

Abstract
Epigenetic modifiers often harbor loss-of-function mutations in lung cancer, but their tumor-suppressive roles are poorly characterized. Here we show that lung-specific loss of the gene encoding the histone methyltransferase MLL4 (alias KMT2D; a COMPASS-like enzyme), which is ranked the most highly inactivated epigenetic modifier in lung cancer, strongly promotes lung adenocarcinoma in mice. Mll4 loss upregulated tumor-promoting programs, including glycolysis. The pharmacological inhibition of glycolysis preferentially impeded tumorigenic growth of human lung cancer cells bearing MLL4-inactivating mutations. Mll4 loss widely impaired epigenomic signals for super-enhancers and enhancers, including the super-enhancer for the circadian rhythm repressor gene Per2, and decreased Per2 expression. Per2 downregulated several glycolytic pathway genes. These findings uncover a distinct tumor-suppressive epigenetic mechanism in which MLL4 enhances Per2-mediated repression of pro-tumorigenic glycolytic genes via super-enhancer activation to suppress lung adenocarcinoma tumorigenesis and also implicate a glycolysis-targeting strategy as a therapeutic intervention for the treatment of MLL4- mutant lung cancer.
Introduction
Lung cancer is the leading cause of cancer death in men and women. Lung cancer patients suffer from a low overall 5-year survival rate (∼18.1%). Lung cancer is often characterized by gain-of-function mutations and gene amplification of oncogenic kinases (e.g. K-RAS and EGFR) as well as loss-of-function alterations in tumor suppressor genes (e.g. TP53 and LKB1/STK11) 1-3. Much research for lung cancer has focused on kinase signaling pathways, leading to the development of the kinase-targeted therapies, such as EGFR mutant inhibitors and the ALK inhibitors. However, a vast majority of lung cancer patients, who are treated with the kinase-targeted therapies, later undergo tumor relapse and drug resistance 3, 4. Recently, the use of immune checkpoint inhibitors (e.g., anti-PD1 and anti-PD-L1) has provided significant survival benefit for lung cancer patients with high PD-L1. However, the overall survival rate of lung cancer patients still remains low 5, 6. Moreover, a majority of lung cancer patients do not have a well-defined drug target 6, 7. For these reasons, there is a great need for a new mechanistic understanding of lung cancer to be used for a therapeutic approach for lung cancer treatment.
Epigenetic alterations, which represent heritable aberrations in gene expression or cellular phenotype without alterations of DNA sequences, have emerged as a major type of cancer-driving events 8. Covalent modifications of DNA and histones play a key role in epigenetic regulation of gene expression and include DNA methylation, histone acetylation, and histone methylation. Interestingly, histone methylation, which can occur at lysine and arginine residues in histones, is linked to either gene activation or silencing, depending on the methylation residues within histones 9. Notably, histone methylation modifiers and other epigenetic modifiers are often mutated in lung tumors, including lung adenocarcinoma (LUAD) and lung squamous cell carcinoma (LUSC) responsible for 40%–50% and 25%–30% of lung cancer, respectively 1, 2, 10-12. In fact, a substantial portion of such mutations results in the loss-of-function, suggesting tumor-suppressive functions of epigenetic modifiers. However, their tumor-suppressive roles in lung cancer are largely unknown. In the present study, our results show that the histone methyltransferase MLL4 (a COMPASS-like enzyme13; also called KMT2D, MLL2, and ALR), which is ranked the most highly inactivated epigenetic modifier in lung tumors, is a key lung tumor suppressor against LUAD. Our findings uncover the molecular mechanism by which the epigenetic modifier MLL4 suppresses LUAD and suggest a mechanism-based therapeutic strategy for treatment of LUAD bearing low MLL4 levels or MLL4’s loss-of-function mutations.
Results
Lung-specific loss of Mll4 strongly promotes K-RasG12D-induced LUAD in mice
To search an epigenetic modifier with tumor-suppressive function in lung cancer, we examined which epigenetic modifier highly undergoes genomic alterations related to loss-of-function mutations (i.e., truncations, missense mutations, and insertions/deletions). Our analysis of Pan-lung cancer data (LUAD and LUSC) in the TCGA database showed that the gene encoding MLL4 harbored such genomic alterations in about 14% of lung tumor samples (Fig. 1a; Supplementary Fig. S1a, b). Although the mutation frequencies of MLL3/KMT2C (16%) were similar to that of MLL4, MLL4 was ranked higher because truncating mutations were more frequent in MLL4 than in KMT2C (Fig. 1a). It should be noted that most of the MLL4 truncations result in loss-of-functions because the catalytic SET domain (5397– 5513 aa) is located at the C-terminus of MLL4. Interestingly, MLL4 was altered in substantial portions of both LUAD and LUSC samples (Supplementary Fig. S2a, b). Because these results indicate that MLL4 is a candidate lung tumor suppressor with high genomic alterations, we chose to characterize the role of MLL4 in lung cancer in subsequent analyses.

