

Abstract
Loss of the RUNX1 transcription factor leads to epithelial-to-mesenchymal transition (EMT), but mechanisms by which RUNX1 stabilizes the mammary epithelial phenotype are not known. Here, we report RUNX1 gene bookmarking during mitosis as one of the key epigenetic mechanisms to convey regulatory information for coordinate control of mammary cell proliferation, growth, and identity through successive cell divisions. Genome-wide RUNX1 occupancy profiles for asynchronous, mitotically-enriched, and G1 breast epithelial cells reveal RUNX1 is retained during mitosis on RNA Pol I- (i.e., ribosomal RNA) and II-transcribed protein coding (e.g., HES1) and long noncoding RNA (e.g., NEAT1) genes controlling proliferation, growth, and mammary epithelial phenotype maintenance. Disruption of RUNX1 DNA binding and target gene occupancy alters cell morphology, global protein synthesis, and phenotype-related gene expression. Together, these findings demonstrate that RUNX1 mitotic bookmarking contributes to maintenance of the normal mammary epithelial phenotype. Compromising RUNX1 DNA binding initiates EMT, an essential first step in the onset of breast cancer.
Summary Statement This study elucidates mitotic gene bookmarking as a novel epigenetic mechanism in breast epithelial cells which impacts cell growth and phenotype and has potential implications in breast cancer onset.
Breast cancer arises from a series of acquired mutations and epigenetic changes that disrupt normal mammary epithelial homeostasis and create multi-potent cells that can differentiate into biologically unique clinically distinct subtypes. Epithelial-to-mesenchymal transition (EMT) – a trans-differentiation process through which mammary epithelial cells acquire an aggressive mesenchymal phenotype - is a key driver of breast cancer progression, invasion and metastasis 1. Transcription factors Snail, Slug, Twist, and Zeb1/2 contribute to EMT during early, normal development and have also been implicated in invasion 2-5. Despite accumulating evidence for a broad understanding of EMT regulation and maintenance of the epithelial phenotype, the mechanism(s) by which mammary epithelial cells maintain their biological phenotype is unknown.
Runt-Related Transcription Factor 1 (RUNX1/AML1) is required for hematopoietic lineage specification during development and hematopoiesis throughout life 6-19. In addition to the recognized role in hematological malignancies, RUNX1 has been recently identified as a key player in breast cancer development and tumor progression 20-24. RUNX1 is significantly mutated in breast tumors 25-27. Findings from our group, reinforced by studies from others, have shown that RUNX1 maintains breast epithelial phenotype and prevents EMT through transcriptional regulation of genes involved in key cellular pathways 23. This regulation is reflected by RUNX1 control of E-cadherin expression, a key cell adhesion protein and marker of EMT 28. Consequently, there is a requirement to understand epigenetic mechanisms by which RUNX1 stabilizes the normal mammary epithelial phenotype.
Mitotic gene bookmarking, i.e. transcription factor binding to target genes during mitosis for transcriptional regulation following cell division, is a key epigenetic mechanism to convey and sustain regulatory information for cell proliferation, growth, and cell identity from parent to progeny cells 29-31. Phenotypic transcription factors that include GATA1, RBPJ, FoxA1, SOX2, OCT4, and KLF4, bookmark target genes during mitosis 32-36. We have established that RUNX proteins as well as other phenotypic transcription factors that include MYOD and CEBPα bookmark RNA Pol I- and II-transcribed genes during mitosis for coordinate control of cell growth, proliferation and phenotype. 37-40
We addressed the hypothesis that RUNX1 maintains the breast epithelial phenotype by mitotic bookmarking of genes that support mammary epithelial proliferation, growth, and phenotype during mitosis for expression immediately after cell division. Immunofluorescence confocal microscopy revealed that RUNX1 is present on chromosomes throughout mitosis and colocalizes with upstream binding transcription factor (UBF), a subunit of RNA Pol I transcriptional machinery. To identify genes occupied by RUNX1, we performed chromatin immunoprecipitation coupled with high throughput sequencing (ChIP-Seq) using a RUNX1-specific antibody on mitotic, G1, and asynchronous normal mammary epithelial MCF10A cells. ChIP-Seq revealed that, in mitosis, RUNX1 associates with RNA Pol II regulated genes specifically involved in maintenance of the epithelial phenotype and EMT progression. Interestingly, ribosomal RNA genes, regulated by the RNA Pol I transcriptional machinery, were occupied by RUNX1. A fluorescence-based, global protein synthesis assay showed reduced protein synthesis when RUNX1 DNA binding was perturbed using a small molecule inhibitor. Strikingly, inhibition of RUNX1 resulted in loss of the epithelial phenotype and acquisition of mesenchymal properties. These findings establish mitotic gene bookmarking as a key mechanism for RUNX1 stabilization of the normal breast epithelial phenotype. Importantly, disruption of RUNX1 mitotic bookmarking initiates EMT, an early event in the onset of breast cancer.
RESULTS
RUNX1 associates with mitotic chromatin and occupies target genes
To investigate subcellular localization of RUNX1 in normal mammary epithelial cells, we performed immunofluorescence microscopy in actively proliferating MCF10A cells. We observed that RUNX1 is distributed in punctate subnuclear domains throughout interphase nuclei (Fig. 1; the Interphase panel). Interestingly, RUNX1 is localized on mitotic chromatin at all topologically identified substages of mitosis (Fig 1; top panels). Two distinct types of foci are detectable on mitotic chromosomes: 2-8 large punctate foci that appear to be allelic as well as numerous smaller foci that were distributed across the chromosomes (Fig 1; bottom panels, white arrowheads). In agreement with our previous findings, RUNX1 foci are equally distributed into resulting progeny cells 40. Presence of RUNX1 at all stages of mitosis indicates that the protein is stable during cell division.
