

Abstract
Genus Frankia is comprised primarily of nitrogen-fixing actinobacteria that form root nodule symbioses with a group of hosts known as the actinorhizal plants. These plants are evolutionarily closely related to the legumes, which are nodulated by the rhizobia. Both host groups utilize homologs of nodulation genes for root-nodule symbiosis, derived from common plant ancestors. However the corresponding endosymbionts, Frankia and the rhizobia, are distantly related groups of bacteria, leading to questions of their symbiotic mechanisms and evolutionary history. To date, a stable system of genetic transformation has been lacking in Frankia. Here, we report the successful electrotransformation of Frankia alni ACN14a, by means of replicating plasmids expressing chloramphenicol-resistance for selection, and the use of GFP as a marker of gene expression. We have identified type IV methyl-directed restriction systems, highly-expressed in a range of actinobacteria, as a likely barrier to Frankia transformation and circumvented this barrier by using unmethylated plasmids, which allowed the transformation of F. alni as well as the maintenance of the plasmid. During nitrogen limitation, Frankia differentiates into two cell types: the vegetative hyphae and nitrogen-fixing vesicles. When the plasmid transformation system was used with expression of egfp under the control of the nif gene cluster promoter, it was possible to demonstrate by fluorescence imaging the expression of nitrogen fixation in vesicles but not hyphae in nitrogen-limited culture.
Importance To date, the study of Frankia-actinorhizal symbioses has been complicated by the lack of genetic tools for manipulation of Frankia, especially stable genetic transformation. The transformation system reported here, particularly coupled with marker genes, can be used to differentiate patterns of gene expression between Frankia hyphae and vesicles in symbiosis or in free-living conditions. This will enable deeper comparisons between Frankia-actinorhizal symbioses and rhizobia-legume symbioses in terms of molecular signaling and metabolic exchange that will broaden understanding of the evolution of these symbioses and potentially make possible their application in agriculture. The development of transformation methods will allow further down-stream applications including gene knock-outs and complementation that will, in turn, open up a much broader range of experiments into Frankia and its symbioses.
Introduction
Bacteria in the genus Frankia form nitrogen-fixing root nodule symbioses with a group of host plants, the actinorhizal plants. The actinorhizal plants are evolutionarily closely related to the legumes within the Nitrogen-fixing Clade (NFC; Soltis et al., 1995). The bacterial symbionts are only distantly related however: Frankia belongs to the phylum Actinobacteria, comprised of high-GC-content gram-positive bacteria, whereas the rhizobia, symbionts of the legumes, are gram-negative proteobacteria. Additionally, Frankia is a multicellular, hyphal genus whereas the rhizobia are single-celled. In most of the actinorhizal symbioses, and in nitrogen-limiting conditions in vitro, Frankia differentiates into two cell types: vegetative hyphae and nitrogen-fixing vesicles. Vesicles are surrounded by a lamellar hopanoid lipid envelope (Berry et al., 1993) that increases in number of layers in response to oxygen tension thereby likely reducing the flow of oxygen into the vesicle interior to protect the nitrogenase complex from deactivation (Benson and Silvester, 1993). Thus, unlike the rhizobia which require their hosts to regulate the flow of oxygen for nitrogen fixation in symbiosis (Appleby, 1984), Frankia vesicles are capable of fixing nitrogen in atmospheric oxygen conditions (Benson and Silvester, 1993).
Biological nitrogen fixation has potential applications in the development of minimal input strategies for more sustainable agriculture globally and in nutrient-deficient soils through genetic improvement of crops (Gutierrez, 2012). In efforts to understand the mechanisms of root nodule symbioses, phylogenetic studies of the Nitrogen-Fixing Clade have shown a shared predisposition underlying the evolution root nodule symbioses between the actinorhizal plants and legumes (Werner et al., 2014; Battenberg et al., 2018). The actinorhizal and legume hosts share the Common Symbiotic Pathway, a signaling pathway that leads to the development of the nodule in response to the symbiont (reviewed in Oldroyd 2013) as well as several key gene orthologs in the process of nodule development (Battenberg et al., 2018; Griesmann et al., 2018). Clear differences also exist in the genetics of nodulation in the two bacterial groups, leaving major questions concerning the evolution and development of root-nodule symbioses in the bacterial partners. The rhizobial signaling molecules that trigger nodulation in many of the legume hosts, called Nod factors, are synthesized by nod genes and have been extensively studied (Oldroyd, 2013). The majority of Frankia genomes do not contain clear sets of nod gene homologs with the exception of several of the cluster II Frankia: genomes of these Frankia contain homologs of the rhizobial nodABC genes that are expressed in symbiosis (Persson et al., 2015). Other mechanisms have been found to trigger legume nodulation by rhizobia, including an effector protein injected into the host by a type III secretion system (Okazaki et al., 2013); however genes responsible for these pathways lack homologs in Frankia genomes as well. Signals from Frankia in clusters I and III that elicit host root responses have been detected experimentally, but the chemical composition has not been defined (Ceremonie et al., 1999; Cissoko et al., 2018).
The major advances in knowledge of rhizobial symbioses have been made possible by the development of genetic tools for dissecting the metabolic and regulatory pathways in the microsymbiont-host interactions. Long et al. (1982) originally demonstrated the role of the nod genes in symbiosis by complementing nodulation-deficient mutants of Sinorhizobium meliloti with nod genes encoded on a replicating, broad host-range, plasmid. More recently, expression of marker genes in rhizobia has enabled a wider-range of experiments including the tracking of microsymbionts during the host infection process (Gage, 2002) and the identification of regulatory networks involved in symbiotic interactions (Spaepen et al., 2009). However, to date, there has been no stable genetic transformation system for Frankia (Kucho et al., 2009). As reported here, a transformation system in Frankia will thus enable functional inquiries into the diverse mechanisms involved in establishing root-nodule symbiosis and maintaining biological nitrogen fixation in actinorhizal symbioses. These inquiries will, in turn, contribute to a wider understanding of the origins of root nodule symbioses and mechanisms of Frankia and rhizobia interaction with hosts in the NFC.