(a) Comparison of alterations of epigenetic modifiers (histone acetyltransferases and deacetylases, histone methyltransferases and demethylases, and DNA modifiers) in TCGA Pan-lung cancer. Other mutations represent missense mutations and inframe mutations. (b) Representative micro-CT (top panels) and H&E (middle and bottom panels) images of wild-type (WT), Mll4fl/fl, p53fl/fl, p53fl/fl;Mll4fl/+, p53fl/fl;Mll4fl/fl. K-RasLSL-G12D, and K-RasLSL-G12D;Mll4fl/fl mice. The lungs in mice were infected with Ad5-CMV-Cre viruses. (c) Comparison of tumor area (%) per mice in the indicated groups of mice (n = 6). (d) IHC analysis for the cell proliferation marker Ki-67 in K-Ras and K-Ras:Mll4-/- lung tumors. Ki-67-positive cells in ten random fields of three different tumors of K-Ras and K-Ras:Mll4-/- groups were quantified. (e) Kaplan-Meier survival analysis of in the indicated groups of mice. (f) Kaplan-Meier survival analysis for MLL4 mRNA levels (probe set, 227527_at) in LUAD patient samples using KM Plotter database (http://kmplot.com/analysis). Lower quartile was used as a cutoff to divide the samples into low and high groups. Scale bars in b, 2 mm (middle panels) and 200 µm (bottom panels). *p < 0.05 and **p < 0.01.
MLL4 alterations frequently co-occur with p53 and K-RAS mutations in lung cancer. For example, about 71% of MLL4 mutations co-occur with p53 alterations in lung tumors, and about 22% of MLL4 mutations coincide with K-RAS mutation in LUAD (www.cbioportal.org) (Supplementary Fig. S3a). Therefore, we sought to determine whether lung-specific loss of Mll4 cooperates with p53 inactivation or K-Ras activation for lung tumorigenesis. We first generated several genetically engineered mouse models (GEMMs), including p53fl/fl;Mll4fl/fl and K-RasLSL-G12D;Mll4fl/fl (Supplementary Fig. S3b–d). We then induced Cre-mediated deletion of loxP sites by infecting the lungs of 6–10 weeks old GEMM mice with Adeno-Cre viruses via the commonly used intra-tracheal intubation. Lung-specific single loss or co-loss of Mll4 and p53 by Adeno-Cre-mediated deletion neither induced any detectable tumor in mouse lungs for up to 16 months post-infection nor changed survival times of mice (Fig. 1b,c; Supplementary Table S1). This may be partly because p53 loss alone rarely induces lung tumors in mice 14-16. For K-Ras-induced lung tumorigenesis, a K-Ras LSL-G12D model (hereinafter referred to as K-Ras model) was used because it is a well-established GEMM for LUAD 17. K-Ras activation was induced by lung-specific deletion of the LSL (loxP-STOP-loxP) cassette via Adeno-Cre virus infection, as previously described 18. We monitored potential tumor formation for up to 130 days post-infection at 6–8 week intervals using a micro CT-scan.
Our micro-CT and H&E data showed that lung-specific loss of Mll4 promoted K-RasG12D-induced lung tumorigenesis (Fig. 1b, c). Specifically, our H&E data demonstrated that a higher percentage of the pulmonary parenchyma was effaced by tumors in the K-Ras;Mll4-/- group of mice than in the K-Ras group of mice (Fig. 1c; Supplementary Fig. S4a, b). Consistent with tumorigenicity enhanced by Mll4 loss, immunohistochemisty (IHC) analysis showed that levels of the cell proliferation marker Ki-67 were increased by Mll4 loss (Fig. 1d). Similar to K-Ras lung tumors, K-Ras:Mll4-/- lung tumors were positive for the well-known LUAD marker TTF-1 (alias NKX2.1), indicating their LUAD characteristics (Supplementary Fig. S4c). In contrast, expression of Keratin 5 (an LUSC marker) was weak in K-Ras;Mll4-/- similar to K-Ras tumors (Supplementary Fig. S4c). Our survival analysis demonstrated that Mll4 loss significantly decreased the survival time of mice bearing K-Ras pulmonary tumors in the lungs (Fig. 1e). Because these results showed that Mll4 loss accelerated LUAD’s tumorigenicity, we decided to focus on understanding how lung-specific loss of Mll4 promotes LUAD. In addition, low MLL4 mRNA levels were associated with poor prognosis in LUAD but not LUSC patients (Fig. 1f; Supplementary Fig. S4d).
Glycolysis program upregulated by Mll4 loss has human LUAD relevance
To understand how lung-specific loss of Mll4 promotes LUAD, we isolated tumor lesions from K-Ras;Mll4-/- and K-Ras lungs and examined the effect of Mll4 loss on expression profiles of K-Ras tumors using RNA-seq. Our RNA-seq data confirmed Cre-mediated deletion of loxP in the Mll4 gene as RNA peaks corresponding to Exon 16–19 of the Mll4 gene were greatly reduced. (Supplementary Fig. S5a). Bioinformatic analyses of RNA-seq data using Gene Set Enrichment Analysis (GSEA) and DAVID tools commonly showed that glycolysis and oxidative phosphorylation pathways were upregulated by Mll4 loss (Fig. 2a–d). Interestingly, many glycolytic genes were also enriched in MYC and mTORC1 pathways. In contrast, these two analyses did not share any common pathway that were significantly downregulated by Mll4 loss, as GSEA analysis did not indicate any significantly downregulated pathway (Supplementary Fig. S5b). In line with our RNA-seq results, analysis of the TCGA lung cancer database showed that glycolysis and oxidative phosphorylation were enriched in human MLL4-low/mutant LUAD samples compared with LUAD samples bearing MLL4-high levels, indicating that our K-Ras:Mll4-/- tumor model mimics human MLL4-low/mutant lung tumors (Fig. 2e–2g). Glycolytic genes include Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1, whose upregulation by Mll4 loss was confirmed by quantitative RT-PCR (Fig. 2h). In contrast, most genes for oxidative phosphorylation were weakly upregulated by Mll4 loss (Fig. 2h). Therefore, we delved into the regulation of glycolytic genes by MLL4.