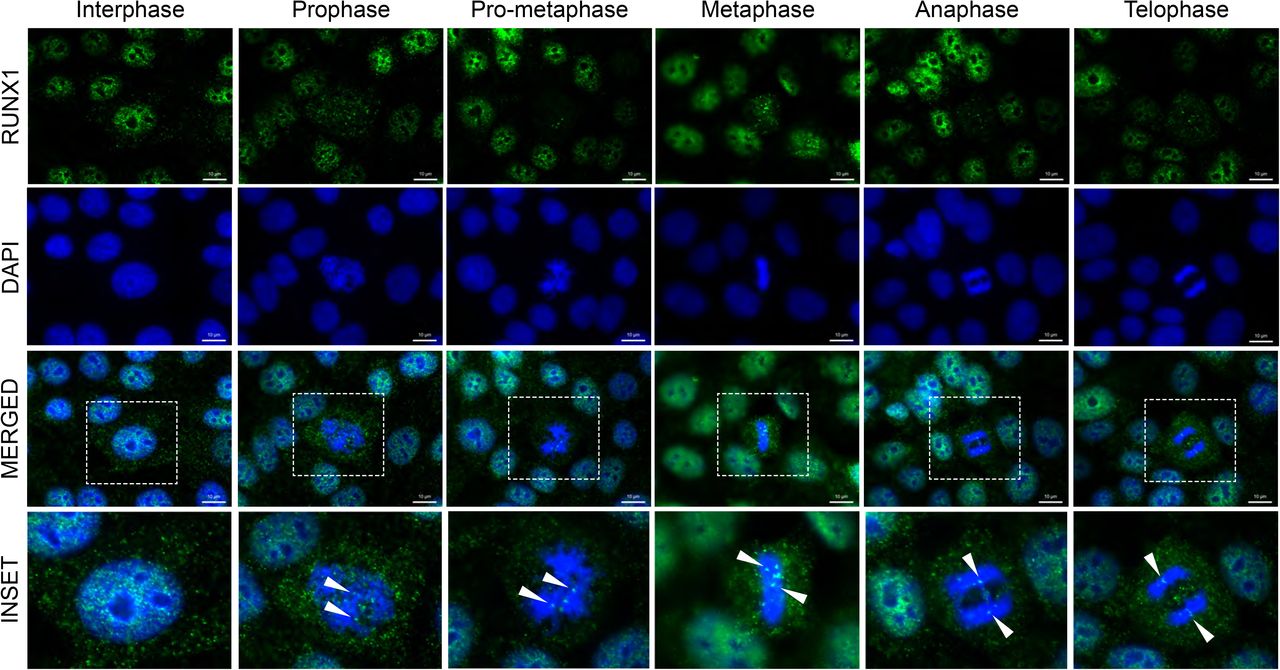
Representative immunofluorescent images of interphase and mitotic MCF10A breast epithelial cells. Mitotic cells were further classified into substages of mitosis based on DAPI topology. RUNX1 - Green (top row), DAPI - Blue (second row from top). Merged channel images (third row from top) contain an outlined region magnified in the bottom row labeled “inset”. White arrows highlight major Runx1 foci on chromatin.
To experimentally address RUNX1 occupancy of target genes, MCF10A cells were synchronized in mitosis using nocodazole (50ng/mL); additionally, cells in the G1 cell cycle stage was collected following a 3hr release from the block (Fig. 2A). Mitotic purity of harvested cells was confirmed by the presence of H3pS28 (>99%; data not shown). Western blot analysis of whole cell lysates from the three cell populations show expected levels of expression for cell cycle-specific proteins Cyclin B and CDT1 (Fig. 2B). FACS profiles of cell populations confirmed the characteristic enrichment of blocked cells in mitosis (Fig 2C; Mitotic) and release into G1 upon media replacement (Fig 1C; G1) when compared to asynchronous cells (Fig 2C; Asynch). Consistent with immunofluorescence observations, RUNX1 was present in all three cell populations (Supplement Fig. 1). Together, these results demonstrate that RUNX1 is stable through mitosis and localizes to mitotic chromatin.
We next determined if RUNX1 remains bound to target genes during mitosis, ChIP-Seq was performed on Asynch, Mitotic, and G1 MCF10A cells using a RUNX1 specific antibody (Fig. 2D). Sequencing datasets were mapped to the latest human genome build (hg38) using Bowtie2. Enriched regions were determined using Model-Based Analysis of ChIP-Seq (MACS) and were analyzed at p<10−5 significance level with an irreproducible discovery rate (IDR) of 0.05. Heatmaps of RUNX1-occupied genes in all three cell populations were generated by seqsetvis (Bioconductor) (Fig 2D). Comparison of the three cell populations revealed subsets of genes that were either shared (354 genes) across the three groups or were specific for each, indicating dynamic binding of RUNX1 during and immediately after mitosis (Fig 2D). Peak calling identified RUNX1 occupancy of both protein coding and long non-coding RNA (lncRNA) genes. Specifically, RUNX1 occupied 2020 genes in Asynch population (Fig 2D; green bar) and 1095 genes G1-enriched cells (Fig 2D; light brown bar). Importantly, RUNX1 occupied 551 genes (413 protein coding and 138 lncRNAs) in mitotically-enriched MCF 10A cells (Fig 2D; blue bar).

A) Experimental schematic depicting mitotic arrest and harvest of each treated MCF10A cell populations: Asynchronous - A, Mitotic - M, and Released - G1. B) Western blot of each harvested MCF10A population for cell cycle specific markers to evaluate mitotic arrest and synchronization procedure. C) Fluorescently activated cell sorting (FACS) analysis of harvested A, M, and G1 MCF10A cells to determine mitotic purity and DNA content (n=3 biological replicates per group). D) Heatmaps showing peaks called between A, M, and G1 MCF10A cells (left, middle, and right respectively). E) Venn diagrams illustrating the number of protein coding genes (left diagram) and lncRNAs (right diagram) identified within and between A, M, and G1 MCF10A populations. F) Motif analysis of A, M, and G1 MCF10A cells
Functional relevance of RUNX1 occupancy in the three cell populations was determined by comparing RUNX1-occupied genes with those that are differentially regulated upon shRNA-mediated RUNX1 knockdown23. Critically important to our central hypothesis that RUNX1 mitotically bookmarks genes for regulation immediately after cell division and as shown in Fig 2D, 399 of 1268 RUNX1-bookmarked genes in the M and G1 populations were deregulated upon RUNX1 depletion. These findings reveal that several hundred target genes are bookmarked by RUNX1 during mitosis and transcriptionally regulated in normal mammary epithelial cells. To identify cellular processes and pathways that comprise of RUNX1-bookmarked genes, we performed gene set enrichment analysis (GSEA) on genes bound by RUNX1 during mitosis or G1, or not bound in either cell cycle stage (Fig 2E). Interestingly, most genes bookmarked by RUNX1 during mitosis were associated with negative regulation of gene expression and metabolic process (Fig 2E; blue box). Consistent with cellular requirement to reattach and enter the next cell cycle and fully resume transcription, genes bound during G1 were primarily enriched in biological processes involving cell anchorage, protein localization and positive regulation of gene expression (Fig 2E; brown box). ChIP-seq results were further validated by motif analysis of RUNX1-bound peaks, which showed that RUNX motif was the top enriched motif in all three cell populations (Fig 2F). Importantly, RUNX1-bound genomic regions were also enriched in motifs for transcription factors known to cooperate with RUNX1 for gene regulation41 (Fig 2F). Together, these findings indicate that RUNX1 bookmarks genes involved in cell proliferation, growth, and phenotype in normal mammary epithelial cells.