Frankia has several barriers to transformation. Actinobacteria in general have low rates of homologous recombination due to competition between their homologous recombination pathway and a Non-Homologous End-Joining pathway (Zhang et al., 2012). Additionally, natural vectors for Frankia transformation are limited as no Frankia phages have been discovered (Simonet et al., 1990). Finally, its multi-cellular hyphae require that all cells be transformed in order for experiments to be viable. Despite these barriers, it has been demonstrated that DNA can be electroporated into Frankia cells (Myers and Tisa, 2004), providing a potential avenue for genetic transformation. Furthermore, Kucho et al. (2009) reported initial success in integrating a non-replicating plasmid into the chromosome of Frankia sp. CcI3. However the recombined plasmid was lost in the following generation, limiting its use in experiments.
Restriction enzymes pose a barrier to successful transformation in bacteria due to their role in defense by digesting foreign DNA. In some actinobacteria, the use of unmethylated plasmids has increased transformation efficiency (Ankri et al., 1996; Molle et al., 1999), potentially due to the presence of Type IV methyl-directed restriction enzymes. In the majority of bacterial taxa, DNA is methylated during replication by the methyltransferase Dam to mark parent strands for DNA repair and excision of misincorporated bases from the daughter strand (Sanchez-Romero et al., 2015). The majority of actinobacteria, however, lack dam homologs (Sanchez-Romero et al., 2015) and an investigation of genomes of Streptomyces, Rhodococcus, and Micromonospora spp. found that these genomes were not methylated in the canonical Dam pattern (Novella et al., 1996). While actinobacteria have recently been shown to have a unique mismatch repair pathway (Castaneda-Garcia et al., 2017) it is not yet known how this pathway utilizes methylation, if at all.
In this study we report successful genetic transformation of Frankia alni ACN14a by using an unmethylated plasmid to circumvent methylation-targeting restriction enzymes encoded in the ACN14a genome. This permitted the maintenance of plasmids derived from one with a very broad host-range origin of replication (Kurenbach et al., 2003) in F. alni cultures. Additionally, we show that gfp expressed from a replicating plasmid can be used to label gene expression differences between Frankia cell types during nitrogen fixation, opening up molecular genetics-based experiments on Frankia symbioses in the future.
Results
Identification of Restriction Enzymes in Frankia and Transcriptome Analysis
Type I, II and IV restriction enzymes were identified in several published Frankia genomes (Figure 1). Examining the transcriptome of Frankia alni ACN14a in (+)N culture (Alloisio et al., 2010), we found that three restriction enzyme genes were highly expressed: one type I enzyme (FRAAL4992, 92nd percentile), one type II enzyme (FRAAL0249, 91st percentile) and one type IV enzyme (FRAAL3325, 85th percentile) (Figure 1). The type IV enzyme was annotated in REBASE as a “Type IV Methyl-directed restriction enzyme” of the Mrr methyladenine-targeting family. Other Frankia genomes contained likely homologs of this type IV enzyme as well. The genome of Frankia casuarinae CcI3 contained five type IV enzymes, the most of any Frankia genome examined (Figure 1). In the Frankia sp. CcI3 transcriptome, type IV Mrr enzymes were very highly expressed, up to the 96th percentile. One of the five enzymes in CcI3 (Francci3_2839) was annotated as a Mcr type IV enzyme, which targets 5-methylcytosine instead (REBASE) and its expression was very low, in only the 18th percentile of genes in the transcriptome. Of the complete genomes examined, only Candidatus Frankia datiscae Dg1 did not contain any putative type IV restriction genes.
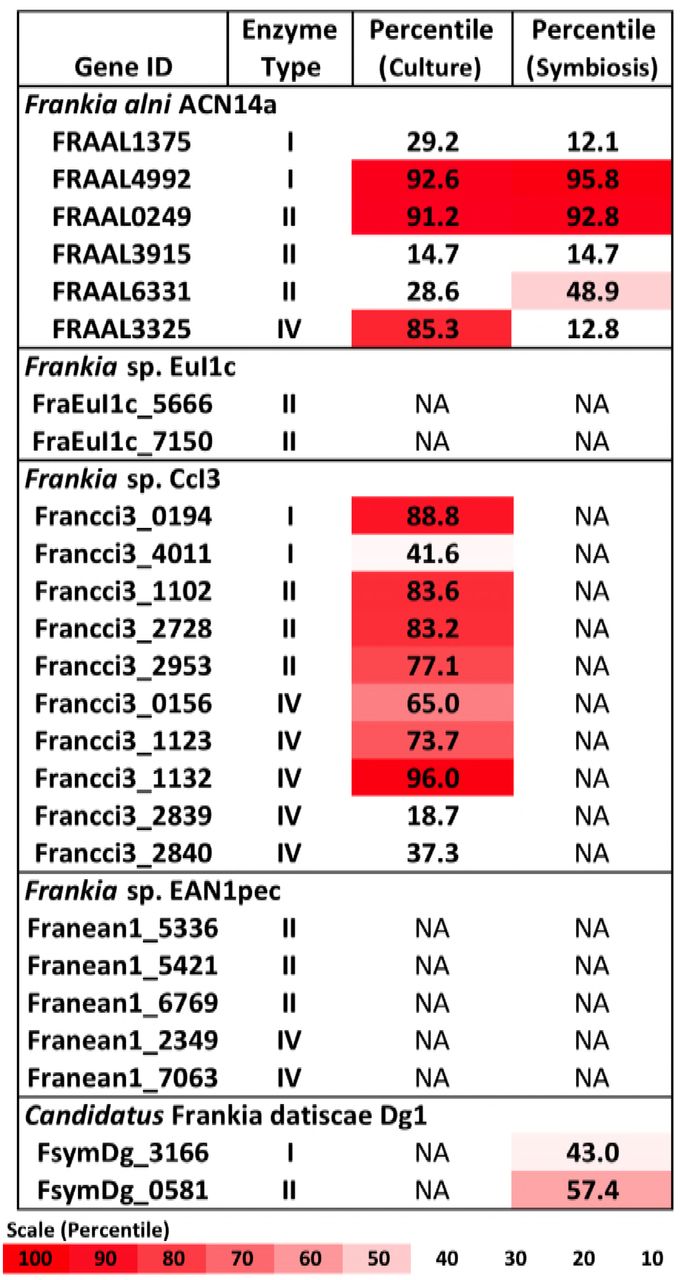
Restriction enzymes annotated in the Frankia genomes and expression levels of each gene in the transcriptome in culture when available (Alloisio et al., 2010 and Bickhart and Benson, 2011). NA: Transcriptome not available.