(a) Ontology analysis of genes upregulated in K-Ras:Mll4-/- tumors in comparison with K-Ras tumors. The functional annotation tool DAVID was used for the analysis. (b) GSEA analysis of genes upregulated by Mll4 loss. (c and d) Enrichment plot of glycolysis (c) and oxidative phosphorylation (d) for genes that were upregulated by Mll4 loss in mouse LUAD. Each of the black bars represents a gene in the pathway. (e) GSEA analysis of genes that were upregulated by in human LUAD samples bearing low MLL4 expression (n = 20) or MLL4 inactivation (n = 4) as compared with human LUAD samples (n = 24) bearing high MLL4 expression. (f and g) Enrichment plot of glycolysis (f) and oxidative phosphorylation (g) in human LUAD samples bearing low MLL4 expression or MLL4 inactivation as compared with human LUAD samples bearing high MLL4 expression. (h) Analysis of mRNA levels of glycolytic genes and oxidative phosphorylation-associated genes in K-Ras and K-Ras:Mll4-/-lung tumors using quantitative RT-PCR. Data are presented as the mean ± SEM (error bars) of at least three independent experiments or biological replicates. *p < 0.05 and **p < 0.01.
It has been shown that upregulation of glycolysis by ENO119, PGK120, PGAM121, LDHA22, and GAPDH23 promotes tumorigenesis 24. CDK1 is a cell cycle gene25 and increases glycolysis26. Glycolytic enzymes are considered cancer-therapeutic targets 19, 27. In line with the above mRNA expression data, IF and IHC analysis showed that ENO1, PGK1, and PGAM1 levels were increased by Mll4 loss whereas MLL4 levels were decreased (Fig. 3a–c). Our analysis of human LUAD database showed that ENO1, PGK1, PGAM1, LDHA, and GAPDH mRNA levels anti-correlated with MLL4 mRNA levels in human LUAD samples (Fig. 3d). Moreover, high mRNA levels of ENO1, PGK1, PGAM1, LDHA, and GAPDH were associated with poor survival in LUAD patients (Fig. 3e). These results from analysis of human LUAD database indicate human relevance of our findings that Mll4 loss upregulates expression of glycolytic genes in mouse LUAD.

(a and b) Immunofluorescence (IF) analysis for ENO1, PGK1, and PGAM1 in K-Ras and K-Ras:Mll4-/- lung tumor tissues. Representative IF images are shown (a), and IF images were quantified (b). TTF1 is an LUAD marker. (c) Immunohistochemistry (IHC) analysis for MLL4 in K-Ras and K-Ras:Mll4-/- lung tumor tissues. (d) Anti-correlation of MLL4 mRNA levels with ENO1, PGK1, PGAM1, LDHA, and GAPDH mRNA levels in human TCGA LUAD database (n = 517). (e) Kaplan-Meier survival analysis for ENO1, PGK1, PGAM1, LDHA, and GAPDH mRNA levels in human LUAD patients in the KM Plotter database. ENO1 probe set, 201231_at; PGK1 probe set, 227068_at; PGAM1 probe set, 200886_at; LDHA probe set, 200650_s_at; GAPDH probe set, 212581_at; Scale bar, 50 μm. **p < 0.01 and ***p<0.001
Mll4 loss globally reduces super-enhancer and enhancer signals in mouse LUAD
We and others have shown that MLL4 can catalyze mono-, di, and trimethylation at H3K4 28-32. Monomethyl H3K4 (H3K4me1), together with acetyl H3K27 (H3K27ac), marks enhancers, which spatiotemporally activate gene expression in various locations 33. MLL4 is required for enhancer formation regulated by the H3K27 acetyltransferases CBP and p300 34, 35. We and others have also shown that MLL4 interacts with the H3K27me3 demethylase UTX 28, 36, 37. For these reasons, we performed ChIP-seq for H3K4me1, H3K27ac, H3K4me3, and H3K27me3 using K-Ras tumors and K-Ras;Mll4-/- tumors to analyze the effect of Mll4 loss on epigenomic landscapes and chromatin states. In the ChIP-seq experiment, we also included H3K79me2 and H3K9me3, because H3K79me2 is a commonly analyzed epigenomic mark for gene transcription and H3K9me3 is widely used to study gene repression38, 39. Our comprehensive analysis of combinatorial patterns of the 6 histone modifications using the analysis tool ChromHMM 40 showed three prominent transitions from K-Ras tumors to K-Ras:Mll4-/- tumors: 1) Active enhancer/State 2 to weak enhancer/State 3; 2) Weak enhancer/State 3 to very low signal/State 9; and 3) Transcribed enhancer/State 7 to H3K4me1-low/State 5 (Fig. 4a,b; Supplementary Fig. S6a–d). Further analysis confirmed that Mll4 loss did not have any obvious effect on H3K4me3 and H3K27me3 signals (Supplementary Fig. S7a–f). In line with these results, Western blot, IHC, and IF analyses showed that global levels of H3K4me1 and H3K27ac but not H3K4me3 were downregulated by Mll4 loss (Supplementary Fig. S8a,b). These results indicate that Mll4 loss highly and negatively impacts enhancer states.