RUNX1 mitotically bookmarks RNA Pol I-transcribed genes that control cell growth
Our ChIP-Seq results revealed that RUNX1 occupies rDNA repeats in MCF10A mammary epithelial cells; all three MCF10A cell populations (Asynch, Mitotic and G1) exhibited significant fold enrichment within the promoter region of hrDNA (Fig 4A), suggesting a potential role for RUNX1 in regulating rRNA genes in MCF10A cells. We confirmed this finding in actively proliferating MCF10A cells by immunofluorescence microscopy for antibodies specific against RUNX1 and upstream binding factor (UBF), a transcriptional activator that remains bound to rRNA genes during mitosis. We observed large RUNX1 foci colocalizing with UBF throughout each stage of mitosis (Fig 3A; bottom panels). Colocalization between RUNX1 and UBF was validated by confocal microscopy. Line scans of MCF10A cells show that although RUNX1 and UBF occupy distinct nuclear microenvironments in interphase (n=15), both proteins substantially colocalize in metaphase (n=15) (Fig 3B). Taken together, these findings establish RUNX1 binding to ribosomal DNA repeat regions identified by ChIP-Seq (Fig 4A) and confirmed at the cellular level by confocal microscopy (Fig 3).
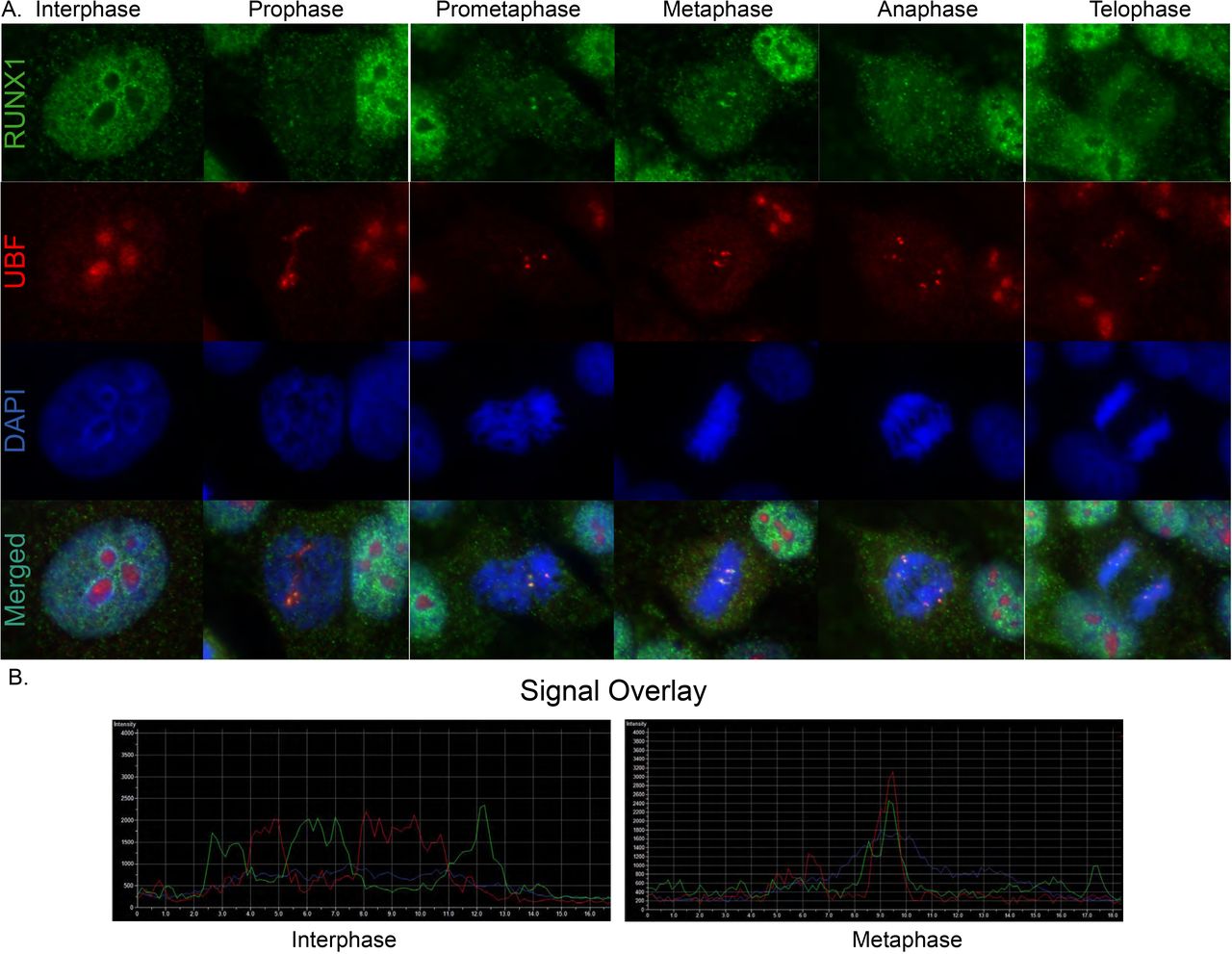
A) Immunofluorescence microscopy images of RUNX1 (green - top row), UBF (red - 2nd row from top), DAPI (blue - 2nd row from bottom), and the three channels merged (bottom row) in MCF10A cells. Images were captured of spontaneously dividing MCF10A cells in different substages of mitosis. B) Representative images of line profiles taken on interphase vs metaphase cells (n=15 each).

A) ChIP-Seq tracks of A, M, and G1 (top, middle, bottom respectively) MCF10A cells mapped against rDNA repeat regions. B) Representative immunofluorescence images of the active compound (AI-14-91)-treated MCF10A cells in prophase and metaphase are shown. A substantial decrease in smaller RUNX1 foci (green) during mitosis is observed when compared to the inactive (AI-4-88) compound. Large RUNX1 foci that colocalize with UBF (red) are detectable at all substages of mitosis (white arrows) in the presence of either active or inactive compounds. C) qRT-PCR data of pre-rRNA in actively proliferating MCF10A cells treated with either active (AI-14-91) or inactive (AI-4-88) compounds for 6, 12, 24, or 48hrs. Expression of pre-rRNA was normalized relative to Beta Actin expression. D) Representative fluorescence microscopy images of global protein synthesis occurring within MCF10A cells treated with either AI-4-88 (left) or AI-14-91 (right) for 24hr at 20μM. Intensity of red fluorescence at 580nm emission indicates nascent protein synthesis. All images were taken with 1000ms exposures.
We experimentally addressed the hypothesis that RUNX1 regulates ribosomal gene expression by using a pharmacological inhibitor of RUNX1. The small molecule inhibitor—AI-14-91—interferes with RUNX1-CBFβ interaction and disrupts RUNX1 DNA binding 42,43. We first determined the effect of RUNX1 inhibitor on mitotic retention of the protein. Actively proliferating MCF10A cells were treated with the inhibitor (AI-14-91) for 6hr, 12hr, 24hr, and 48hr at 20μM; a structurally equivalent inert compound (AI-4-88) was used as a control under identical conditions. Cells were subjected to immunofluorescence microscopy followed by detection of RUNX1 and UBF as described above. Although RUNX1 signal was detected in all mitotic sub-stages (data not shown), we observed a substantial decrease in RUNX1 signal intensity on mitotic chromosomes (white arrows; Fig 4B), indicating that RUNX1-Cbfβ interaction and RUNX1 DNA binding activity plays a key role in mitotic gene bookmarking. These changes were more pronounced for smaller RUNX1 foci and were not observed in control-treated cells; appreciable signal for large RUNX1 foci that colocalize with UBF (Fig 3) remained detectable in all sub-stages of mitosis (Fig 4B and data not shown).
