

Abstract
Sex differences in cancer occurrence and mortality are evident across tumor types; men exhibit higher rates of incidence and often poorer responses to treatment. Targeted approaches to the treatment of tumors that account for these sex differences require the characterization and understanding of the fundamental biological mechanisms that differentiate them. Hepatocellular Carcinoma (HCC) is the second leading cause of cancer death worldwide, with the incidence rapidly rising. HCC exhibits a male-bias in occurrence and mortality. Most HCC studies, to date, have failed to explore the sex-specific effects in gene regulatory functions. Here we have characterized the regulatory functions underlying HCC tumors in sex-stratified and combined analyses. By sex-specific analyses of differential expression of tumor and tumor adjacent samples, we uncovered etiologically relevant genes, pathways and canonical networks differentiating male and female HCC. While both sexes exhibited activation of pathways related to apoptosis and p53 signaling, pathways involved in innate and adaptive immunity showed differential activation between the sexes. Using eQTL analyses, we discovered germline regulatory variants with differential effects on tumor gene expression between the sexes. We discovered eQTLs overlapping HCC GWAS loci, providing regulatory mechanisms connecting these loci to HCC risk. Furthermore, we discovered genes under germline regulatory control that alter survival in patients with HCC, including some that exhibited differential effects on survival between the sexes. Overall, our results provide new insight into the role of genetic regulation of transcription in modulating sex differences in HCC occurrence and outcome and provide a framework for future studies on sex-biased cancers.
Author Summary Sex differences in cancer occurrence and mortality are evident across tumor types, and targeted approaches to the treatment male and female tumors require the characterization and understanding of the fundamental biological mechanisms that differentiate them. Hepatocellular Carcinoma (HCC) is the second leading cause of cancer death worldwide, with the incidence rapidly rising. HCC exhibits a male-bias in occurrence and mortality. Here, we have characterized the regulatory functions underlying male and female HCC. We show that while HCC shares commonalities across the sexes, it shows demonstrable regulatory differences that could be critical in terms of prevention and treatment: we detected differential activation of pathways involved in innate and adaptive immunity, as well as differential genetic effects on gene expression, between the sexes. Furthermore, we have detected regulatory variants overlapping known HCC GWAS risk loci, providing regulatory mechanisms connecting these risk variants to HCC. Finally, we discovered genes under germline regulatory control altering the overall survival of patients with HCC, including some that showed differential effects between the sexes. Overall, our findings create a paradigm for future studies on sex-biased cancers.
Introduction
Differences in cancer occurrence and mortality between sexes are evident across tumor types; males exhibit higher rates of cancer incidence and often poorer response to treatment, including some forms of chemotherapy and immunotherapy [1,2]. While differences in risk behaviors and environmental exposure may explain some portion of the sex-bias, cellular and molecular differences are also likely to be important. The sexes differ in their endocrinological profile, immunological functions and genetic makeup [3–5]. However, sex differences are rarely considered in the development of cancer therapies, and the contribution from sex chromosome variation to tumor etiology remains poorly understood. Sex-biased gene expression and regulatory functions may underlie differences between the sexes in disease prevalence and severity [6,7]. Analyses of sex-specific regulation of gene expression are essential for understanding sources of sex difference in cancer incidence, as well as sex-specific mechanisms affecting tumor etiology, disease progression and outcome.
Hepatocellular carcinoma (HCC) exhibits sex-bias in occurrence, with a male-to-female incidence ratio between 1.3:1 and 5.5:1 across populations [8,9]. HCC is the second leading cause of cancer mortality worldwide, accounting for 8.2% of all cancer deaths [10]. HCC risk is influenced by genetic susceptibility, environmental factors such as oncogenic viral infections, metabolic syndrome, and alcohol use, and shaped by numerous biological processes, resulting in a high degree of genetic and transcriptional heterogeneity [11]. HCC incidence in the US has doubled in the last 3 decades, attributable to increased rates of obesity [9].
While sex-bias in HCC is partly attributed to sex-specific differences in risk behaviors and environmental exposure, the relationship of these factors and sex as a biological variable has not been systematically investigated. Previously, sex differences in HCC were explored in conjunction with 12 additional Cancer Genome Atlas (TCGA) tumor types by Yuan et al [12]. They discovered extensive sex-biased signatures in gene expression in HCC and other strongly sex-biased cancers.
HCC shows evidence of molecular subtypes, though their role in sex-bias has yet to be explored. More specifically, clustering analyses of gene expression data have produced a characterization of four distinct HCC subtypes that differ in terms of gene expression, pathway enrichment, and median survival [13]. A study utilizing a weighted co-expression network analysis method discovered hub genes associated with HCC outcome [14]. On the other hand, genetic variants affecting the expression of TNFRSFI0 [15], PAKS [16], and PVT1 [17] have been found to alter HCC disease progression and outcome, highlighting the role of regulatory variants in HCC. How these progression altering subtypes or variants are related to HCC sex-bias has not been examined.