A symbiotic transcriptome of F. alni ACN14a from root nodules is available in addition to transcriptomes of free-living culture (Alloisio et al., 2010). In symbiosis with Alnus glutinosa the mrr homolog was down-regulated relative to the rest of the transcriptome, from the 85th percentile in culture to an expression level at maximum no higher than 12 percent of transcriptome genes, while the other restriction enzymes identified in the F. alni genome were not down-regulated in symbiosis (Figure 1). In the other Frankia sp. with a symbiotic transcriptome available, the uncultured cluster II species Frankia sp. Dg1 (Persson et al., 2015), expression of its two restriction enzymes, neither of which was a Type IV enzyme, occurred at very low levels (Figure 1).
Of the actinobacteria outside of genus Frankia with published transcriptomes in pure culture, Mycobacterium smegmatis, Streptomyces avermitilis, and Rhodococcus jostii also highly expressed type IV restriction enzyme genes, in the upper-70th percentiles of their respective transcriptomes (Figure 2). In comparison, the transcriptomes of proteobacteria and firmicutes examined expressed their corresponding mrr genes around the 50th or 60th percentiles. One notable exception was the actinobacterium Mycobacterium tuberculosis, which expressed its single gene for a methyladenine-directed restriction enzyme (Mrr) at extremely low levels, around the 6th percentile in culture.
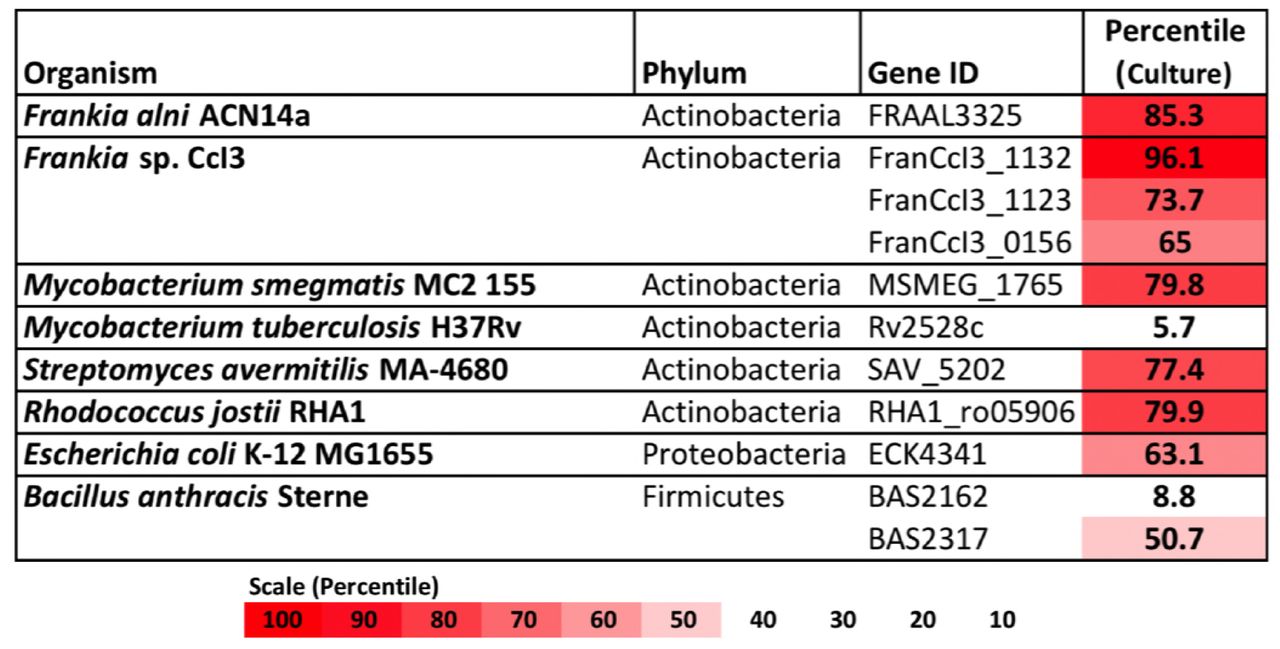
Restriction enzymes annotated in the genomes of Frankia, other actinobacteria, and proteobacteria and firmicutes for comparison. For each, expression levels for available transcriptomes growing in pure culture are provided.
Genetic Transformation of Frankia alni ACN14a with an Unmethylated Replicating Plasmid
After electroporation F. alni cells formed visible hyphae in culture after approximately ten days (data not shown). When sub-cultured into chloramphenicol-selective media F. alni cultures transformed with unmethylated plasmid pSA3 were able to grow, whereas those transformed with methylated plasmid were not. When imaged under 488nm wavelength of excitation in the confocal microscope, green fluorescence typical of GFP was observed in hyphae as shown in Figure 3. Wild-type F. alni hyphae displayed autofluorescence around 575nm (data not shown), as observed by Hahn et al. (1993), but no fluorescence was observed in the 500-550nm range (Figure 3).

Transformation of Frankia alni ACN14a with a plasmid expressing egfp. Top: Wild-type F. alni ACN14a control. Bottom: Transformed F. alni ACN14a with plasmid pIGSAF. Images were obtained on a confocal microscope with both brightfield and epifluorescence and then overlaid.