We next examined the effect of RUNX1 inhibitor on pre-rRNA expression and found that pre-rRNA expression was significantly increased at 12hr and 48hr timepoints after treatment of asynchronous cells with specific RUNX1 inhibitor but not inactive compound, indicating that RUNX1 suppresses rRNA gene expression in normal mammary epithelial cells (Fig 4C). Because levels of rRNA directly correlate with global protein synthesis, a fluorescent-based detection method was used to measure newly synthesized proteins. Cells treated with AI-14-91 for 24hr or 48hr showed a moderate change in levels of global protein synthesis in comparison to control-treated cells under identical conditions (Fig 4D). Together, our results demonstrate that RUNX1 bookmarks RNA Pol I regulated rRNA genes during mitosis and transcriptionally represses them with moderate impact on global protein synthesis in normal mammary epithelial cells.
RUNX1 mitotically bookmarks RNA Pol II-transcribed genes involved in hormone-responsiveness and cell phenotype
Using RUNX1-bookmarked genes, gene set enrichment analysis (GSEA) was performed to identify regulatory pathways (Fig 5A). In agreement with known roles of RUNX1 44-48, top 10 pathways identified were those involved in regulation of G2M Checkpoint, E2F targets, p53, and DNA repair (Fig 5A). Consistent with our finding that RUNX1 bookmarked and regulates rRNA genes, one of the pathways identified is mTOR signaling, a pathway that is required for cell growth and is a therapeutic target in breast cancers 49,50. Relevant to the normal mammary epithelial phenotype, both early and late estrogen response signaling gene sets significantly overlap with RUNX1 mitotically bookmarked genes (Fig 5A). Because estrogen plays vital roles in promoting proliferative phenotypes of mammary epithelial cells 51-53, we interrogated RUNX1 bookmarked genes to identify those bound by RUNX1 and ERα (Fig 5B) 54. Using publicly available datasets of ERα genome-wide occupancy and estradiol-regulated gene expression, we find that a subset of genes mitotically bookmarked by RUNX1 is also bound by ERα, and either up or down regulated in response to estradiol. These findings indicate that RUNX1-bookmarked genes are involved in pathways that control hormone-responsiveness, proliferation and growth of normal mammary epithelial cells (Fig 5B).
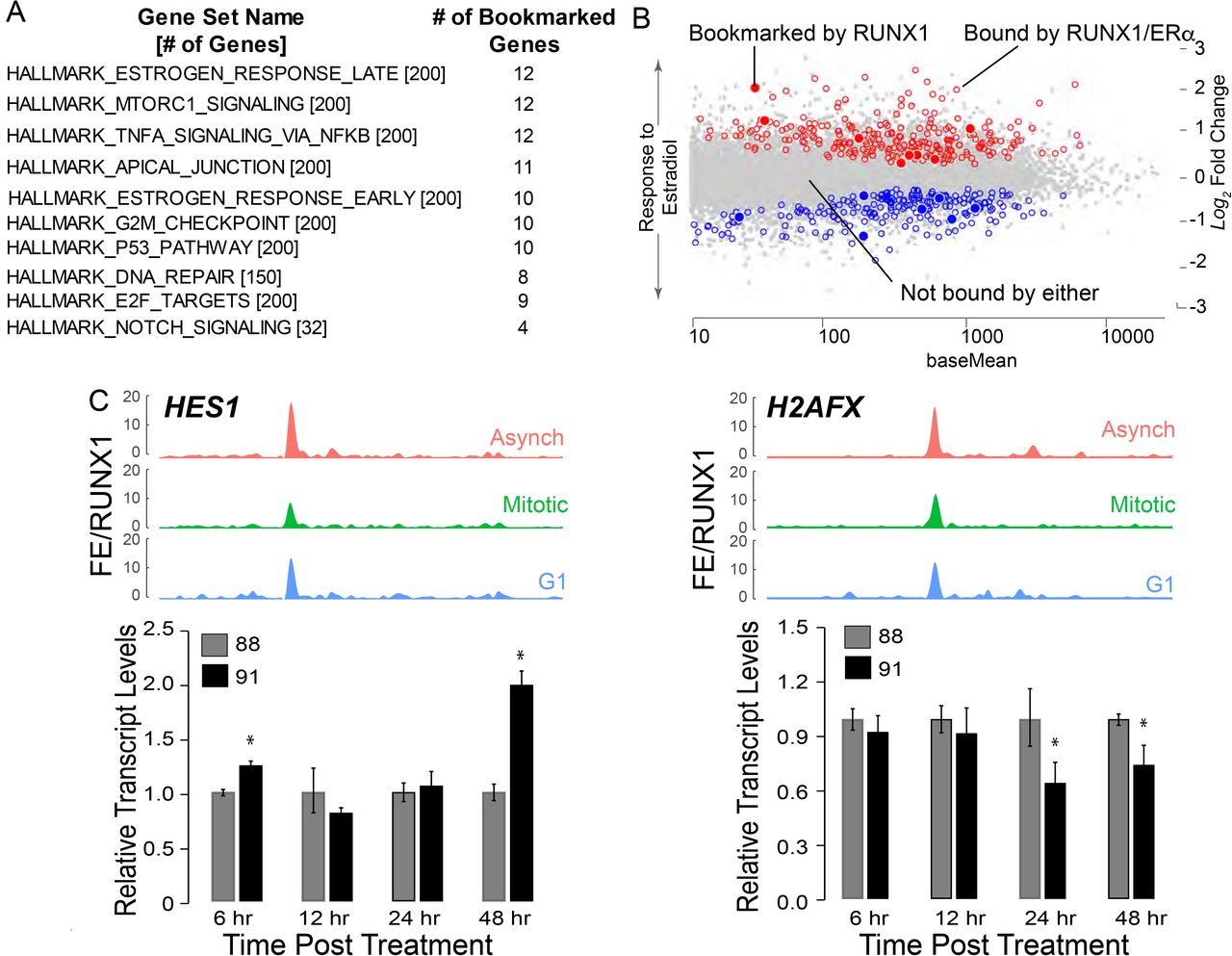
A) Gene Set Enrichment (GSE) analysis from interrogating mitotically bookmarked genes (i.e. RUNX1 mitotically occupied) against Hallmark Gene sets from Molecular Signatures Database (MSigDB). The top 10 most significantly overlapping gene sets are shown from top to bottom. B) Scatter plot of genes identified to be up or down regulated in response to estradiol treatment, that are also bound by estrogen receptor α (ERα) and RUNX1 (empty circles, blue for downregulated and red for upregulated). Scatter plot also illustrates up or down regulated genes in response to estradiol treatment that are bound by ERα and mitotically bookmarked by RUNX1 (filled in circles, blue for downregulated and red for upregulated). C) Top panel: ChIP-Seq tracks of HES1 (left) and H2AFX (right) from asynchronous (top-red), mitotic (middle-green), and G1 (bottom-blue). Bottom panel: qRT-PCR data of HES1 (left) and H2AFX (right) in asynchronous MCF10A cells treated with either active (AI-14-91) or inactive (AI-4-88) inhibitors for 6hr, 12hr, 24hr and 48hr at 20μM. Expression of target genes were normalized relative to beta actin.