Sex-specific analyses can reveal genetic and regulatory mechanisms of sexual dimorphism in HCC susceptibility, progression, and mortality. Targeted approaches to the treatment of male and female HCC will require the characterization and understanding of the fundamental biological mechanisms that differentiate them. Here, we analyzed data from The Genotype-Tissue Expression (GTEx) project and The Cancer Genome Atlas (TCGA) to examine the sex-specific patterns of gene expression and regulation in HCC and healthy liver tissue. We find etiologically meaningful differences in regulatory functions in HCC between males and females, providing insight into the mechanisms underlying sex-bias in cancer. Additionally, we observed genes under germline regulatory control that alter survival in sex-biased way in patients with HCC.
Results
Sex-specific patterns of gene expression HCC
We found sex-differences in gene expression in normal liver, tumor adjacent, and tumor tissues (Fig 1). We found 13 genes showing a consistent sex-bias in expression in liver, HCC adjacent tissue and HCC. Notably, we find that X-inactivation and X-linked dosage compensation appear to be functioning in the healthy liver, HCC adjacent and HCC tissues. This is concluded from the bulk RNAseq measurements where we observe XIST expression only in female samples. As XIST is primarily involved in X-inactivation [18], this is further supported by the observation of very little or no sex differences in expression of other X-linked genes. We find Y-linked genes expressed in male samples in all three tissues. Many of these Y-linked genes have functional X-linked homologs. When examining the combined expression levels of these homologous gene pairs, we did not detect notable differences between the sexes (Fig S1).

X-linked genes are indicated in red, Y-linked in blue, and autosomal genes in black. An FDR-adjusted p-value threshold of 0.01 and absolute log2(FC) threshold of 2 were used to select significant genes. Multiple transcripts of the long non-coding RNA XIST are independently expressed.
Interestingly, we identify 9 genes, including 6 protein-coding genes (CYP4F22, HRCT1, ZCCHC16, DTX1, ATF5, CD24), 2 non-coding RNAs (CTD-2325A15.3, RP11-495P10.9) and one pseudogene (ASB9P1) that show sex-differences in expression in HCC, but not in HCC adjacent or healthy liver. Notably, Notch-regulating DTX1, activating transcription factor 5 (ATF5) and signal transducer CD24 were downregulated in male HCC.
To further investigate the regulatory mechanisms underlying HCC of males and females, we detected differentially expressed genes (DEGs) between tumor and tumor adjacent normal samples in males and females, as well as in a joint analysis of both sexes (Fig 2A). Substantially more DEGs were detected in sex-specific analyses than in the unstratified analysis (Fig 2B). Specifically, DEGs that showed different magnitudes in fold change between the sexes were detected in the sex-specific analyses, while DEGs with similar fold changes across all comparisons were detected in the unstratified analysis as well as the sex-specific analyses (Fig S2). DEGs that were only detected in the unstratified analysis, and not in sex-specific analyses, showed a large variance in expression and were thus not detected as statistically significant DEGs in sex-specific analyses (Fig S2). Tumor-infiltrating immune cells may produce spurious signals in DEG analyses, which is evident from the detection of various immunoglobulin genes in tumor vs. tumor adjacent comparisons (Table S4-6). However, cell content analyses based on gene expression data did not exhibit sex differences in the prevalence of tumor-infiltrating immune cells (Fig S3), and thus such spurious signals are unlikely to affect male-female comparisons.

A: Volcano plots of DEGs in tumor vs. tumor adjacent comparisons in the sex-specific analyses and in joint analysis of both sexes. X-linked genes are indicated as red, Y-linked as blue, and autosomal as black. Significant genes were selected based on an FDR-adjusted p-value threshold of 0.01 and absolute log2(FC) threshold of 2. B: Venn-diagram of the overlap of DEGs in the sex-specific analyses and in joint analysis of both sexes. C: Overlap of functional pathways and canonical networks enriched in males, females, and in the joint analysis of both sexes. C: Examples of sex-specific and sex-shared pathways.
In the joint analysis of male and female samples, we detected 581 DEGs, 23 of which were X-linked (Table S4). In male and female specific analyses, we detected 606 and 416 DEGs, respectively (Supplementary Tables 5 and 6). In both sex-specific analyses, 25 X-linked genes were detected. Some of these X-linked genes had similar expression patterns in male and female comparisons: including the MAGE (Melanoma-associated antigen) gene family members MAGEA1, MAGEA12, MAGEA6, MAGEC2, MAGEA3, XAGE (X Antigen) family members XAGEA1 and XAGEB1, PAGE (Prostate-associated) family member PAGE2, as well as SXX (synovial sarcoma X) family member SSX1 were overexpressed in both male and female HCC.
Interestingly, prostate-associated antigen family member PAGE4 was only overexpressed in male HCC.
To further investigate the sex-specific functions of HCC tumors, we analyzed the lists of male- and female-specific DEGs for the enrichment of functional pathways and canonical networks (Fig 2C and 2D). We found that sex-specific DEGs were enriched in pathways relevant to oncogenesis and cancer progression (Supplementary Tables 7 and 8). Most of the top pathways were nonoverlapping, strongly indicating that male and female HCC are driven by different mechanisms and processes.