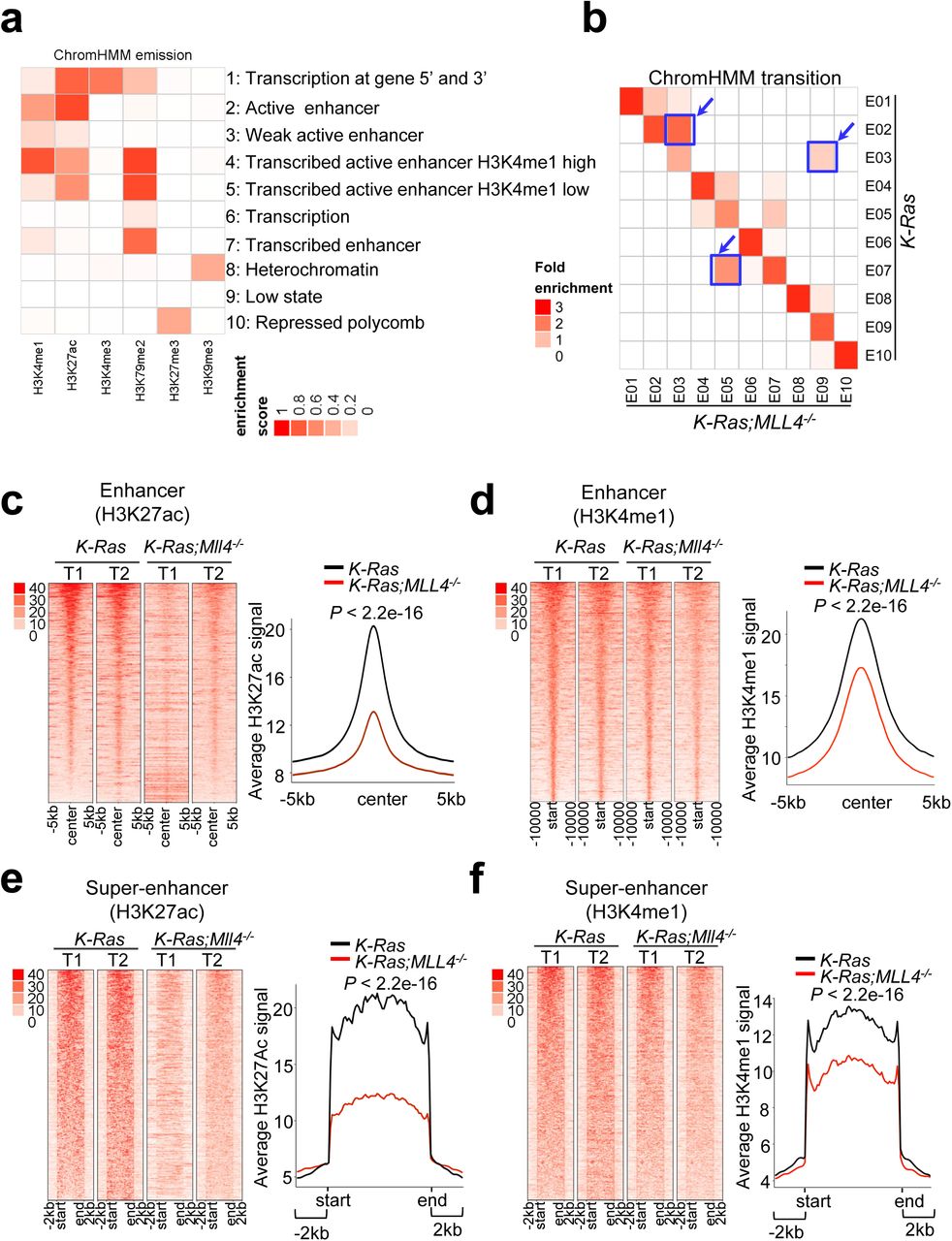
(a) Emission probabilities of the 10-state ChromHMM model calculated from six histone modification profiles in K-Ras and K-Ras:Mll4-/- lung tumors. Each row represents one chromatin state. The 10 states predicted using ChromHMM represent various enhancer states (States 2, 3, 4, 5, and 7), promoter state (State 1), transcription (State 1, 6, and 7), polycomb-repressed (state 10), and heterochromatin (State 8). Each column corresponds to a histone modification. The intensity of the color in the scale from 0 (white) to 1 (red) in each cell reflects the frequency of occurrence of each histone mark in the indicated chromatin state. (b) Heat map showing the fold enrichment of transitions of chromatin states from K-Ras lung tumors to K-Ras:Mll4-/- lung tumors. The color intensities represent the relative fold enrichment. (c and d) Heat maps (left panels) and average intensity curves (right panels) of ChIP-Seq reads (RPKM) for H3K27ac (c) and H3K4me1 (d) at typical enhancer regions. Enhancers are shown in a 10kb window centered on the middle of the enhancer) in K-Ras and K-Ras:Mll4-/- lung tumors. (e and f) Heat maps (left panel) and average intensity curves (right panels) of ChIP-Seq reads for H3K27ac (e) and H3K4me1 (f) at the super-enhancer regions plus their flanking 2kb regions in K-Ras and K-Ras:Mll4-/- lung tumors.
It has been documented that enhancers can be categorized into super-enhancers and typical enhancers 41. Super-enhancers are characterized by clusters of enhancers that are much larger than typical enhancers with a median size of 0.7–1.3 kb and highly activate gene expression 41, 42. Average super-enhancer signals were broader than average typical enhancer signals (Supplementary Fig. S9a,b). Interestingly, Mll4 loss strongly decreased average H3K4me1 and H3K27ac levels in super-enhancers and to a lesser extent typical enhancers (Fig. 4c–f; Supplementary Fig. S9c,d). Expression levels of lung-enriched, super-enhancer-associated genes were downregulated (Supplementary Fig. S9e,f).