Differential Regulation of egfp Under the Control of the Frankia alni ACN14a nif Cluster Promoter
When grown in (–)N culture, transformants carrying the pIGSAFnif plasmid showed significant up-regulation of the egfp gene conjugated to the nif cluster promoter, approximately 100-fold relative to (+)N culture (Table 1). The nifH gene, used as a positive control for nitrogen fixation, was also significantly up-regulated approximately 9-fold in (–)N media compared with (+)N cultures. Thus, the up-regulation of the egfp gene is consistent with the previous report that plasmid pSA3 is maintained at approximately 10 copies per cell (Dao and Ferretti, 1985). Expression of the rpoD housekeeping gene, used as a negative control, was not significantly different between (+)N and (–)N cultures.
qPCR verification of differential regulation of egfp in (+)N and (–)N media. qPCR tested fold-change of egfp as well as nifH as a nitrogen-fixation positive control and rpoD as a housekeeping negative control.
F. alni containing pIGSAFnif grown in (–)N media fluoresced predominantly in the vesicles (Figure 4). Little to no fluorescence was observed in hyphae. Fluorescence in the vesicles was present both in the spherical portion as well as in the stalk connecting the vesicle to the hyphae. No fluorescence was observed in hyphae or vesicles of wild-type F. alni grown in (–)N media.

Transformation of Frankia alni ACN14a with a plasmid expressing egfp under the control of the nif cluster promoter region from the F. alni genome. Transformed cultures were grown in both (+)N and (–)N media to compare fluorescence with and without nitrogen fixation. Top: F. alni ACN14a transformed with plasmid pIGSAFnif grown in (+)N media. Middle: F. alni ACN14a with plasmid pIGSAFnif grown in (–)N media. Bottom: Close-up of a vesicle showing fluorescence in the stalk. Images were obtained on a confocal microscope with both brightfield and epifluorescence and then overlaid.
Stability of Plasmid pIGSAF in F. alni ACN14a in the Absence of Selection
In cultures grown without chloramphenicol selection, qPCR analysis did not find a significant difference in the amount of plasmid relative to genomic DNA after one, two, or three rounds of sub-culturing (Figure 5). Only after the fourth round of sub-culturing was a significant decrease from the initial plasmid concentration detected by qPCR (Figure 5), however, fluorescence throughout the hyphae was still observed even after four weeks without selection (Figure 6).

Maintenance of plasmid pIGSAF in Frankia alni in culture without chloramphenicol selection over the course of four weeks. Each week the culture was sub-cultured into fresh media and genomic DNA was extracted to measure relative plasmid concentrations via qPCR. Groups ‘a’ and ‘b’ indicate time points that are not significantly different from each other (p<.05).

Fluorescence of a Frankia alni hyphal colony transformed with plasmid pIGSAF grown with chloramphicol selection (top) and without selection for four weeks (bottom).
Discussion
Genetic Transformation of Frankia alni ACN14a with an Unmethylated Replicating Plasmid
We have shown that F. alni can be stably transformed with an unmethylated plasmid introduced by electroporation. A homolog of an Mrr Type IV methyladenine-targeting restriction enzyme was highly expressed in F. alni in culture (Figure 1), suggesting that DNA with methylated adenine bases would be degraded in this organism. Actinobacteria, especially Frankia, expressed Type IV methyl-directed restriction enzyme genes more highly than proteobacteria and firmicutes (Figure 2), a finding that correlates with previous reports of higher transformation efficiencies with unmethylated plasmids in Corynebacterium (Ankri et al., 1996) and Streptomyces spp. (Molle et al., 1999). Genomes of the majority of actinobacteria are missing homologs of the dam methyltransferase gene (Sachez-Romero et al., 2015) whose product is used to mark parent DNA strands during replication, and also are missing mutS and mutL that form a complex for the removal and repair of mismatched bases on the daughter strand determined by the methylation of adenine residues (Sachadyn, 2010). Together, these factors suggest a difference in preference for unmethylated over methylated DNA among most of the actinobacteria relative to other bacterial phyla.
Type IV restriction enzymes have been suggested to have evolved as a counter to phage methylation systems that themselves evolved to evade host restriction systems by methylating restriction sites (Westra et al., 2012). Phage genomes adopt the methylation patterns of their previous host (Loenen and Raleigh, 2014) thus increasing the likelihood of digestion by actinobacterial enzymes if replicated in a dam+ host. The expression of type IV restriction enzymes in actinobacteria therefore could represent an adaptation to prevent infection by DNA phages based on the methylation state of their genomes. Differences in methylation patterns between actinobacteria and other bacterial phyla (Novella et al., 1996) potentially constitute a barrier to horizontal gene transfer between these groups, including phage-mediated gene transfer. Of particular interest to the evolution of root nodule symbioses is the transfer of genes between Frankia and the rhizobia and vice versa. It has been suggested that the nodA gene involved in Nod factor synthesis evolved in the actinobacteria, including some Frankia, and was then horizontally transferred to the rhizobia (Persson et al., 2015). With type IV restriction enzymes creating a barrier to horizontal transfer into actinobacteria from dam+ bacteria including proteobacteria, this direction would be more likely than the reverse.
However, F. alni was observed to down-regulate its type IV mrr gene substantially in symbiosis (Figure 1). As roots contain much lower concentrations of bacteriophage than the surrounding soil (Ward and Mahler, 1982) this likely represents a decreased necessity for restriction enzymes as a defense mechanism during symbiosis. A potential side-effect of this down-regulation, however, is that the barrier to horizontal transfer posed by type IV enzymes is likely lowered during symbiosis, promoting horizontal transfer to Frankia from other endophytic bacteria.
M. tuberculosis showed much lower transcription of its annotated type IV methyladenine targeting restriction enzyme than other actinobacteria. M. tuberculosis expresses an adenine methyltransferase in hypoxic conditions that regulates the expression of genes likely involved with survival during macrophage infection (Shell et al., 2013). For this reason it is likely that M. tuberculosis responds to methylated DNA differently than other actinobacteria, indeed electrotransformation of M. tuberculosis can be readily achieved with methylated plasmids replicated in E. coli DH5α (Pelicic et al., 1997), suggesting that methylated DNA is not digested in M. tuberculosis.