Differential cis-eQTL effects in male and female HCC
We used eQTL analyses to detect germline genetic effects on tumor gene expression in males, females, and in both sexes (Fig 3A). Due to the small number of samples from females, we were unable to reliably detect female-specific eQTL associations. However, we detected 24 male-specific eGenes (Fig 3B), which were not detected in the female-specific or in the unstratified analysis. Since these associations were not detected in the unstratified analysis, they are likely not a result of differential power to detect associations due to different sample sizes or allele frequencies between the sexes, but exhibit differential effects between the sexes. None of the male-specific eGenes were differentially expressed between male and female HCC, indicating that the male-specific associations are not a driven of differences in overall gene expression levels between males and females, but are likely to arise from factors such as chromatin accessibility, transcription factor activity, and hormone receptors [19,20]. Genomic annotations show that most of the detected regulatory variants were located on the non-coding regions (Fig 3C).

A: QQ-plot of eQTL associations in the joint analysis of both sexes (grey), male HCC (blue) and female HCC (red). B: Overlap of eGenes detected in joint and stratified analyses. C: Genomic annotations of eVariants detected in joint and stratified analyses. CDS: coding sequences. D: An example of a male-specific eQTL. POGLUT1 expression is modulated by a germline variant in cis in male HCC, but not in female HCC nor in joint analysis of both sexes.
Overlap of eQTLs and HCC risk GWAS loci
eQTL mapping can be used to find a regulatory mechanism explaining GWAS risk loci by identifying associations between genotypes and intermediate molecular phenotypes (gene expression levels). A number of variants in the HLA region in chr6, as well as variants near Signal Transducer and Activator of Transcription 4 (STAT4) in chr4 and Aspartylglucosaminidase (AGA) in chr2, have been identified as HCC risk loci [21–25], but the regulatory mechanisms connecting these risk variants to the HCC phenotype are not clear. We discovered sex-shared regulatory variants tightly linked with known HCC risk loci altering gene expression levels in HCC tumors (Table 1).
GWAS study citation is denoted in the risk variant column. Adjacent genes: If the GWAS variant is located within a gene, that gene is listed. If the variant is intergenic, the upstream and downstream genes are listed. Asterisk denotes instances where eVariants outside the LD block associated with reported or adjacent genes were found. eVariant sites have been annotated for genes and putatively regulatory regions (Methods, Table S12). Variant positions and distance are denoted as base pairs.
Germline genetic effects on HCC tumor gene expression and survival
We used Cox’s proportional hazard model to test for effects of eGene expression on patient survival in the TCGA Liver Hepatocellular Carcinoma (LIHC) cohort. Out of the 1204 sex-shared eGenes, 50 were associated with joint survival (FDR-adjusted p-value <0.05, Table 2). No female-specific survival associations were detected, likely due to the low number of female samples affecting statistical power to detect survival effects. However, we detected 65 sex-shared eGenes associated with male survival (Table 3). 15 of these were not associated with joint survival, clearly indicating differential effects on overall survival between the sexes.
Cox’s proportional hazard ratio: likelihood ratio χ2, FDR-adjusted p-value threshold 0.05.
Cox’s proportional hazard ratio: likelihood ratio χ2, FDR-adjusted p-value threshold 0.05. Genes that were not associated with joint survival are indicated with an asterisk.
Discussion
Sex-specific patterns of gene expression in liver and HCC
It is well established that patterns of gene expression vary between the sexes. Previous studies have confounded these differences with those which may be etiologically important in cancer. For example, Yuan et al. previously reported extensive sex-biased signatures in gene expression in HCC and other strongly sex-biased cancers [12]. From the results presented here, it is possible to distinguish the differences detected in comparisons of male and female HCC from those reflecting normal sex-differences.
We report etiologically meaningful differences in gene expression between male and female HCC cases. In addition to X- and Y-linked genes, 8 autosomal genes were expressed in a sex-biased way in HCC tumors. Three of these genes - all of which were underexpressed in male HCC compared to female - are of particular interest in the context of HCC: Notch-regulating DTX1 has been identified as a putative tumor suppressor gene in head and neck squamous cell carcinoma [26]. ATF5 is highly expressed in the liver but downregulated in HCC [27]. ATF5 is known to inhibit hepatocyte proliferation [28] and re-expression of ATF5 in HCC inhibits proliferation and induces G2/M arrest of the cell cycle [28]. ATF5 also may act as a negative regulator of IL-1ß signaling pathway in hepatocytes and thus act as a regulator of IL-1β-mediated immune response [29]. CD24 has a crucial role in T cell homeostasis and autoimmunity [30]. In breast cancer, high CD44/CD24 ratio is an indicator for malignancy [31]. The opposing roles of CD24 expression in cancer and autoimmune diseases raise interesting questions on the role of sex differences in immunity underlying female prevalence in autoimmunity [32] and male prevalence in cancer. More studies are needed to better understand the differential regulation of immune functions between the sexes, and how these differences contribute to the observed biases in disease occurrence.