The pharmacological inhibition of glycolysis impedes the proliferation and tumorigenic growth of LUAD cells with MLL4-inactivating mutations
As described above, Mll4 loss upregulated glycolysis program but decreased H3K27ac levels. This promoted us to test whether the inhibition of glycolytic pathways or H3K27 deacetylation may impede tumorigenic ability of lung tumors bearing MLL4 loss. For glycolysis inhibition, we used 2-deoxy-D-glucose (2-DG, a hexokinase inhibitor and a glucose analog, and clinical trial phase I/II) 43, 44, POMHEX (a new enolase inhibitor) a, koningic acid (KA, a selective inhibitor of GAPDH)45, Lonidamine (a hexokinase and mitochondrial respiration inhibitor and clinical trial phase II) 46, and Dinaciclib (an inhibitor of CDK1 and CDK2/5/9 and clinical trial phase III) 47, 48. To increase histone acetylation via HDAC inhibition, we used AR-42 (Clinical trial phase I) and FDA-approved SAHA/Vorinostat 49, 50. We compared the inhibitory effects of these inhibitors on cell-proliferative abilities between human LUAD cell lines with wild type MLL4 (A549, H1792, H1437, H23, and H358) and those bearing MLL4’s truncating mutations (i.e., H1568 with nonsense at E758, DV-90 with the truncating mutation P2118fs, and CORL105 with the truncating mutation R2188fs) (Supplementary Table S2). In the MLL4-mutant cell lines compared with the MLL4-normal cell lines, there were some increased trends of glucose uptake and lactate excretion (Supplementary Fig. S10a,b). Our inhibitor experiments showed that 2-DG, POMHEX, KA, and to a lesser extent SAHA and AR-42 inhibited the proliferation of the MLL4-mutant cell lines more than the MLL4-normal cell lines (Fig. 5a–f). In contrast, Lonidamine and Dinaciclib did not exert any selective inhibition against the proliferation of the MLL4-mutant cell lines (Supplementary Fig. S10c, d).

(a–f) Growth inhibition curves of LUAD cell lines bearing wild type (WT) MLL4 (A549, H1792, H23, H1437, and H358) and those bearing MLL4 truncation mutants (H1568, DV-90, and CORL105). Cells were treated with various concentrations of 2-DG (a and b), POMHEX (c), KA (d), Vorinostat (e), and AR-42 (f). (g & h) The effect of 2-DG on spheroid growth of shLuciferase (shLuc)-infected cells and MLL4-depleted LKR-10 cells in a 3D culture. Representative images are shown (g). Spheroid sizes were measured (h, left panel), and relative sizes of spheroids were plotted (h, right panel). (i–k) The effect of tumorigenic growth of H1568 cells bearing an MLL4 truncation mutation and of H358 cells bearing wild type MLL4 in a mouse subcutaneous xenograft model. Shown are the treatment schedule of mice with 2-DG (i). The sizes of xenograft tumors from H1568 and H358 after the treatment with 2-DG or vehicle control were measured (j). Tumors were dissected from the mice after the treatment with 2-DG (500mg/Kg) or vehicle control for 20 days (k). Data are presented as the mean ± SEM (error bars) of at least three independent experiments or biological replicates. *p < 0.05, **p < 0.01, and ***p<0.001.
Because 2-DG selectively inhibited the MLL4-mutant LUAD cell lines over several MLL4-normal cell lines and is currently under clinical trials, we further tested the effect of 2-DG on the growth of the lung cancer cell line LKR10 that is derived from mouse K-Ras tumors. A 3D culture system was used, because 1) it better mimics an in vivo situation than 2D culture system does, 2) MLL4 knockdown increased the sizes of the spheroids formed from LKR10 cells in the 3D culture (Supplementary Fig. S10e), and 3) MLL4 knockdown decreased cell proliferation in the 2D culture system (data not shown). Similar to its inhibitory effect on proliferation of human LUAD cells, 2-DG inhibited more spheroid growth of MLL4-depleted LKR-10 cells than that of control (shLuc)-treated LKR-10 cells in the 3D culture system (Fig. 5g, h). In addition, we compared the inhibitory effect of 2-DG on tumorigenic growth between H1568 cells harboring a MLL4 truncation and H358 cells harboring wild type MLL4 in a mouse xenograft model. Notably, 2-DG (500 mg/kg per day) preferentially inhibited tumorigenicity of H1568 cells compared with H358 cells (Fig. 5i–k).
MLL4 is required for the activity of the Per2 super-enhancer, and Per2 represses glycolytic genes
Because MLL4 is a transcriptional co-activator, Mll4 loss may not directly upregulate tumor-promoting glycolysis program. Thus, we hypothesized that glycolysis program upregulated by Mll4 loss may be repressed by a tumor-suppressive, transcription-repressive regulator encoded by an MLL4-activated gene (i.e., a gene downregulated by Mll4 loss). To identify such tumor-suppressive, transcription-repressive regulator, we first searched the overlapping genes between genes downregulated by Mll4 loss (n = 522) and genes associated with significant decreases in H3K27ac levels (n = 3751), because Mll4 loss strongly downregulated the enhancer mark H3K27ac to decrease gene expression (Fig. 6a and Supplementary Fig. S11a). To further reduce the list for such regulator, we then incorporated human lung cancer aspects into our analysis. Specifically, we examined which Mll4-loss-downregulated genes correlate with MLL4 mRNA levels in more than 0.3 of the correlation coefficient value (r) in human LUAD samples in the TCGA database. This analysis resulted in 14 overlapping genes (Supplementary Fig. S11b,c). Of these 14 genes, expression levels of five genes were lower in LUAD tumors than in adjacent normal tissues, and their low levels correlated with poor prognosis in lung cancer patients (Fig. 6b–d; Supplementary Fig. S12a–e). Interestingly, these genes were occupied by wide H3K27ac and H3K4me1 signals that were decreased by Mll4 loss (Fig. 6e; Supplementary Fig. S13a–d).