A subset of RUNX1-bookmarked genes relates to regulatory pathways involved in cellular phenotype including TNFα, Apical Junction and Notch signaling (Fig 5A). Furthermore, NEAT1 and MALAT1, lncRNAs often deregulated in breast cancer 55,56, were also mitotically bookmarked by RUNX1. Of the 413 RUNX1 bookmarked protein coding genes, TOP2A, MYC, HES1, RRAS, H2AFX, and CCND3 are representative of RNA Pol II-transcribed genes involved in phenotype maintenance and cell fate decisions (See Supplemental Table 1 for complete list). Recently, HES1 and H2AFX have been identified as regulators of breast epithelial phenotype 57-59. In our ChIP-seq dataset, HES1 and H2AFX show significant fold enrichment of RUNX1 occupancy between the three populations of MCF10A cells (Fig 5C; top panels). Expression of HES1 increased upon inhibition of RUNX1 DNA binding (Fig 5C; left panel—bar graph), indicating that RUNX1 represses HES1. In contrast, H2AFX expression at 24hr and 48hr of inhibitor treatment was decreased, suggesting RUNX1 activates H2AFX expression (Fig 5C; right panel— bar graph). These results indicate that by bookmarking both protein coding and noncoding genes that are critical determinants of lineage identity, RUNX1 stabilizes the mammary epithelial phenotype.
Inhibition of RUNX1 DNA binding causes epithelial to mesenchymal transition
To experimentally address whether disruption of RUNX1 bookmarking leads to a change in epithelial phenotype, we treated cells with RUNX1 DNA binding inhibitor and monitored changes in cell morphology (Fig 6). Consistent with RUNX1 bookmarking and regulation of genes critical for epithelial phenotype (Fig 5), disruption of RUNX1 DNA binding resulted in mesenchymal morphology. We next examined whether long-term inhibition of RUNX1 caused a permanent change in cell phenotype. Longer term treatment (5 days) of actively proliferating MCF10A cells showed significant apoptosis, although a small sub-population of cells survived and exhibited an altered phenotype (Fig 6B). The surviving sub-population at day 5 was recovered by culturing cells in media without the inhibitor. By day 3-4 following media replacement, cells clearly showed a mesenchymal morphology (Fig 6B), indicating that interfering with RUNX1 mitotic bookmarking causes loss of the normal mammary epithelial phenotype. Consistent with changes in cell morphology, we find alterations in expression and localization of the cytoskeletal F-actin protein (Fig 6C). These observations were confirmed by examining the expression of epithelial markers (e.g., E Cadherin (Fig 6D)), as well as mesenchymal markers (e.g., SNAl2 (Fig 6E)). E-cadherin was partially downregulated, while SNAl2 expression was significantly increased, confirming an epithelial-to-mesenchymal transition upon inhibition of the RUNX1-Cbfβ interaction. The p21 gene, a known target that is repressed by RUNX1, was included as a control and, as expected, showed an increased expression with the inhibitor treatment (Fig 6E; right panel). Together, these findings show that RUNX1 mitotic bookmarking of epithelial cell growth, proliferation, and lineage-related genes is a key epigenetic mechanism required to stabilize the normal mammary epithelial phenotype. Disruption of RUNX1 gene bookmarking results in an epithelial-to-mesenchymal transition, a key first event at the onset of breast cancer.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A) Phase contrast microscopy images of MCF10A cells treated with AI-4-88 or AI-14-91 for 48hr at 20μM. Left panel - 4X magnification, middle panel - 20X magnification, right panel - 40X magnification. Outlined square in middle panel is the resulting 40X magnification in the right panel. B) Top Panel: Experimental schematic depicting treatment schedule for the “crisis” and “recovery” stages. Bottom Panel: Phase contrast microscopy images from Day 0, 1, and 2 of crisis where MCF10A cells were treated with AI-14-91 at 20μM (top - left, middle, right respectively). Phase contrast images from Day 0, 4, and 7 of recovery following a media replacement. C) Morphological changes upon inhibition of RUNX1-CBFβ interaction are confirmed by examining localization of the cytoskeletal protein F-actin. When compared to inactive compound (top panel), cells treated with active compound show substantial alterations in cytoarchitecture (bottom panel). D) Western blot for epithelial marker E-Cadherin (top row), mesenchymal marker Vimentin (middle row), and loading control beta actin (bottom row) in MCF10A whole cell lysates harvested from cells treated with either inactive AI-4-88 (88) or active AI-14-91 (91) inhibitors at 20μM for either 6, 12, 24, or 48hrs. E) qRT-PCR of target genes p21 (RUNX1 regulated) and SNAI2 (EMT inducing transcription factor) within asynchronous MCF10A cells treated with either inactive AI-4-88 (88) or active AI-14-91 (91) inhibitors at 20μM for either 6, 12, 24, or 48hrs. Expression of target genes were normalized relative to beta actin.
DISCUSSION
This study identifies RUNX1 mitotic bookmarking as a novel epigenetic mechanism for coordinate regulation of RNA Pol I- and II-transcribed genes that are critical for mammary epithelial proliferation, growth, and phenotype maintenance. Pharmacological inhibition of RUNX1 DNA binding causes transition to a mesenchymal phenotype, indicating that RUNX1 bookmarking of target genes contributes to stabilizing the normal breast epithelial phenotype.
Our findings are the first to identify coordinate control of cell growth-related ribosomal RNA (rRNA) genes and a large subset of cell proliferation/phenotype-related genes by RUNX1 in normal mammary epithelial cells. In addition to RUNX1 bookmarking of RNA Pol I-transcribed rRNA genes, RUNX1 is mitotically retained on RNA Pol II-transcribed genes that are important in breast epithelial cell growth and phenotype. One target gene of interest is hairy and enhancer of split-1 (HES1). Hes1 is a transcription factor which represses genes involved in cellular development, and is regulated primarily by NOTCH signaling, one of our top ten overlapping hallmark gene sets bookmarked by RUNX1 (Fig 5) 60,61. HES1 was recently shown to have a prominent role in proliferation and invasion of breast cancer cells, and its silencing led to a downregulation of p-Akt signaling and ultimately prevented EMT57. Our findings indicate that RUNX1 stabilizes the normal mammary epithelial phenotype, in part, by bookmarking HES1 and suppressing its expression.