In both sex-specific analyses, 25 X-linked genes were detected including the MAGE family (Melanoma-associated antigen), the XAGE (X Antigen) family members, the PAGE (Prostate-associated) family, and the SXX (synovial sarcoma X) family. While most of the 25 X-linked genes exhibited similar patterns between the sexes, PAGE4 was overexpressed only in male HCC. PAGE4 is expressed in the human fetal prostate during development as well as malignant prostate and prostate cancer, but not in healthy adult prostate [33]. The role of PAGE4 in prostate cancer has been studied in considerable detail, and it has been indicated as a promising target for therapies [33]. However, its role in other cancers has not been thoroughly explored. MAGE, PAGE, and SSX genes are members of a large family of cancer testis antigens (CTAs). CTAs are expressed in tumors, but not in normal tissue, with the exception of testis and placenta, and recognized by cytotoxic T lymphocytes, making them ideal targets for immunotherapeutic approaches [34]. MAGE expression has been associated with response to checkpoint inhibition therapy in metastatic melanoma [35]. Given the observed differences here, CTA expression may prove valuable in determining the potential response for checkpoint inhibition therapy in HCC [36].
Sex-differences in the pathway and network activation strongly indicate that male and female HCC are driven by distinct functional pathways. Males and females differed in oncogenic processes with females showing RAS pathway enrichment while males showed PI3K/AKT pathway enrichment. Additionally, female DEGs were highly enriched in genes involved in innate immunity Toll-like receptor (TLR) signaling (Table S9), while male DEGs exhibited high enrichment of genes involved in adaptive immunity and Notch signaling (Table S8). TLR signaling has a crucial role in the regulation of the immune system by evoking an inflammatory response [37]. Interestingly, TLR signaling may activate Notch signaling [38], and Notch signaling may feedback modulate TLR signaling pathway to modulate inflammatory response through extracellular signal-regulated kinase 1/2-mediated nuclear factor κB activation [39]. Both of these processes regulate macrophage functions and are likely to have a major role in cancer progression by modulating the inflammatory tumor microenvironment [37–39]. TLR and Notch signaling pathways are notable targets for anti-cancer and anti-metastasis therapies [40,41]. Furthermore, while both sexes exhibited an enrichment of genes involved in cell cycle, apoptosis and p53 signaling, only males showed an activation of pathways regulating natural killer cell cytotoxicity, and females showed an activation of pathways involved in NF-kB (nuclear factor kappa-light-chain-enhancer of activated B cells) activation (Fig 2).
Sex-specific regulatory effects of germline variants
We detected 24 genes under germline regulatory control in male HCC only (Fig 3B). Functional annotations of these male HCC specific eGenes provide insight into possible regulatory mechanisms contributing to the observed male-bias in HCC. Protein O-glucosyltransferase 1 (POGLUT1) was found to be under germline regulation in male HCC, but not in female HCC or in joint analysis of both sexes (Fig 3D). The eVariant associated with POGLUT1 is located on a promoter region of its target (Table S14). POGLUT1 is an enzyme that is responsible for O-linked glycosylation of proteins. Glycosylation is a major type of post-translational modification, during which proteins are decorated with oxygen (O) or nitrogen (N) atoms of amino acids. These modifications may critically alter the proteins’ folding, localization, and function [42]. Altered glycosylation of proteins has been observed in many cancers [43,44], including liver cancer [45,46]. Cell surface receptor-mediated growth and apoptosis have been proposed as mechanisms explaining the role of O-glycosylation in cancer [47]. Interestingly, altered glycosylation of tumor proteins may also allow the tumor cells to evade natural killer cell immunity [48].
POGLUT1 is an essential regulator of Notch signaling and is likely involved in cell fate and tissue formation during development. Genes involved in Notch signaling were also found to be expressed in a sex-biased way in HCC tumors and highly enriched in DEGs detected in tumor vs. tumor adjacent normal comparison of male samples, pointing to a possible role of Notch in the development of HCC in males. The role of Notch signaling in various biological processes has been thoroughly studied in a variety of organisms and tissues, particularly in Drosophila [49]. Notch signaling is of particular interest in the context of HCC, as it is involved in liver development [50,51], the development of sexually dimorphic traits [52], and tumorigenesis [49]. The role of Notch signaling in HCC may differ between early and late-stage tumors and among molecular subtypes, and further studies are necessary to understand the possible oncogenic properties of Notch among HCC subtypes and between the sexes.
Chromatin states are a major determinant of sex-biased gene expression and sex-specific regulatory functions in mouse liver [53] and in human peripheral blood mononuclear cells [19]. Other plausible mechanisms underlying sex-specific regulatory effects are differential transcription factor activity and hormonal regulation. Further studies are needed to explore the role of these mechanisms in male and female HCC.
