscope, and the number of endocytosed organisms per high-power field was determined, counting at least 100 organisms per coverslip. Each experiment was performed at least 3 times in triplicate.
Cytokine and chemokine measurements in vitro
Cytokine levels in culture supernatants were determine as previously described (15). Briefly OKF6/TERT-2 cells in a 96- well plate infected with C. albicans at a multiplicity of infection of 5. After 8 h of infection, the medium above the cells was collected, clarified by centrifugation and stored in aliquots at −80 °C. The concentration of inflammatory cytokines and chemokines in the medium was determined using the Luminex multipex assay (R&D Systems).
Epithelial cell damage
The effects of gefitinib on extent of epithelial cell damage caused by C. albicans was determined by our previously described 51Cr release assay (20). OKF6/TERT-2 cells in a 24-well plate were loaded with 51Cr overnight. The next day, they were incubated with gefitinib or diluent and then infected with C. albicans at a multiplicity of infection of 10. After 8 h of infection, the medium above the epithelial cells was collected and the epithelial cells were lysed with RadiacWash (Biodex). The amount of 51Cr released into the medium and remaining in the cells was determined with a gamma counter, and the percentage of 51Cr released in the infected cells we compared to the release by uninfected epithelial cells. The experiment was performed 3 times in triplicate.
Mouse model of oropharyngeal candidiasis
6 week old male BALB/c mice were purchased from Taconics. OPC was induced in mice as described previously (20, 21). Starting on day -2 relative to infection, the mice were randomly assigned to receive gefitinib or no treatment. Gefitinib was administered by adding the drug to the powdered chow diet at final concentration of 200 parts-per-million. For inoculation, the animals were sedated, and a swab saturated with 2 × 107 C. albicans cells was placed sublingually for 75 min. Mice were sacrificed after 1 day of infection. The tongues were harvested, weighed, homogenized and quantitatively cultured. The researchers were not blinded to the experimental groups because the endpoints (oral fungal burden, cytokine levels, and leukocyte numbers) were an objective measure of disease severity.
Cytokine and chemokine measurements in vivo
To determine the whole tongue cytokine and chemokine protein concentrations, the mice were orally infected with C. albicans as above. After 1 day of infection, the mice were sacrificed, and their tongues were harvested, weighed and homogenized. The homogenates were cleared by centrifugation and the concentration of inflammatory mediators was measured using a multiplex bead array assay (R&D Systems) as previously described (15, 37).
Flow cytometry of infiltrating leukocytes
The number of phagocytes in the mouse tongues were characterized as described elsewhere (38). Briefly, mice were orally infected with C. albicans as described above. After 1 d of infection, the animals were administered a sublethal anesthetic mix intraperitoneally. The thorax was opened, and a part of the rib cage removed to gain access to the heart. The vena cava was transected and the blood was flushed from the vasculature by slowly injecting 10 mL PBS into the right ventricle. The tongue was harvested and cut into small pieces in 100 μL of ice-cold PBS. 1 mL digestion mix (4.8 mg/ml Collagenase IV; Worthington Biochem, and 200 μg/ml DNase I; Roche Diagnostics, in i× PBS) was added after which the tissue was incubated at 37°C for 45 min. The resulting tissue suspension was then passed through a 100 μm cell strainer. The single-cell suspensions were incubated with rat anti-mouse CD16/32 (2.4G2; BD Biosciences) for 10 min in FACS buffer at 4°C to block Fc receptors. For staining of surface antigens, cells were incubated with fluorochrome-conjugated (FITC, PE, PE-Cy7, allophycocyanin [APC], APC-eFluor 780,) antibodies against mouse CD45 (30-F11; BD Biosciences), Ly6C (AL-21; BD Biosciences), Ly6G (1A8, BioLegend), CD11b (M1/70; eBioscience), and CD90.2 (30-H12; BioLegend). After washing with FACS buffer, the cell suspension was stained with a LIVE/DEAD fluorescent dye (7-AAD; BD Biosciences) for 10 min. The stained cells were analyzed on a 2-laser LSRII flow cytometer (BD Biosciences), and the data were analyzed using FACS Diva (BD Biosciences) and FlowJo software (Treestar). Only single cells were analyzed, and cell numbers were quantified using PE-conjugated fluorescent counting beads (Spherotech).
Phagocyte killing assay
The effects of gefitinib on neutrophil killing of C. albicans were determined by our previously described method (20). Briefly, neutrophils were isolated from the blood of healthy volunteers and incubated with gefitinib or diluent in RPMI 1640 medium plus 10% fetal bovine serum for 1 h at 37°C. Next, the neutrophils were mixed with an equal number of serum-opsonized C. albicans cells. After a 3 h incubation, the neutrophils were lysed by sonication, and the number of viable C. albicans cells was determined by quantitative culture.
To obtain bone marrow-derived macrophages, bone marrow cells from BALB/c mice (Taconics) were flushed from femurs and tibias using sterile RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 2 mM EDTA onto a 50 ml screw top Falcon tube fitted with a 100 μm filter (39). 6×106 bone marrow cells per 75 cm2 were seeded in RPMI 1640 supplemented with 20% FBS, 100 μg/ml streptomycin, 100 U/ml penicillin, 2 mM Glutamine, and 25 ng/ml rHu M-CSF (PeproTech). After 7 days, the bone marrow derived macrophages (BMDMs) were treated with gefitinib or the diluent and then incubated with serum-opsonized C. albicans cells (multiplicity of infection 1:20) for 3 h. Next, the BMDMs were scraped, lysed by sonication, and the number of viable C. albicans cells was determined by quantitative culture.
Indirect Immunofluorescence
To detect phosphorylation of EphA2, and EGFR in the tongue of C. albicans infected mice, 2-μm-thick sections of OCT-embedded organs were cut with a microtome and then fixed with cold acetone. Next, the cryosections were rehydrated in PBS and then blocked. They were stained with primary antibodies against phosphorylated EphA2 (#6347, Cell Signaling) and phosphorylated EGFR (#2234, Cell Signaling) primary antibodies and then rinsed and stained with an Alexa Fluor 488 secondary antibody. To detect C. albicans, the sections were also stained with an anti-Candida antiserum (Biodesign International) conjugated with Alexa Fluor 568 (Thermo Fisher Scientific). The sections were imaged by confocal microscopy. To enable comparison of fluorescence intensities among slides, the same image acquisition settings were used for each experiment. To determine corrected total cell fluorescence the integrated density was subtracted by the selected cell area which was multiplied by mean fluorescence of background readings using ImageJ (40).
Statistics
At least three biological replicates were performed for all in vitro experiments unless otherwise indicated. Data were compared by Mann-Whitney corrected for multiple comparisons using GraphPad Prism (v. 6) software. P values < 0.05 were considered statistically significant.
Data Availability
The raw data that support the findings of this study are available from the corresponding authors upon request.
Competing interests
S.G.F. is a co-founder of and shareholder in NovaDigm Therapeutics, Inc., a company that is developing a vaccine against mucosal and invasive Candida infections.
Acknowledgments
This work was supported in part by NIH grant K99DE026856 to M.S, and R01DE022600 and R01AI124566 to S.G.F. M.D.L. was supported by the LA BioMed Summer Fellowship Program. We thank S.W. French and E. Vitocruz for histopathology, and members of the Division of Infectious Diseases at Harbor-UCLA Medical Center for critical suggestions. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References