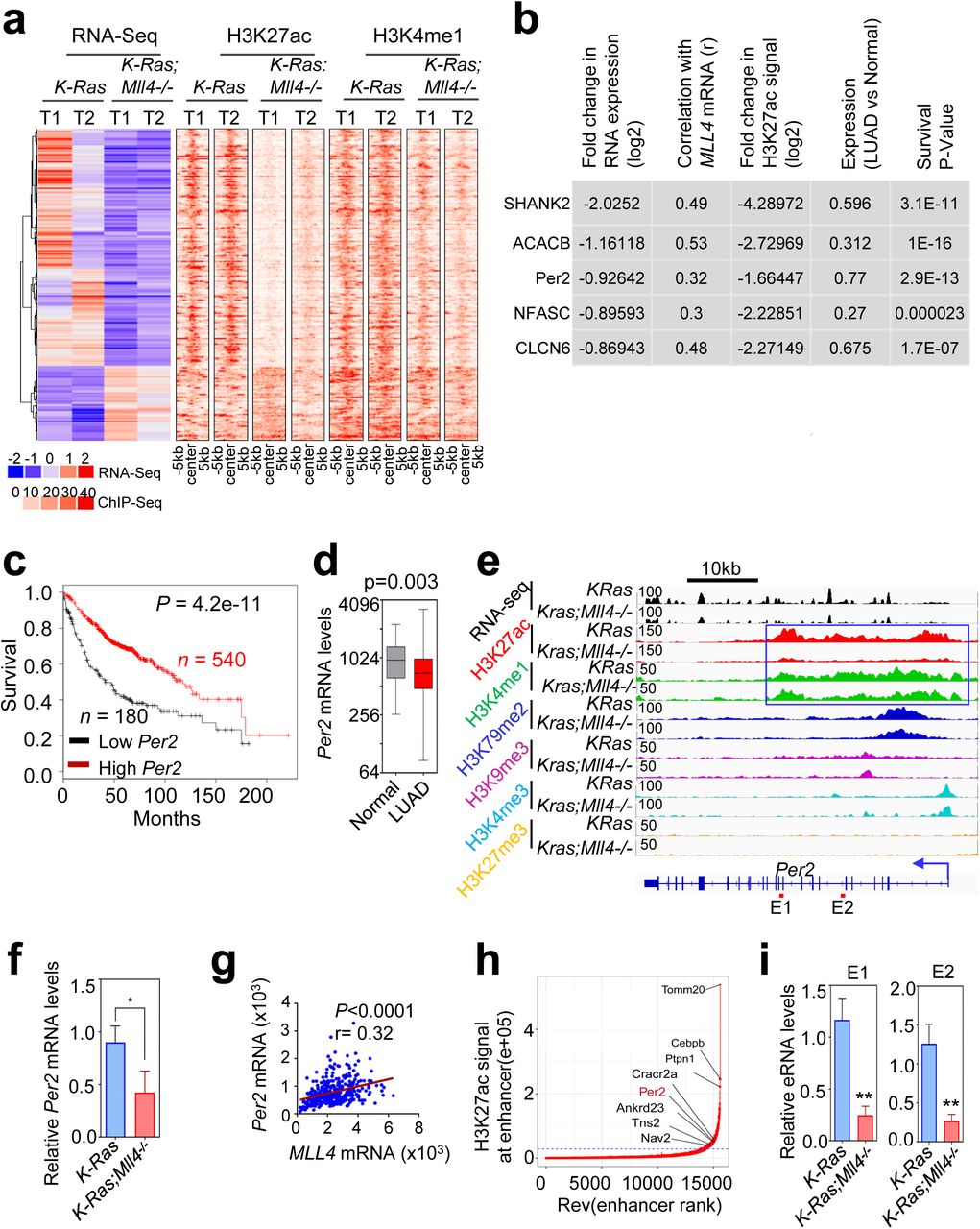
(a) Heat maps showing differentially expressed genes and their H3K27ac and H3K4me1 signals in K-Ras and K-Ras:Mll4-/- lung tumors. (b) The top five hits on the basis of 5 different parameters indicated. (c) The Kaplan-Meier survival analysis showing the correlation of low Per2 mRNA levels with poor survival in lung cancer patients. The auto cutoff was used to divide samples into low and high groups in the KM Plotter database (http://kmplot.com/analysis). Lower quartile was used as a cutoff to divide the samples into low and high groups. PER2 probe set, 205251_at. (d) Box plots showing downregulation of PER2 mRNA levels in LUAD samples (n = 517) compared with their adjacent normal samples (n = 54) in TCGA dataset. (e) Genome browser view of normalized signals of six chromatin marks (H3K27ac, H3K4me1, H3K79me2, H3K9me3, H3K4me3, and H3K27me3) at the Per2 locus in K-Ras and K-Ras:Mll4-/- lung tumors. All the tracks were average of two biological replicates that were normalized to their inputs. The Per2-associated super-enhancer is indicated using a blue line box. (f) Analysis of relative Per2 mRNA levels in K-Ras and K-Ras:Mll4-/- lung tumors using quantitative RT-PCR. (g) A scatter plot showing a correlation between MLL4 and PER2 mRNA levels in TCGA LUAD dataset. (h) A plot indicating super-enhancers identified on the basis of H3K27ac signals. (i) Analysis of eRNA levels for two different regions (E1 and E2) of the Per2 super-enhancer using quantitative RT-PCR. Data are presented as the mean ± SEM (error bars) of at least three independent experiments or biological replicates. *p < 0.05 and **p < 0.01.