Regulatory mechanisms underlying HCC risk variants
Genetic variants in the Human Leukocyte Antigen (HLA) region have been found to be associated with hepatitis B related HCC in Asian populations [22,25]. While some of these risk variants were found to be associated with HLA expression in normal liver tissue [25] or differentially expressed between HCC tumors and tumor adjacent tissue [22], direct evidence of regulatory mechanisms connecting the risk variants to HCC was lacking. We discovered regulatory variants tightly linked with known HCC risk loci associated with HLA expression in HCC tumors (Table 1). Polymorphisms in the HLA region may influence immune responses and oncogenesis by altering the epitope binding properties and/or expression levels of HLA molecules. Loss of HLA expression, associated with an immune escape by avoiding T-cell recognition, is commonly found in malignant cells [54]. Tumor immune escape is one of the hallmarks of cancer, and it has been shown to have negative effects on the clinical outcome of cancer immunotherapies [55]. HLA expression is critical in tumor rejection, and HLA re-expression has been recognized as a major potential target in the development of immunotherapies [54]. HLA allele frequencies and LD relationships among these variants vary between population, and HLA polymorphisms are likely partially responsible for the ethnic disparities in HBV persistence among ethnicities [56], and thus likely contribute to the observed biases in HCC occurrence between populations. Interestingly, sex modulates the degree to which HLA molecules propagate the selection and expansion of T cells [57]. Sex-specific regulatory functions discovered in this study may partially underlie the sex-biases in diseases with immune system involvement. HLA associated shaping of the immune system points to a promising target for future studies to develop more targeted therapies accounting for patients’ germline genetic makeup.
Genes under germline regulatory control alter patient survival in HCC
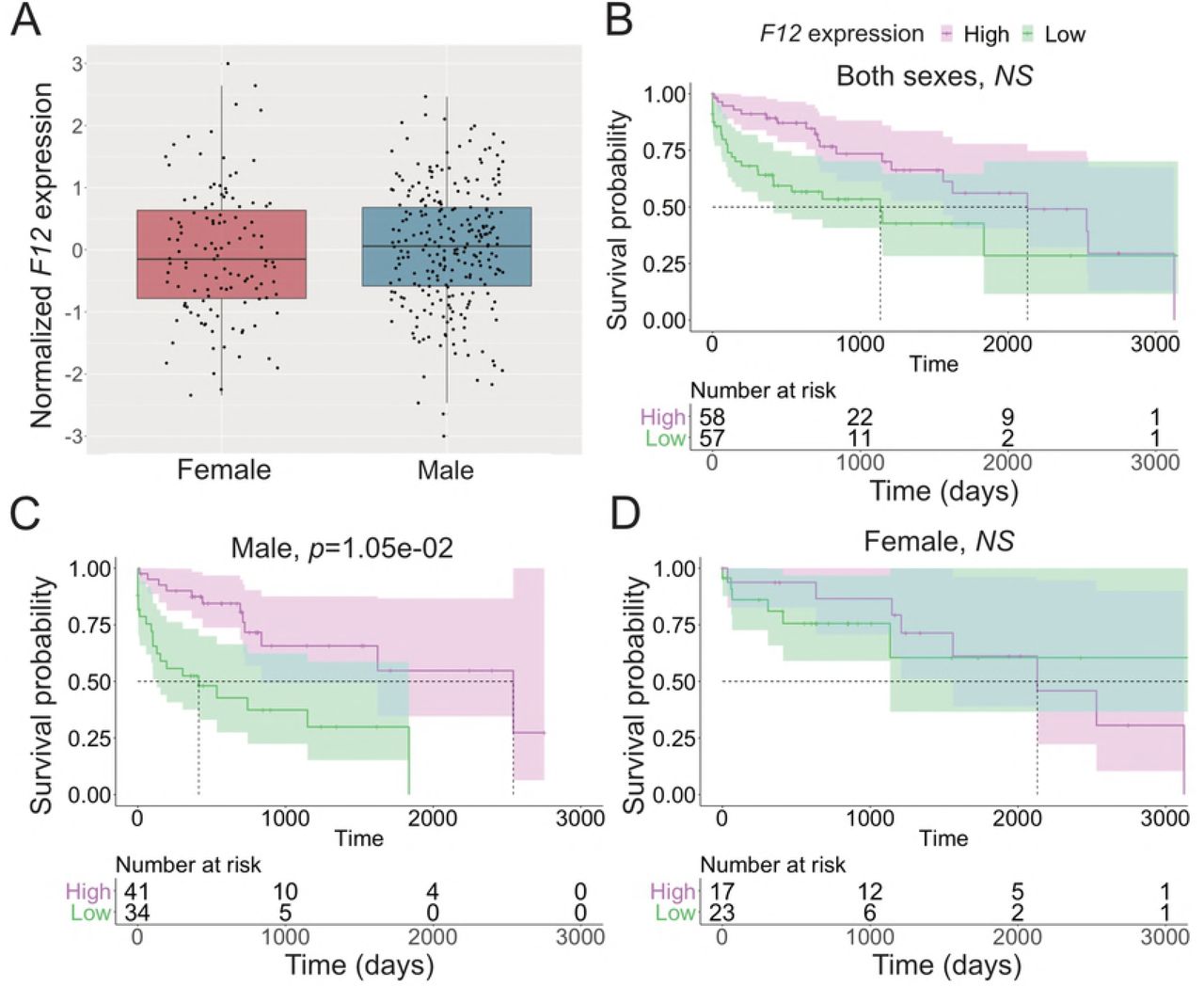
In addition to the discovery of HCC risk variants, characterizations of patterns and levels of gene expression in HCC tumors have led to the discovery of biomarkers associated with HCC etiologies, disease progression and outcome [58–60]. We discovered genes under germline regulatory control altering overall survival in the TCGA LIHC cohort, 15 of which showed differential survival effects between the sexes (Tables 2 and 3). Some of the 15 male-specific survival associated eGenes have been found to affect disease progression and outcome in other cancer types, indicating a pan-cancer role of these genes. In breast cancer, lower Coagulation Factor XII (F12) expression is a favorable prognostic marker [61]. Interestingly, low F12 expression was associated with poor survival among males in the TCGA LIHC cohort (Fig 4). In some population studies, variants in Cytochrome P450 2D6 (CYP2D6) have been found to be associated with recurrence in breast cancer patients treated with Tamoxifen ([62] but see [63] and [64]), and it is highly expressed in the liver, where its expression may affect drug clearance, efficacy, and safety [65]. G protein-coupled receptor kinase 6 (GRK6) deficiency promotes angiogenesis, tumor progression, and metastasis in murine models of human lung cancer [66]. Variations in the miRNA binding sites of the Zymogen Granule Protein 16B (ZG16B) gene are associated with prognosis in patients with colorectal cancer [67]. Variants at the Dynactin Subunit 5 (DCTN5) gene are associated with ovarian cancer mortality [68].