Another important RNA Pol II-transcribed gene mitotically bookmarked by RUNX1 and critical for maintaining cellular phenotype is histone variant H2AFX (H2AFX). Silencing H2AFX in breast epithelial cells leads to induction of EMT through activation of SNAIL2/SLUG and TWIST159. We find a decrease in H2AFX expression and a concomitant, significant increase in SNAIL2/SLUG expression upon inhibition of the RUNX1-Cbfβ interaction. These data identify RUNX1 as a novel upstream regulator of H2AFX expression; RUNX1 bookmarking and activation of H2AFX and subsequent suppression of SNAIL2/SLUG prevents EMT in breast epithelial cells.
Several groups have shown that RUNX1 interacts with ERα at both enhancer regions and transcriptional start sites (TSSs) for regulation of specific genes 22,54. Our ChIP-Seq results, coupled with publicly available data sets, reveal a novel finding: RUNX1 bookmarking of a subset of ERα-occupied, hormone-responsive genes, during mitosis may be critical for maintenance of breast epithelial phenotype. Future studies will be required to investigate mechanistic significance of this observation.
Our findings are the first to demonstrate that mitotic gene bookmarking contributes to stabilizing the mammary epithelial phenotype. Equally important, our study shows that inhibition of RUNX1 DNA binding specifically elicits an epithelial-to-mesenchymal transition, indicating that mitotic gene bookmarking is a central epigenetic mechanism by which RUNX1 maintains the epithelial phenotype. These findings are further supported by RUNX1 mitotic occupancy of cell growth-related rRNA genes, and together highlight key role(s) of RUNX1 in coordinating cell proliferation, growth and phenotype. Another novel contribution of the current study is mitotic bookmarking of lncRNAs by a transcription factor. RUNX1 was recently shown to regulate lncRNAs NEAT1 and NEAT2 (MALAT1)55,62, lncRNAs with critical roles in the onset and progression of breast cancer56. Our findings show that, in addition to bookmarking protein coding genes, RUNX1 bookmarks several lncRNAs for post-mitotic regulation. It will be important to identify G1-specific roles of RUNX1-bookmarked lncRNAs in maintaining the normal mammary epithelial phenotype and/or in the onset and progression of breast cancer.
In summary, this study establishes a novel epigenetic mechanism where RUNX1 mitotically bookmarks RNA Pol I- and II-transcribed genes for coordinate regulation of normal mammary epithelial proliferation, growth, and phenotype. Disruption of RUNX1 DNA binding leads to epithelial-to-mesenchymal transition, a key event in breast cancer onset, and validates the contribution of RUNX1 bookmarking to physiologically sustain the mammary epithelial phenotype.
MATERIALS AND METHODS
Cell Culture Techniques
Breast epithelial (MCF10A) cells were cultured in DMEM/F-12 50/50 mixture (CorningTM, Corning, NY). Culturing media was also supplemented with horse serum to 5% (GIBCO, Grand Island, NY), human insulin to 10μg/mL (Sigma Aldrich, St. Louis, MO), human epidermal growth factor to 20ng/mL (PeproTech, Rocky Hill, NJ), cholera toxin to 100ng/mL (Thomas Scientific, Swedesboro, NJ), hydrocortisone to 500ng/mL (Sigma Aldrich, St. Louis, MO), Penicillin-Streptomycin to 100U/mL (Thermo Fisher Scientific, Ashville, NC), and L-Glutamine to 2mM (Thermo Fisher Scientific, Ashville, NC).
For mitotic arrest of parental MCF10A cells, culturing media was supplemented with 50ng/mL of Nocodazole (Sigma Aldrich, St. Louis, MO) and incubated with cells for 16hrs. Supplementing culturing media with equivalent volumes of DMSO (Sigma Aldrich, St. Louis, MO) served as a negative control. For DMSO-treated and mitotically arrested populations of MCF10A cells, harvests were conducted following the 16hr incubation. For G1 (released from mitotic arrest) populations of MCF10A cells, the nocodazole-supplemented culturing media was replaced with normal culturing media and incubated with cells for 3hrs. Following the 3hr incubation, released populations of cells were harvested for subsequent analysis.
Protein Expression and Localization
SDS-PAGE was performed to visualize protein expression within MCF10A cells. 8% SDS resolving gels and 4% stacking gels were prepared in-house (National Diagnostics, Atlanta, GA). Cell harvests were resuspended in RIPA buffer and incubated on ice for 30min. Following incubation, cell lysates were sonicated using Q700 Sonicator (QSonica, Newtown, CT). Total sonication time for samples were 70 seconds, with 7 programed cycles of 10 seconds sonication at power setting 30 followed by 30 seconds of no sonication. Sonicated lysates were centrifuged at 15,000 rpm for 30min at 4°C. Protein concentration in the remaining supernatant was quantified using a PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific, Ashville, NC). Electrophoresis was performed at 160V for 15min followed by 200V for 45min. Overnight wet transfer of protein into PVDF membranes was performed at 30V for 18hr in 4°C. PVDF membranes were blocked at room temperature in 5% BSA in 1X TBST. Primary antibodies used for protein visualization were diluted 1:1000 and raised against UBF (sc-13125, Santa Cruz Biotechnology, Dallas, TX), RUNX1 (4334S, Cell Signaling Technologies, Danvers, MA), Cyclin B (4138S, Cell Signaling Technologies, Danvers, MA), Beta-Actin (3700S, Cell Signaling Technologies, Danvers, MA), and CDT1 (ab70829, AbCam, Cambridge, UK). Lamin B1 (ab16048, AbCam, Cambridge, UK) primary antibody was used at 1:2000 dilution for protein visualization. Primary antibodies were diluted in 5% BSA in 1XTBST and incubated with blots overnight at 4°C. Blots were washed four separate times with PBST or TBST. Goat anti-mouse IgG HRP conjugated (31460, Invitrogen, Carlsbad, CA) secondary antibody was incubated with blots at 1:5000 and incubated for 1hr at room temperature with mild agitation. Goat anti-rabbit IgG HRP conjugated (31430, Thermo Fisher Scientific, Ashville, NC) secondary antibody was incubated with blots at 1:1000, 1:2000, or 1:5000 and incubated for 1hr at room temperature with mild agitation. Blots were developed using Clarity Western ECL Substrate (Bio-Rad, Hercules, CA) following manufacturer’s instructions. Blots were exposed to visualize protein and images were captured using Molecular Imager® Chemi docTM XRS+ Imaging System (Bio-Rad, Hercules, CA). Captured images were processed using Image Lab Software Version 5.1 (Bio-Rad, Hercules, CA).