In this study plasmid pSA3 and its derivatives were capable of replication in F. alni. This shows that the broad host-range origin is capable of replication in Frankia and supports its use as a vector for the manipulation of Frankia spp. The parent plasmid of pSA3, pIP501, replicates in a very broad range of bacteria including Streptomyces lividans and E. coli (Kurenbach et al., 2003) indicating the potential for transformation of additional actinobacteria with these plasmids.
Differential Regulation of egfp Under the Control of the Frankia alni ACN14a nif Cluster Promoter
The expression of the egfp gene of plasmid pIGSAFnif was up-regulated in (–)N media compared with expression in (+)N media, at proportional levels similar to the expression of the nifH nitrogenase gene (Table 1), demonstrating for the first time that expression of reporter genes can be manipulated in Frankia. This transformation system resulted in the ability to visualize the expression of nitrogen-fixation genes in vitro by fluorescence microscopy (Figure 4). Interestingly, fluorescence was detected in both the spherical portion of the vesicle as well as in the stalk that connects to the hyphae, suggesting that nitrogen fixation genes are expressed in both parts of the vesicle. Previous studies have shown that the vesicle envelope is deposited around the stalk as well (Lancelle et al., 1985), supporting the observation that nitrogen fixation occurs in the stalk.
Although the fluorescence observed when egfp was expressed under the control of the nif cluster promoter was predominantly in the vesicles, some fluorescence was occasionally observed in hyphae under nitrogen-fixing conditions, whereas in (+)N media there was no observable fluorescence (Figure 4). This suggests that there is some expression of nif genes in the hyphae as well as the vesicles induced by nitrogen limitation. Frankia spp. in symbiosis with members of the Casuarinaceae have been reported to fix nitrogen in hyphae, since no vesicles are differentiated (Murry et al., 1985); this correlated with the formation of a lignified host cell wall in the symbiotic tissue that likely excludes oxygen (Berg and McDowell, 1987). In liquid culture, there may be zones of low pO2 that develop in portions of the Frankia hyphal colony where nitrogen fixation could be induced.
Frankia spp. in symbiosis have been suggested to be more autonomous than rhizobial microsymbionts due to their ability to control the flow of oxygen with the formation of vesicles as well as the expression of more metabolic pathways in the microsymbiont. These factors allow Frankia to be more metabolically independent from their hosts (Alloisio et al., 2010; Berry et al., 2011). As an example, both Frankia and the rhizobia share a second glutamine synthetase gene, named glnII, which is not present in most other bacteria (Ghoshroy et al., 2010). glnII is up-regulated in F. alni in symbiosis relative to nitrogen-replete culture (Alloisio et al., 2010) but not differentially expressed or required for effective nodule formation by rhizobia such as Sinorhizobium meliloti (Becker et al., 2004; de Bruijn et al., 1989). The development of genetic tools for the manipulation of Frankia will allow further experimentation into these and other distinctive molecular aspects of actinorhizal symbioses, which will, in turn, inform analyses of the evolution of root nodule symbiosis.
Future Directions
Even in the absence of selection plasmid pIGSAF was found to be stable in F. alni cultures for at least three weeks (Figure 5) and cultures continued to fluoresce after at least four weeks (Figure 6). Three to four weeks is sufficient to nodulate F. alni hosts (Alloisio et al., 2010), suggesting that transformants can be used to inoculate plants in studies of the role of Frankia and its interactions with hosts during nodule establishment and symbiosis. In future this system could be modified using recombination or viral integrases to anchor genes within the genome. Differential regulation of reporter genes such as egfp can be used to localize the expression of genes identified by genomics and transcriptomics in specific Frankia cell types, in different growth conditions, and in symbiosis. Replicating plasmids may also enable the study of gene function by constitutive expression of selected genomic genes, by promoter switching or by knock-down experiments expressing anti-RNAs to genes of interest (Gillaspie et al., 2009). Circumventing the natural restriction systems of Frankia will also increase the transformation rate of non-replicating plasmids and enable higher efficiency recombination for gene knock-out experiments as attempted by Kucho et al. (2009).
Materials and Methods
Restriction Enzyme Analysis
Genes annotated as restriction enzymes (Restriction enzyme types I, II, III, or IV) present in the annotations of all completely sequenced actinobacterial genomes and their annotations were downloaded from the REBASE database (Roberts et al., 2010). Bacterial transcriptomes used in this study were downloaded from the NCBI GEO database (Supplementary Table 1). Transcriptomes were chosen based on the following criteria: 1) transcriptomes were made from pure cultures in log-phase growth, 2) organism did not have genetic manipulations including mutations or exogenous plasmids, and 3) cultures did not have additional experimental compounds added including antibiotics or complex carbon sources. For comparisons between transcriptomes, expression levels for each gene were calculated as the percent of genes with lower expression than the gene of interest in each transcriptome.
Culture Conditions
E. coli strains DH5α and GM48 (dam- and dcm-) were grown in 50ml Difco 1.5% (w/v) Luria Broth (LB) (Catalog #241420, Becton Dickinson, Franklin Lakes, NJ), pH 6.8, in 250mL flasks at 37°C with shaking at 150 rpm overnight. Plates were made with 1.5% (w/v) Bacto Agar (Becton Dickinson, Franklin Lakes, NJ, catalog #214010) in 1.5% LB, and incubated overnight at 37°C. All media were first sterilized by autoclaving for 30 minutes at 121°C.