Of the five genes, the circadian transcriptional repressor gene Per2 was particularly interesting by the following reasons: 1) Per2 loss in mouse lung promotes lung tumorigenesis 51; 2) Per2 loss increased glucose metabolism, implicating a role for Per2 in regulating glycolysis 51; and 3) disruption of circadian rhythm increases tumorigenicity 52, 53. As mentioned above, Mll4 loss decreased Per2 expression levels (Fig. 6f) and Per2 mRNA levels significantly correlated with MLL4 mRNA levels (Fig. 6g). Interestingly, Per2 was occupied by a large cluster of H3K4me1 and H3K27ac peaks that are indicative of a super-enhancer (Fig. 6e,h). In enhancer regions, RNAs called enhancer RNAs (eRNAs) are bidirectionally transcribed by RNA Polymerase II 54. Enhancer activities can be assessed by eRNA levels 54. Therefore, we measured the effect of Mll4 loss on eRNA levels in the Per2–associated super-enhancer. Mll4 loss decreased eRNA levels in the super-enhancer region, suggesting that Mll4 loss reduces the activity of the Per2 super-enhancer (Fig. 6i).
To assess whether Per2 represses glycolytic genes that are upregulated by Mll4 loss, we used the mouse K-Ras LUAD line LKR10. We first examined whether MLL4 knockdown in LKR10 cells recapitulates the effect of Mll4 loss on glycolytic genes in K-Ras tumors. In fact, MLL4 knockdown in LKR10 cells upregulated expression levels of several glycolytic genes (e.g., Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1) while downregulating Per2 expression (Fig. 7a). In line with this, MLL4 knockdown increased glucose uptake and lactate excretion (Fig. 7b,c). We then examined the effect of Per2 knockdown and Per2 overexpression on expression of these glycolytic genes in LKR10 cells. Per2 knockdown increased expression levels of Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1 genes, whereas Per2 overexpression decreased their expression levels (Fig. 7d,e). Per2 expression levels anti-correlated with expression of several glycolytic genes (ENO1, PGK1, PGAM1, LDHA, and CDK1) in LUAD samples in the TCGA database (Fig. 7f). Finally, we examined the effect of ectopic expression of Per2 on the spheroid growth of LKR10 cells that is increased by MLL4 depletion in the 3D culture system. Per2 re-expression decreased the spheroid growth of MLL4-depleted LKR10 cells (Fig. 7g). These results indicate that tumor-suppressive function of MLL4 is dependent, at least in part, on the upregulation of Per2 expression by MLL4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(a) Analysis of the effect of MLL4 knockdown on Per2, Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1 mRNA levels in mouse LKR-10 cells bearing K-RasG12D using quantitative RT-PCR. (b and c) The effect of MLL4 knockdown on glucose uptake (b) and lactate excretion (c) in LKR10 cells. (d) Analysis of the effect of Per2 knockdown on Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1 mRNA levels in LKR-10 cells using quantitative RT-PCR. (e) Analysis of the effect of ectopic Per2 expression on Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1 mRNA levels in LKR-10 cells using quantitative RT-PCR. (f) Scatter plots showing inverse correlations of PER2 mRNA levels with ENO1, PGK1, PGAM1, LDHA or CDK1 mRNA levels in human LUAD TCGA dataset (n = 517). (g) The effect of ectopic expression of Per2 on spheroid sizes of MLL4-depleted LKR-10 cells in 3D-culture. Representative images of spheroid are shown (left panel). The sizes of the spheroids were quantified (right panel). shLuc and pLenti-GFP-infected cells were used as controls. Scale bars represent 200 µm. Data are presented as the mean ± SEM (error bars) of at least three independent experiments or biological replicates. shLuc, shLuciferase *p < 0.05.
Discussion
It has been reported that MLL4 is required for the formation of acute myeloid leukemia by the MLL-AF9 oncogene 55. In contrast, results reported here showed that lung-specific deletion of Mll4 significantly promoted K-Ras–driven lung tumorigenesis in mice and reduced the survival of mice bearing K-Ras-driven tumors, suggesting that Mll4 loss cooperates with other oncogenic aberrations to increase LUAD tumorigenicity. The lung tumor-suppressive function of MLL4 is further supported by our following additional results: 1) Mll4 loss upregulated expression of tumor-promoting glycolytic genes, such as ENO1, PGK1, LDHA, PGAM1, and CDK1; 2) Mll4 loss downregulated tumor-suppressive genes, such as Per2; and 3) MLL4 depletion increased spheroid sizes of lung cancer cells in the 3D culture system. In line with tumor-suppressive function of MLL4, we have recently shown that brain-specific Mll4 loss alone induces spontaneous medulloblastoma56. Our additional study (the manuscript co-submitted by Rai and colleagues) indicates that MLL4 acts as a tumor suppressor in melanoma tumorigenesis. Other recent studies have demonstrated that genetic ablation of Mll4 in B cells enhanced B-cell lymphoma genesis, also indicating MLL4’s tumor-suppressive function 57, 58. Thus, the anti- or pro-tumorigenicity of MLL4 may be cell-type-dependent, although MLL4 may have a tumor-suppressive function against tumorigenesis in a majority of tissues.