Immunofluorescent microscopy was performed to observe distribution and localization of protein expression within MCF10A cells throughout all stages of mitosis and interphase. MCF10A cells were plated within a 6 well plate at 175,000 cells/mL on coverslips coated in gelatin (0.5% w/v solution in 1XPBS) and allowed to grow overnight. Coverslips were washed twice with sterile-filtered PBS at 4°C. Coverslips were then placed in room temperature fixative solution (1% MeOH-free Formaldehyde in PBS) for 10min. After a sterile-filtered PBS wash, coverslips were transferred to permeabilization solution (0.25% Triton X-100 in PBS) for 20min on ice. Following another sterile-filtered PBS wash, coverslips were then blocked in sterile-filtered PBS supplemented with bovine serum albumin (PBSA) at 0.5% w/v (Sigma Aldrich, St. Louis, MO). Coverslips were then incubated with primary antibody for 1hr at 37°C in a humidified chamber. Primary antibodies were specific for RUNX1 at a dilution of 1:10 (4334S, Cell Signaling Technologies, Danvers, MA) and Upstream Binding Transcription Factor (UBF) at a dilution of 1:200 (F-9 sc-13125, Santa Cruz Biotechnology, Dallas, TX). Coverslips were washed four separate times in sterile-filtered PBSA following primary antibody incubation. Coverslips were then placed in secondary antibody for 1hr at 37°C within a humidified chamber. Secondary antibodies used were goat anti-rabbit IgG conjugated with Alexa Fluor 488 (A-11070, Life Technologies, Carlsbad, CA) and goat anti-mouse IgG conjugated with Alexa Fluor 594 (A-11005, Life Technologies, Carlsbad, CA) diluted 1:800. Coverslips were then washed four times in sterile-filtered PBSA. Staining of the coverslips for DNA was performed with 1.0μg DAPI in 0.1% Triton X-100 and sterile-filtered PBSA for 5min on ice. Stained coverslips were washed once in 0.1% Triton X-100 in sterile-filtered PBSA, then two times with sterile filtered PBS. Coverslips were mounted onto slides using ProLong Gold Antifade Mountant (Thermo Fisher Scientific, Ashville, NC). Images were captured using a Zeiss Axio Imager.Z2 fluorescent microscope and Hamamatsu ORCA-R2 C10600 digital camera. Images were processed using ZEN 2012 software.
Confocal microscopy was performed on slides prepared as described above. MCF10A breast epithelial cells were initially imaged with a Zeiss LSM 510 META confocal laser scanning microscope (Carl Zeiss Microscopy, LLC., Thornwood, NY, USA) for a preliminary study to assess potential colocalization. At a later time, additional samples were imaged with a Nikon A1R-ER laser scanning confocal microscope (Nikon, Melville, NY,USA) for complete colocalization analysis. Images were acquired with the resonant scanner at a frame size of 1024 × 1024 pixels with 8X averaging. Fluorescently labeled samples were excited by laser lines sequentially imaged in channel series mode. The DAPI signal was excited with a 405 nm laser and collected with a 425-475 nm band pass filter, Alexa 488 was excited with a 488 nm laser and collected with a 500-550 nm band pass filter, and Alexa 568 with a 561 nm laser and collected with a 570-620 nm band pass filter. Images were captured with a Plan-Fluor 40X (1.3 NA) objective lens. The confocal pinhole was initially set to 1.2 Airy Unit diameter for the 561 nm excitation giving an optical section thickness of 0.41 μm. Images were acquired at 12-bit data depth, and all settings, including laser power, amplifier gain, and amplifier offset were established using a lookup table to provide an optimal gray-scale intensities. All images were acquired using matching imaging parameters.
Images were acquired with at 40X objective were subject to colocalization analysis via Volocity version 6.3.0 (Perkin Elmer, Waltham, MA, USA). Images were opened in the colocalization tab. Cell nuclei, indicated by the DAPI signal, were circled via the ROI tool. At least 15 interphase and 15 metaphase cells were identified within captured images and appropriate thresholds were manually determined to eliminate background fluorescence for calculating Pearsons and Manders correlation coefficients between RUNX1 and UBF.
Images were also viewed in NIS Elements version 5.02.01 and analyzed using the line profiling tool. Overlaying DAPI, RUNX1, and UBF fluorescent intensities from individual channels along the line profile revealed overlapping peak intensities between the RUNX1 and UBF channels, thus indicating colocalization.
Core binding factor - Beta (CBFβ) inhibitors AI-4-88 and AI-14-91 were given to us from John H. Bushweller (University of Virginia) and used to evaluate RUNX1 DNA-binding inhibition in MCF10A cells. Protein synthesis evaluation by immunofluorescence was conducted following manufacturer protocol (K715-100, BioVision, San Francisco, CA).
Molecular Techniques
Total RNA was isolated from MCF10A cells using TRIzolTM Reagent (Invitrogen, Carlsbad, CA) and Direct-ZolTM RNA MiniPrep isolation kit (Zymo Research, Irvine, CA) following manufacturer instructions. cDNA was created using SuperScript IV® First-Strand Synthesis System for RT-PCR (ThermoFisher, Asheville, NC). Resulting samples were quantified on a Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) and diluted to 500pg/μL. Equal amounts of DNA template were loaded for samples analyzed by qPCR.
Chromatin Immunoprecipitation was conducted on asynchronous (Asynch), mitotically arrested (M), and released from mitosis (G1) MCF10A breast epithelial cells. Cells were fixed with 1% v/v MeOH-free Formaldehyde in 1XPBS for 10min at room temperature. Formaldehyde fixation was neutralized using 2.5M Glycine and incubated with cells for 5min at room temperature. Two washes with 1XPBS supplemented with cOmpleteTM EDTA-free Protease Inhibitor Cocktail (Sigma Aldrich, Saint Louis, MO) and MG-132 (Calbiochem-Millipore Sigma, Burlington, MA) were performed. For asynchronous and G1 populations of cells, culture dishes were scraped to collect fixated lysate. Mitotic cells were isolated using a mitotic shake off. Mitotic cells were spun down at 1500rpm × 5min, resuspended in 1% v/v MeOH-free Formaldehyde in 1XPBS, and neutralized with 2.5M Glycine for 5min. Fixed harvests were centrifuged at 1500rpm × 5min (4°C) and the supernatant was discarded. All fixed cell pellets were flash frozen in liquid nitrogen and stored at −80°C until lysis.