Frankia alni ACN14a (Normand and Lalonde, 1982) was cultured in 5ml liquid BAPP media modified from Murry et al., 1984 by the addition of 5mM pyruvate and 5mM MOPS, adjusted to pH 6.7, in sterile 25ml glass test tubes with tetracycline added to a final concentration of 10µg/ml to prevent contamination. (+)N media included 5mM ammonium chloride as a nitrogen source whereas (–)N media had no added nitrogen source. For sub-culturing, hyphae were collected in sterile 2ml microcentrifuge tubes, centrifuged at 10,000rpm for ten minutes in a tabletop microcentrifuge (model #5424, Eppendorf, Hamburg, Germany), and re-suspended in 1mL fresh BAPP media. Cultures were then homogenized by passage through a 21G needle six times, then homogenate equivalent to 50ul packed-cell volume as measured after centrifugation was added to 4ml of fresh media and incubated at 28°C without shaking. Cultures were routinely sub-cultured once per week. For selection of transformants, chloramphenicol was added to a final concentration of 25µg/ml.
Frankia Transformation
F. alni cells were grown in culture for one week prior to transformation. Two milliliters of growth media with hyphae was then pelleted as above and the pellet was re-suspended in 500µL of ice-cold deionized (DI) water that had been sterilized by autoclaving. This was repeated two more times, but on the third round of centrifugation the pellet was re-suspended in 300µL ice-cold sterile 10% glycerol instead. Hyphae in the cell suspension were then homogenized by passage through a 21G needle twice.
300µL of the F. alni cell suspension was pipetted into an electroporation cuvette with a 2mm gap (Molecular BioProducts Catalog #5520, San Diego, CA) and mixed with 10µg of plasmid DNA. The cuvette was then incubated on ice for five minutes. Electroporation was carried out in a Bio-Rad Gene Pulser™ with Pulse Controller at 2.5kV, 200Ω resistance, and 25µF of capacitance. The cuvette was then immediately filled with 1ml of ice-cold BAPP media. The cuvette was sealed with Parafilm® (Pechiney Plastic Packaging, Menasha, WI) and incubated overnight without shaking at 28°C.
The following day the F. alni culture was removed from the cuvette and added to 3.5ml of sterile BAPP media with tetracycline as above, in a glass test tube. The culture was incubated at 28°C without shaking until visible hyphae were observed (approximately 10 days after electroporation). At this point F. alni hyphae were sub-cultured into fresh media as above and then once more one week later. The following week (two weeks after visible hyphae were observed) the hyphae were sub-cultured again into BAPP media, this time with chloramphenicol added for selection to a final concentration of 25µg/ml. Chloramphenicol was chosen as the selective antibiotic because all Frankia strains tested in a study by Tisa et al. (1998) were susceptible to it. This process was repeated the following week for an additional round of selection.
DNA Extraction
Plasmids were purified from E. coli using a QIAprep® Spin Miniprep Kit (Catalog #27106, Qiagen). Two milliliters of overnight cultures were pelleted at 13,000rpm for five minutes and used for extraction. Plasmids were resuspended in EB buffer (Qiagen) and quantified on a NanoDrop Microvolume Spectrophotometer (ThermoFisher). Plasmids were further purified by running on a 0.7% agarose gel and extracted with a Zymoclean Gel DNA Recovery Kit (Catalog #11-300, Genesee Scientific, San Diego, CA). To synthesize unmethylated plasmids, methylated plasmids were first extracted from E. coli DH5α and transformed into E. coli GM48 by heat shock in CaCl2 (Sambrook et al., 1989) then cultured and re-extracted as above.
F. alni DNA extraction was carried out with a CTAB protocol (Feil et al., 2012). Briefly, cells were pelleted at 10,000rpm for five minutes in a tabletop centrifuge. Cells were then re-suspended in TE buffer and lysed with lysozyme, SDS, Proteinase K, and CTAB. DNA was extracted with 24:1 chloroform:isoamyl alcohol, and 25:24:1 phenol:chloroform:isoamyl alcohol, washed with isopropanol followed by ethanol, resuspended in 50ul TE buffer, and quantified by NanoDrop.
Plasmid Synthesis
A plasmid designated as pIGSAF was synthesized by ligating a PCR-amplified fragment containing the egfp gene and promoter from plasmid pDiGc (Helaine et al., 2010, Addgene, Cambridge, MA) to plasmid pSA3 (Dao and Ferretti, 1985). Plasmid pSA3 is a broad host-range replicating plasmid developed as a shuttle vector from pIP501. The pIP501 origin of replication has been shown to replicate in bacteria of diverse phyla including Firmicutes, from which it was originally isolated (Horodniceanu et al., 1976), as well as Actinobacteria and Proteobacteria (Kurenbach et al., 2003). Plasmid pSA3 contains chloramphenicol and tetracycline resistance genes and an E. coli-specific origin of replication for propagation (Dao and Ferretti, 1985).