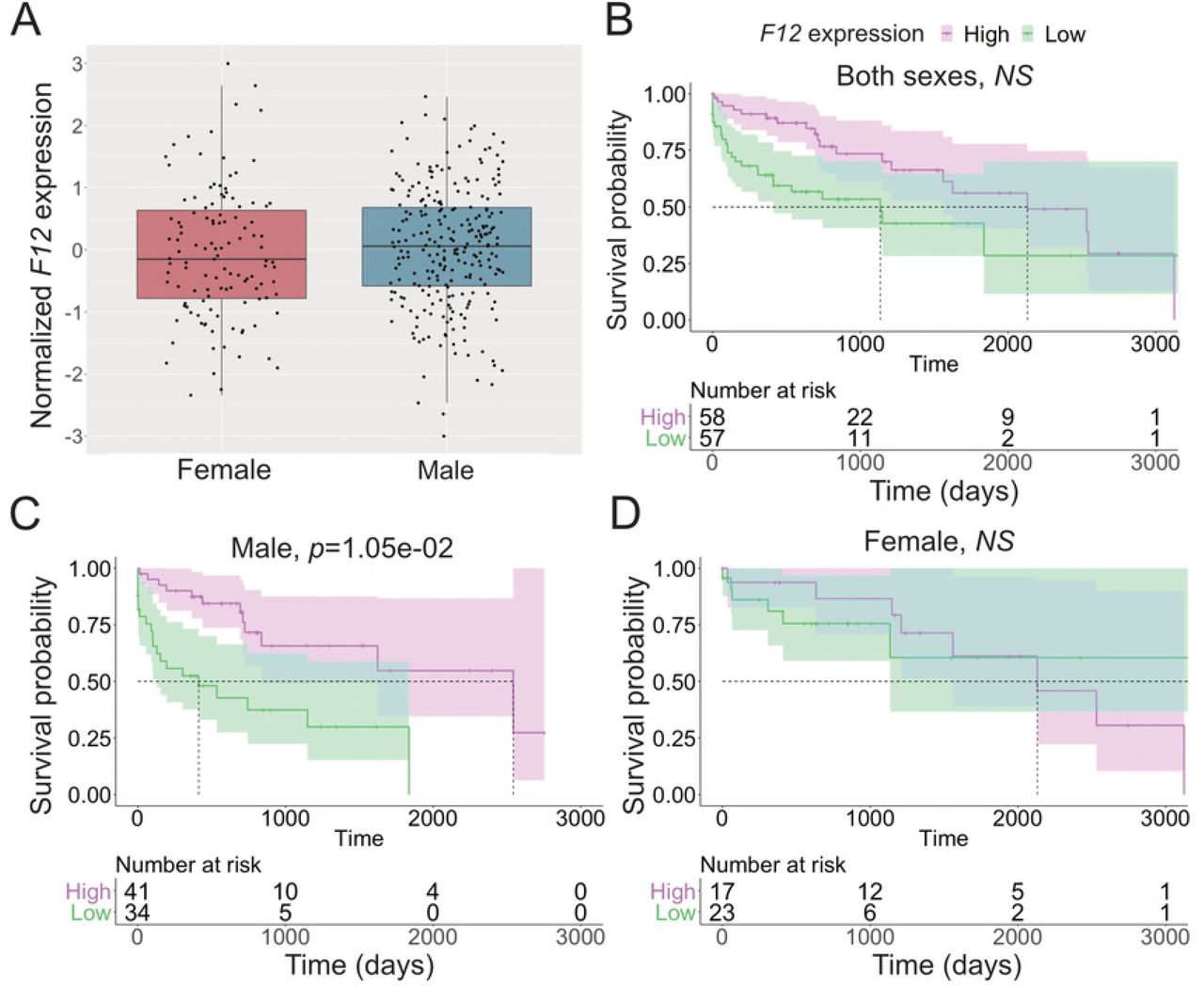
A: Expression of F12 in female and male HCC. B-D: Effect of F12 expression on survival in joint analysis of both sexes (B), female (C), and male (D). Cox’s proportional hazard ratio, FDR-adjusted p-value threshold 0.01. For visualization, samples with normalized expression value >1 were selected as high expression and <-1 as low expression samples.