Fixed cell pellets were thawed on ice. Once thawed, pellets were lysed in a nuclear lysis buffer supplemented with cOmpleteTM EDTA-free Protease Inhibitor Cocktail (Sigma Aldrich, Saint Louis, MO) and MG-132 (Calbiochem-Millipore Sigma, Burlington, MA) with a volume that was approximately 5X the volume of pellet. Pellets were incubated in nuclear lysis buffer for 30min before being flash frozen down in liquid nitrogen. Lysates were thawed at room temperature but not allowed to reach room temperature. Sonication of the lysates were performed using a S220 focused ultra-sonicator (Covaris, Matthews, NC). Sonication parameters for each population of cells was as follows: Peak Watt 140W, Duty Factor 10, Cycle/Burst 200. M and G1 populations of cells were sonicated for 28min total whereas asynchronous populations of cells were sonicated for 36min. All samples were sonicated at 6°C. Following sonication, aliquots were spun down at 15,000rpm × 10min and 4°C. Following the spin, the resulting supernatants were pooled together and analyzed.
Sonicated lysate was boiled in 100°C for 15min with NaCl and elution buffer. Boiled lysate was allowed to cool and treated with RNaseA (10ug/uL) for 10min at 37°C. DNA was isolated using PureLinkTM PCR Purification Kit (K310001, ThermoFisher, Ashville, NC) following manufacturer recommendations. Resulting DNA was quantified via nanodrop and 1.0-2.0ug was run on a 1.5% agarose gel to observe sonication results prior to generating ChIP reactions. Resulting DNA was also quantified via Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) and analyzed by using a High Sensitivity DNA Kit on a Bioanalyzer 2100 (Agilent, Santa Clara, CA).
For chromatin immunoprecipitation (ChIP) reactions, 150ug of sonicated chromatin was incubated with 10ug of RUNX1 antibody (4336BF, Cell Signaling Technologies, Danvers, MA), diluted 1:10 in IP dilution buffer, and incubated overnight (16-18hrs) at 4°C with mild agitation. Following incubation, 150uL of Protein A/G magnetic beads (Thermo Scientific - Pierce, Waltham, MA) per ug of antibody used were added to each IP reaction and incubated for 2-4hrs at 4°C with mild agitation. Beads were isolated from solution using a powerful magnet, and washed two times in two separate IP wash buffers. Lastly, beads were resuspended in an elution buffer and agitated in a thermomixer (Eppendorf, Hamburg, Germany) or vortexer at 1000rpm × 30min at room temperature. This elution step was repeated on the beads. Using a magnet, beads were discarded and the resulting supernatant was incubated with NaCl overnight (16-18hrs) at 67°C to reverse formaldehyde crosslinks. DNA from RUNX1 ChIP samples were purified using PureLinkTM PCR Purification Kit (K310001, ThermoFisher, Ashville, NC) following manufacturer recommendations.
ChIP libraries were generated using Accel-NGS® 2S Plus DNA Library kit (Swift Biosciences, Ann Arbor, MI) following manufacturers protocol. Input and RUNX1 ChIP samples were normalized to 1ng prior to library generation. Libraries were amplified in an optional PCR step for 12 total cycles. Finalized libraries were double size selected using AMPure XP beads (0.8X and 0.2X volume ratios to sample), resulting in the majority fragments sized between 250-400bp. Next generation sequencing of pooled ChIP libraries was performed by the University of Vermont Cancer Center - Vermont Integrated Genomics Resource (VIGR).
Bioinformatics Analyses
Because we were specifically investigating rDNA, a customized build of hg38 was constructed that included normally masked regions of rDNA (Gencode U13369). Since some (although not complete) rDNA sequence is present in the hg38 assembly, we masked all parts of hg38 that would normally be attributed to rDNA sequences (bedtools v2.25.0 maskfasta). Finally, we appended the complete rDNA sequence as a “unique” chromosome (chrU13369.1) to the masked hg38 FASTA resulting in the hg38_rDNA assembly used for analysis.
Single-end, 50bp reads (SE50) were processed pre-alignment by removing adapter reads (Cutadapt v1.6) and trimming low quality base calls from both ends (FASTQ Quality Trimmer 1.0.0; min score >= 20, window of 10, and step size of 1). Resulting reads were aligned to hg38_rDNA (STAR v2.4; splicing disabled with ‘– –alignIntronMax 1’). Next, we called peaks and generated fold-enrichment (FE) bedGraph files (MACS2 v2.1.0.20140616; callpeak at p-value e-5; and bdgcmp with FE method) 63. Irreproducible Discovery Rate (IDR) was conducted using unpooled replicates with all peaks in pooled samples passing an IDR cutoff of 0.5 64. To reduce artificial peaks, we calculated strand cross-correlation for all peaks at a shift of 95 bp (the mean observed fragment size of 180 bp minus the read size of 85bp) and unshifted 65. We eliminated peaks that exhibited low shifted correlation (shifted correlation <.7) and those that exhibited high unshifted correlation relative to shifted (shifted - unshifted correlation < .1). This increased retrieval of the RUNX1 motif and improved agreement with other RUNX1 datasets. Passing peaks were annotated separately to mRNA and lncRNA transcript start sites (TSSs) using GENCODE v27 with a distance cutoff of 5000 bp. Regional distribution of peaks was determined using the same annotation reference limited to the “basic” tag for exons and promoters.
COMPETING INTERESTS
No competing interests declared.
FUNDING
This work was supported by NIH grants NCI P01 CA082834 (to G.S. Stein and J.L. Stein), R01 CA139322 (to G.S. Stein), R37 DE012528 (to J.B. Lian), NCI F32 CA220935 (to A.J. Fritz, G.S. Stein, and J.L. Stein), U01 CA196383 (to J.L. Stein), and the Charlotte Perelman Fund for Cancer Research (to G.S. Stein). The confocal microscopy work described in this manuscript was supported by Award Number 1S10RR019246 from the National Center for Research Resources for purchase of the Zeiss 510 META confocal scanning laser microscope and NIH award number 1S10OD025030-01 for purchase of the Nikon A1R-ER point scanning confocal microscope from the National Center for Research Resources. FACS experiments performed at the Harry Hood Bassett Flow Cytometry and Cell Sorting Facility, University of Vermont College of Medicine were supported by NIH S10-ODO18175.
DATA AVAILABILITY
GEO accession number for the sequencing data generated in this study is GSE121370.
ACKNOWLEDGEMENTS
The authors would like to thank John H. Bushweller, PhD (University of Virginia), who created and gifted us Core binding factor - Beta (CBFβ) inhibitors AI-4-88 and AI- 14-91 to conduct RUNX1 inhibition experiments for this study. Confocal imaging and colocalization analysis were performed by Nicole Bouffard in the Microscopy Imaging Center at the University of Vermont College of Medicine. The authors would also like to thank Scott Tighe, Pheobe Kehoe, and Jessica Hoffman for performing next generation sequencing of samples (Vermont Integrated Genomics Resource (VIGR) at the University of Vermont Cancer Center). The authors also thank Roxana del Rio-Guerra, Ph.D of the UVM Flow Cytometry and Cell Sorting Facility for analysis of samples by FACS. The authors would also like to thank Alan Howe, Ph.D. (University of Vermont) for his Phalloidin reagent used in immunofluorescence microscopy experiments.
REFERENCES