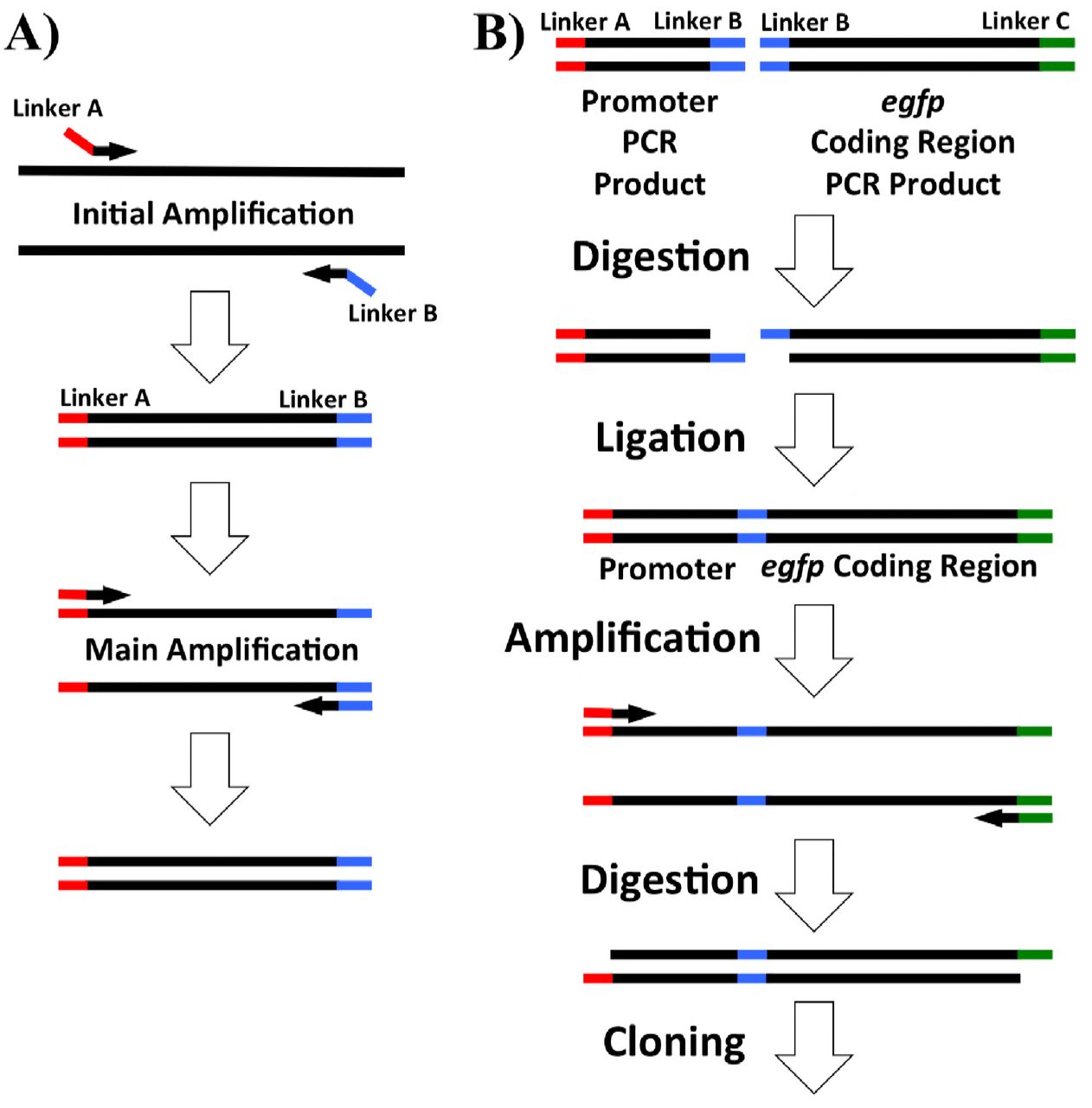
Primers for PCR, listed in Table 2, were designed using NCBI Primer-BLAST with default settings (Ye et al., 2012). For cloning, primers were designed with linkers adding target restriction sites onto their 5’ ends (Table 2) as well as 4-6 additional bases to aid in restriction digestion of the ends. A diagram of the addition of linkers by PCR is shown in Figure 7A. For the synthesis of pIGSAF, primers were designed targeting the egfp coding region as well as the promoter region 200 base pairs upstream. PCR was performed in a Bio-Rad S1000 Thermal Cycler (Bio-Rad, Hercules, CA) using a Qiagen Taq PCR kit (catalog #201223). Products were synthesized as in Figure 7A, first amplified for 10 cycles using the annealing temperature of the region corresponding to the binding site on the target DNA and then an additional 30 cycles using the annealing temperature of the full primer including the linker (Table 2).
Primers designed for this study. Restriction sites added to primers by linker PCR for use in cloning are bolded in the primer sequence and annealing temperatures are listed both for the initial reaction (“Without Linker”) and the main amplification phase of the PCR (“Full Sequence”).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
PCR amplification and digestion and ligation overview for plasmid synthesis used in this study. Linkers including restriction sites are colored red, blue, or green. A) PCR products for cloning were amplified by amplification at a lower initial temperature to allow the incorporation of additional restriction sites on the 5’ end of the primers (shown in red and blue). The annealing temperature was then increased to amplify full-length products with the added restriction sites. B) Addition of the F. alni ACN14a nif cluster promoter region to the egfp coding sequence. Both the promoter region and egfp coding sequence were amplified with EcoRI sites (blue). These were then digested and ligated together followed by amplification of the ligation product. Restriction sites on the ends (red and green) were digested, allowing incorporation of the nif promoter:egfp product into the digested plasmid.
The egfp fragment was synthesized with SalI and BamHI restriction sites on the 5’ and 3’ ends, respectively, with primers EGFP_SalI_F and EGFP_BamHI_R. The PCR product and plasmid pSA3 were then digested with both enzymes. The fragments were purified on an agarose gel as above and then mixed together, denatured by heating for five minutes at 65°C, and then ligated together by incubation with T4 ligase (catalog #M0202S, Qiagen) at 16°C overnight. The resulting ligation was transformed into E. coli DH5α by heat shock (Sambrook et al., 1989). Transformants were selected on LB plates with chloramphenicol at a final concentration of 25µg/ml. Transformed colonies were inoculated into liquid LB media with chloramphenicol and cultured as above. Plasmid pIGSAF was then re-extracted from transformed E. coli and its composition was confirmed by digestion with SalI and BamHI as above.
The presence of plasmids pSA3 and pIGSAF electroporated into F. alni cultures was confirmed with PCR. Genomic DNA was extracted and amplified with primers pairs pSA3_Cm_F/pSA3_CmR for the chloramphenicol resistance gene of plasmid pSA3 and GFP_qPCR_F/GFP_qPCR_R for the egfp gene of plasmid pIGSAF. PCR products were separated on an agarose gel and extracted as above, and sequenced by the UCDNA Sequencing Facility (Davis, CA). Sequences were compared by BLAST against gene sequences obtained from the original plasmids (Supplementary Table 2).