Our results obtained using our K-Ras;Mll4-/- GEMM in the present study indicate that MLL4 acts as an epigenetic LUAD suppressor by positively and highly regulating super-enhancers and to a lesser extent enhancers. Therefore, this GEMM represents a new epigenetic LUAD mouse model. This mouse model would be useful for future additional studies for human LUAD because glycolysis pathway is enriched in both K-Ras;Mll4-/- tumor model and human LUAD tumors with low MLL4 mRNA levels.
The circadian rhythm transcriptional repressor Per2 plays an important role in tumor suppression 52, 53. As our results indicate that the tumor suppressor MLL4 upregulates Per2 expression to subsequently repress tumor-promoting glycolysis, MLL4-mediated Per2 activation represents a previously unknown tumor-suppressive mechanism that links between an epigenetic tumor suppressor and a circadian rhythm regulator. Per2 moves into the nucleus during the evening and downregulates gene expression. During the night, Per2 are gradually phosphorylated and targeted for ubiquitination that leads to proteasomal degradation 59, 60. Distinct from this type of Per2 regulation, our data suggest a new mode of Per2-regulatory mechanism in which MLL4 upregulates Per2 expression by activating the Per2 super-enhancer, providing molecular insights into how Per2 is epigenetically regulated. Super-enhancer formation has been linked to oncogene activation 61. However, our results indicate that the Per2 super-enhancer is associated with tumor-suppressive function in LUAD, consistent with our recent finding that super-enhancer diminution downregulates expression of tumor suppressor genes linked to medulloblastoma genesis 56. Our results also define several tumor-promoting glycolytic genes (e.g., Eno1, Pgk1, Pgam1, Ldha, Gapdh, and Cdk1) as new Per2’s downstream genes. Taken together, our findings reveal a tumor-suppressive mechanism in which MLL4 indirectly represses glycolytic genes by enhancing Per2 expression via super-enhancer activation and thereby suppresses LUAD.
Increased aerobic glycolysis known as the Warburg effect is a major characteristics of cancer cells and provides cancer cells with proliferation advantage by producing ATP as well as glucose-derived metabolites for biosynthesis of nucleotides, lipids and proteins 24. Targeting glycolytic pathways to arrest tumorigenic growth of cancer cells remains an attractive therapeutic intervention. Interestingly, the present study, along with the study co-submitted by Rai and colleagues, showed that Mll4 loss increased expression of glycolytic genes. In addition, we demonstrated that the glycolysis inhibitor 2-DG impeded tumorigenicity of LUAD cells bearing an MLL4 mutation in a mouse xenograft model. Because MLL4 is mutated in other several types of cancer 62 and certain glycolysis inhibitor (i.e., 2-DG) has re-entered into clinical trials, our findings may rationalize glycolysis inhibition as an anti-cancer treatment strategy against human MLL4-inactivated LUAD and other cancer types bearing MLL4-inactivating mutations.
Methods
Methods, including experimental procedures and any associated accession numbers and references, are available as supplementary information.
Author Contributions
H.A. planned and carried out experiments, analyzed data, prepared figures, and wrote the manuscript. M.T. performed the bioinformatics analysis of RNA-seq and ChIP-seq data, prepared bioinformatics figures, and wrote the manuscript. M.M. and S.S.D. performed ChIP-seq experiments. S.A. contributes to data analysis. T-Y.C. contributed to inhibitor experiments. J.C. contributed to IHC experiments and analysis. Y-H.L., F.M., and F.J.D. provided reagents. L.B. analyzed tumor data. K.R. directed ChIP-seq experiments, guided the bioinformatics study, contributed to experiment design, and wrote the manuscript. M.G.L. conceived the study, designed and oversaw the study, evaluated data, and wrote the manuscript.
Acknowledgments
We are thankful to Julien Sage and Tyler Jacks for providing their reagents and also thank Haoqiang Ying, Zhenbo Han, Su Zhang, and Charles Kingsley for their technical assistance. This work was supported by grants to MG Lee from the National Institutes of Health (NIH; R01 CA157919, R01 CA207109, and R01 CA207098) and the Center for Cancer Epigenetics at the UT MD Anderson Cancer Center, by a grant to K Rai from the NIH (CA160578), by a grant to FL Muller from American Cancer Society (RSG-15-145-01-CDD), by a grant to FJ DeMayo from the Intramural Research Program of the National Institute of Environmental Health Sciences (Z1AES103311-10), and by a fellowship to H Alam from the Odyssey program at the UT MD Anderson Cancer Center. The animal imagining and histology work were performed at the Small Animal Imaging Facility and Histopathology Core Lab, respectively, at the UT MD Anderson Cancer Center supported by the NIH National Cancer Institute (P30CA016672).
Footnotes
↵The authors declare no potential conflicts of interest.
a Lin et al., Eradication of ENO1-deleted Glioblastoma through Collateral Lethality bioRxiv 331538; doi: https://doi.org/10.1101/331538
References