Furthermore, several of the male-specific survival associated eGenes have been shown to be involved in HCC and other liver disorders. Family With Sequence Similarity 99 Member A (FAM99A) has previously been shown to be downregulated in HCC [69]. In our study, low FAM99A expression indicated poor survival in males (Table 3). Carboxypeptidase X, M14 Family Member 2 (CPXM2) has been identified as a driver gene in HCC based on patterns of somatic alterations [70]. Alkylglycerol Monooxygenase (AGMO) has been shown to be hypermethylated and transcriptionally repressed in HCC in comparison to cancer-free liver tissue [71]. Mutations in the Leucyl-TRNA Synthetase (LARS) gene have been identified as a cause of infantile hepatopathy [72].
In summary, we discovered differential genetic and regulatory functions in HCC tumors between the sexes. By integrating genotype and gene expression data, we identified putative regulatory mechanisms underlying known HCC risk loci and discovered genes under germline regulatory control modulating the overall survival of patients with HCC. Our results highlight the role of differential immune functions in cancer sex-bias and germline regulatory variants associated with these functions. This work provides a framework for future studies on sex-biased cancers.
Materials and Methods
Data
GTEx (release V6p) whole transcriptome (RNAseq) data (dbGaP accession #8834) were downloaded from dbGaP. TCGA LIHC Affymetrix Human Omni 6 array genotype data, whole exome sequence (WES) and RNAseq data (dbGaP accession #11368) were downloaded from NCI Genomic Data Commons [73]. FASTQ read files were extracted from the TCGA LIHC WES BAM files using XYAlign [74]. We used FastQC [75] to assess the WES and RNAseq FASTQ quality. Reads were trimmed using TRIMMOMATIC IlluminaClip [76], with the following parameters: seed mismatches 2, palindrome clip threshold 30, simple clip threshold 10, leading quality value 3, trailing quality value 3, sliding window size 4, minimum window quality 30 and minimum read length of 50.
Read mapping and read count quantification
Reads were mapped to custom sex-specific reference genomes using HISAT2 [77]. To avoid miss mapping of reads along the sex chromosomes, female samples were mapped to the human reference genome GRCh38 with the Y-chromosome hard-masked. Male samples were mapped to the human reference genome with Y-chromosomal pseudoautosomal regions hard-masked. Gene-level counts from RNAseq were quantified using Subread feature Counts [78]. Reads overlapping multiple features (genes or RNA families with conserved secondary structures) were counted for each feature.
Germline variant calling
BAM files were processed according to Broad Institute GATK (Genome Analysis Toolkit) best practices [79–81]: Read groups were added with Picard Toolkit’s AddOrReplaceReadGroups and optical duplicates marked with Picard Toolkit’s MarkDuplicates (v.2.18.1, http://broadinstitute.github.io/picard/). Base quality scores were recalibrated with GATK (v.4.0.3.0) BaseRecalibrator. Germline genotypes were called from whole blood Whole Exome Sequence samples from 248 male and 119 female HCC cases using the scatter-gather method with GATK HaplotypeCaller and GenotypeGVCFs [79]. Affymetrix 6.0 array genotypes of matching samples were lifted to GRCh38 using the UCSC LiftOver tool [82] and converted to VCF. Filters were applied to retain variants with a minimum quality score >30, minor allele frequency >10%, minor allele count >10, and no call rate <10% across all samples.
Cellular content of tumor samples
To examine the cellular heterogeneity of tumor samples, we performed cell type enrichment analysis for 64 immune and stromal cell types using xCell [83].
Filtering of gene expression data
FPKM (Fragments Per Kilobase of transcript per Million mapped reads) expression values for each gene were obtained using EdgeR [84]. Each expression dataset was filtered to retain genes with mean FPKM>0.5 and read count of >6 in at least 10 samples across all samples under investigation to avoid falsely inferring differential expression between two groups where both are functionally not transcribed.
Differential expression analysis
For differential expression (DE) analysis, filtered, untransformed read count data were quantile normalized and logCPM transformed with voom [85]. From the TCGA LIHC dataset, paired tumor and tumor adjacent samples were available for 22 females and 28 males. From the GTEx liver dataset, 22 female and 28 male samples were randomly selected to be used in the DE analysis. A multi-factor design with sex and tissue type as predictor variables were used to fit the linear model. To make comparisons both between and within subjects in the paired tumor and tumor adjacent samples, duplicateCorrelation was used to treat the individual as a random effect.