For the differential expression of egfp by nitrogen limitation under the control of the nif cluster promoter region of F. alni ACN14a, plasmid pIGSAFnif was synthesized. Ligation of the nif cluster promoter region to the coding region of egfp was carried out as outlined in Figure 7B. The egfp coding region of pDiGc was amplified without the upstream promoter region using primers GFP_CDS_EcoRI_F and GFP_CDS_SalI_R (Table 2). An EcoRI restriction site was added before the start codon and a SalI site was added 200 bases downstream of the stop codon. Separately, the 300 bases upstream of the nif nitrogenase cluster in the F. alni ACN14a genome were amplified with an XbaI site upstream and an EcoRI site downstream using primers nif_promoter_XbaI_F and nif_promoter_EcoRI_R (Table 2). These two PCR products were digested with EcoRI (catalog #R0101S, New England Biolabs) and ligated together. The ligation product was then re-amplified by PCR using the nif promoter forward primer and the egfp reverse primer. The amplified ligation product and plasmid pSA3 were then each digested with SalI and XbaI, ligated together, transformed, and selected on chloramphenicol as above.
qPCR Verification of Differential Regulation
Quantifications of gene expression and fold-changes were obtained based on qPCR amplification. Cultures of transformed cells were grown in 4.0 ml (+)N and (–)N media in sterile six-well plates (catalog #353046, Corning Inc, Corning, NY) with shaking at 50rpm at 28°C. After five days, RNA was extracted by bead-beating, following a protocol adapted from Dietrich et al. (2000): F. alni hyphae were pelleted at 9000rpm for fifteen minutes, resuspended in 1050ul Buffer RLT (Qiagen), and transferred to 2ml tubes containing Lysing Matrix B (catalog #6911-100, MP Biomedicals, Burlingame, CA). Samples were processed with a FastPrep FP120 (Thermo Fisher Scientific, Waltham, MA) for 45 seconds at setting 6.5, then placed on ice for 45 seconds. The processing step was then repeated twice more with cooling on ice between each step. Supernatants were then transferred to Qiagen RNeasy spin columns and purified with a Qiagen RNeasy Mini Kit (catalog #74104). RNA was eluted in RNase-free water and then contaminating DNA was digested with an Invitrogen TURBO DNA- free Kit (catalog #AM1907, Waltham, MA). Finally, cDNA was synthesized with an Invitrogen Superscript III Kit (catalog #18080051) with random hexamer primers. qPCR was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) with Fast SYBR Green qPCR Master Mix (catalog #4385612, Applied Biosystems) and primer pairs rpoD_qPCR_F/rpoD_qPCR_R, nifH_qpCR_F/nifH_qpCR_R, infC_qPCR_F/infC_qPCR_R, and gfp_qPCR_F/gfp_qPCR_R (Table 1). egfp and nifH were used as experimental targets to confirm differential regulation of egfp by nitrogen limitation; housekeeping gene infC was used for normalization as in Alloisio et al. (2010); and sigma factor gene rpoD was used as a negative control.
Confocal Microscopy
For visualization of hyphae and GFP fluorescence, Frankia cells were grown either in (+)N or (–)N BAPP medium as above and immobilized on glass slides with a drop of 3% molten agarose solution: 1.5g molecular biology-grade agarose (catalog #A9539, Sigma, St. Louis, MO) was dissolved in 50ml DI H2O and kept warm in a water bath at 50°C. Slides were pre-heated on a slide warmer (Fisher) at 50°C. Then 15ul of Frankia hyphae were pipetted onto each slide and covered with 35ul of 3% molten agarose. A #1.5 coverslip was added to the Frankia cells in agarose, which were allowed to cool to room temperature. The Frankia preparations were visualized on a Leica TCS SP8 STED 3X confocal microscope with a 100X oil-immersion objective and a HyD detector. For fluorescence imaging, samples were excited with 488nm light. The emission wavelengths were collected from 500-550nm. Images were stored as. lif files from Leica LAS X and then viewed in FIJI (Schindelin et al., 2012). To visualize hyphal three-dimensional structure, Z-stack images were taken and then combined using the highest fluorescent intensity of each pixel (FIJI MAX setting).
Plasmid Stability
In order to test the persistence of a plasmid in transformed F. alni, cultures containing plasmid pIGSAF were grown without selection in non-selective media lacking chloramphenicol. The cells were first pelleted and re-suspended in fresh BAPP media, then sub-cultured as above without the addition of chloramphenicol to the media. These cultures were grown in six-well plates as above for one week. At that time each culture was pelleted and re-suspended in 500ul fresh BAPP media. This suspension was homogenized by passage through a 21G needle twice. 250ul of the homogenate was transferred to fresh BAPP media without chloramphenicol and the remaining 250ul was used for total genomic DNA extraction as above. This process was repeated once per week for four weeks. The amount of plasmid in each sample was quantified by qPCR, performed in duplicate for each of three biological replicates, using egfp primers GFP_qPCR_F and GFP_qPCR_R (Table 2). Relative fold-change of plasmid between each time point was calculated using the ΔΔCt method (Lee et al., 2006) with the infC gene as a control to normalize the amount of DNA in each sample (amplified with primers infC_qPCR_F and infC_qPCR_R, Table 1). To determine significant changes in plasmid abundance, two-tailed Welch’s t-tests were performed in R on normalized ΔCt values calculated (p<.05). Cultures grown with selection and without selection for four weeks were also imaged with fluorescence as above.
Supplementary Figures and Tables
Supplementary Table 1 List of genomes and transcriptomes used in this study. Transcriptomes are listed with their GEO accession number and original reference.
Supplementary Table 2 Sequences from PCR products of Frankia alni transformants corresponding to the chloramphenicol resistance gene (camr) from pSA3 and egfp gene of plasmid plasmid pIGSAF and pDiGc.
Acknowledgements
We thank Dr. Rebecca Parales for providing plasmid pSA3 and Dr. Wolf Heyer for E. coli strain GM48. We also thank Dr. Ingrid Brust-Mascher for technical assistance with confocal microscopy. IG was supported by a UC Davis Department of Plant Sciences graduate student research fellowship. Research funding for this project was provided by UC Davis Henry A. Jastro Research Grants to IG, and by USDA-NIFA-CA-D-PLS-2173-H (AB).
References