Differentially expressed genes (DEGs) between comparisons were identified using the limma/voom pipeline [85]. Linear modeling was conducted using the LmFit and contrast.fit functions. DEGs between sexes and tissue types were identified using contrast designs (e.g., all males for tumor versus all females for tumor, or all tumor samples versus all tumor adjacent samples within the same sex). In all comparisons, DEGs were identified by computing empirical Bayes statistics with eBayes, with an FDR adjusted p-value threshold of 0.01 and an absolute log2 fold-change (log2FC) threshold of 2.
Enrichment of biological functions and canonical pathways
We utilized the Gene Ontology Consortium’s web tool [86] to analyze gene lists for enrichment in regards to molecular function, biological processes, and cellular functions. Fisher’s exact test was used to calculate enrichment scores. We also used the NetworkAnalyst [87] for enrichment and network-based analyses of gene lists in regards to pathway relationships (KEGG and Reactome pathways). An FDR-adjusted p-value threshold of 0.01 was used to select significantly enriched GO terms and canonical pathways.
Accounting for technical confounders and population structure
Gene expression values are affected by genetic, environmental, and technical factors, many of which may be unknown or unmeasured. Technical confounding factors introduce sources of variance that may greatly reduce the statistical power of association studies, and even cause false signals [88]. Thus, it is necessary to account for known and unknown technical confounders. This is often achieved by detecting a set of latent confounding factors with methods such as principal component analysis (PCA), Probabilistic Estimation of Expression Residuals (PEER) [89] or Surrogate Variable Analysis (SVA) [88]. These known and surrogate variables are then used as covariates in downstream analyses, or expression data is adjusted using a regression model or other approach e.g. ComBat, which utilizes an Empirical Bayes method [90]. In eQTL studies, the number of latent factors to adjust for is often selected to maximize the number of significant associations. However, such an approach may remove biologically relevant sources of variation and increase the number of false positive associations. Introducing too many covariates to linear models may also cause a problem of overfitting. Furthermore, confounding factor methods may model the effects of trans-acting broad impact eQTL as confounding variation [91,92].
Our goal was to remove the technical sources of variation while protecting the biologically relevant variation. Because of missing values in technical covariate data, we were unable to adjust the expression data directly for known covariates. We identified 50 PEER factors in the tumor data. Some of these latent factors were associated with known clinical phenotypes or technical covariates (Fig S4). Weights of the PEER factors that did not strongly correlate with biologically relevant phenotypes were used as covariates in the eQTL analysis. To select these PEER factors, FDR adjusted p-value threshold of 0.01 based on linear regression for continuous covariates and ANOVA for categorical covariates were used. To avoid overfitting, the number of PEER factors to include was limited to 10.
We used the R package SNPRelate [93] to perform PCA on the germline genotype data. We accounted for population structure by applying the first three genotype PCs as covariates in the eQTL analysis.
eQTL analysis
Germline genotypes and tumor gene expression data from 248 male and 119 female donors were available for use in the eQTL analysis. Filtered count data was normalized by fitting the FPKM values of each gene and sample to the quantiles of the normal distribution. cis-acting eQTLs were detected with QTLtools v.1.1 [94]. We used the permutation pass with 10,000 permutations to get adjusted p-values of associations between the phenotypes and the top WES and array variants in cis. FDR adjusted p-values were calculated to correct for multiple phenotypes tested. An FDR-adjusted p-value threshold of 0.01 was used to select significant associations.
Annotating genomic locations of putative regulatory variants
We used the R package Annotatr to annotate the genomic locations of eVariants [95]. Variant sites were annotated for promoters, 5’UTRs, exons, introns, 3’UTRs, CpGs (CpG islands, CpG shores, CpG shelves), and putative regulatory regions based on ChromHMM [96] annotations.
Overlap of eQTL loci and HCC GWAS loci
Due to the lack of genome-wide GWAS summary statistics, we were unable to test for colocalization of GWAS and eVariants. To overcome this, we used top HCC GWAS variants and top eVariant to detect high confidence colocalization events based on linkage. Locations of 32 GWAS loci associated with HCC were obtained from the NHGRI-EBI GWAS catalog [97]. To find instances where known GWAS loci and eQTL loci are overlapping or tightly linked, we searched for loci with r2>0.8 of the GWAS risk locus. Linked loci were obtained with the LDLink’s LDProxy tool [98].
Survival analyses
We used the R package survival to test for differences in survival between different eVariant genotypes. To detect possible differences in early and late survival, we used the surv_pvalue function with log-rank, Tarone-Ware and Fleming-Harrington (p=1, q=1) tests. For genotype comparisons, the pairwise_survdiff function from the R package survminer was used to calculate p-values between all pairwise comparisons. To test for the effect of eGene expression on patient survival, we utilized Cox’s proportional hazards regression model with the function coxph. FDR adjusted p-values were calculated with the p.adjust function from the stats R package.
Acknowledgments
This study was supported by ASU Center for Evolution and Medicine postdoctoral fellowship for HMN, ASU School of Life Sciences startup funds for MAW and ASU Center for Evolution and Medicine Venture funds.
Citations
- 1.↵
- 2.↵
- 3.↵
- 4.
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.
- 24.
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.





{kind=link}
{kind=link}
{kind=link}
{kind=link}