

ABSTRACT
Condensins are evolutionarily conserved protein complexes that are required for chromosome segregation during cell division and genome organization during interphase. In C. elegans, a specialized condensin, which forms the core of the dosage compensation complex (DCC) binds to and represses X chromosome transcription. Here, we analyzed DCC localization and effect of DCC depletion and binding on histone modifications, transcription factor binding, and gene expression using ChIP-seq and mRNA-seq. Across the X, DCC binding coincides with gene regulatory sites at active chromatin and not heterochromatin. DCC is required for reducing the levels of active histone modifications, including H3K4me3 and H3K27ac, but not repressive modification H3K9me3 on the X. In X-to-autosome fusion chromosomes, DCC spreading into the autosomal sequences locally reduces the level of H3K4me3 and gene expression. Together, our results suggest that DCC fine-tunes X chromosomal transcription by binding to and modulating the activity of gene regulatory elements through reducing activating histone modifications.
BACKGROUND
Regulation of chromosome structure is essential for the establishment and maintenance of accurate gene expression. A key regulator of chromosome structure across all organisms is condensin, a multi-subunit protein complex that belongs to the family of structural maintenance of chromosomes (SMC) complexes [1, 2]. Condensins are required for chromosome condensation and segregation in all eukaryotes [3]. Condensins are also important for genome organization and have been implicated in gene regulation during interphase [4]. Genome-wide binding experiments indicate that condensins bind to a subset of gene regulatory elements including promoters, enhancers, tRNA genes, and topologically associated domain (TAD) boundaries [5]. However, the link between condensin binding at these sites and its function in gene regulation remains unknown.
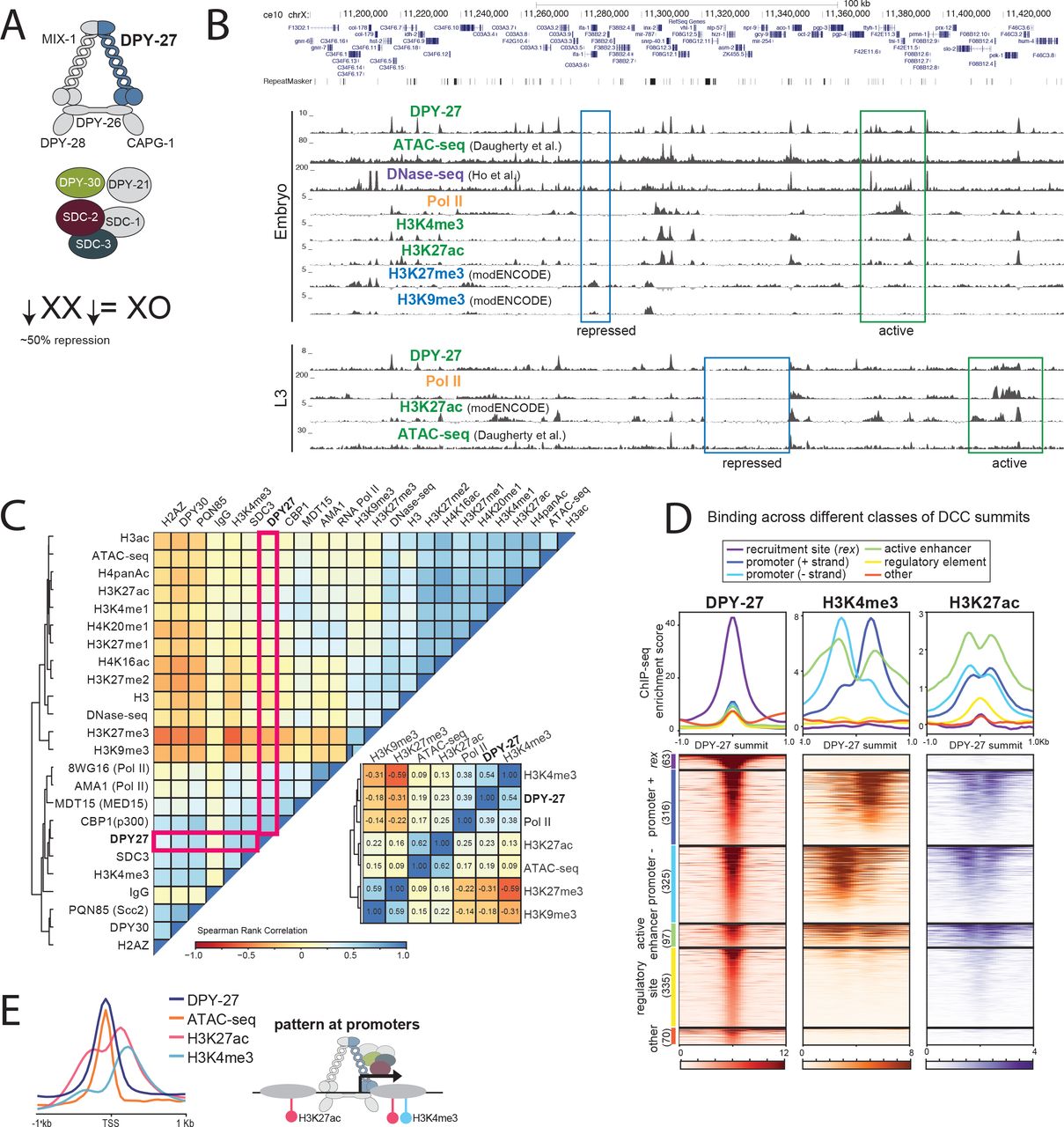
Here we addressed the link between condensin and transcription by using a clear paradigm for the gene-regulatory function of condensins, the C. elegans dosage compensation complex (DCC) [6]. Like most metazoans, C. elegans contain two types of condensins (I and II) that partially differ in their subunit composition, chromosomal binding and function [7]. In addition to the canonical condensins, C. elegans contains a third condensin, condensin DC, that differs from condensin I by a single SMC-4 variant, DPY-27 [7]. Condensin DC interacts with additional subunits necessary for DCC binding and function (Figure 1A). The DCC specifically binds to both hermaphrodite X chromosomes and represses each by half to equalize X chromosomal transcript levels between XX hermaphrodites and XO males [8–10].

(A) The C. elegans dosage compensation complex (DCC) contains a specialized condensin complex (condensin DC) that is distinguished from canonical condensin I by a single SMC-4 variant, DPY-27. The non-condensin DCC subunits SDC-2, SDC-3, and DPY-30 interact with condensin DC, and are required for its recruitment to the X chromosomes. DPY-21 is a histone demethylase that converts H4K20me2 to H4K20me1. DCC binds to and represses X chromosomes in hermaphrodites by approximately two-fold. (B) ChIP-seq, DNase-seq [24] and ATAC-seq [22] profiles at a representative 250kb region of the X chromosome in embryos and L3 larval stage worms. Example active and repressed chromatin regions are labeled in blue and green, respectively. DPY-27 (DCC) binding overlaps with Pol II binding, active chromatin marks, and accessible regions (ATAC-seq). Repressive marks are enriched in less accessible regions lacking DPY-27 (DCC) and Pol II. (C) Spearman rank correlation values are shown for average ChIP-seq scores of histone modifications, ATAC-seq and DNase-seq signals within 1kb contiguous windows across the X chromosome. DCC (DPY-27) binding positively-correlates more with promoter marks (H3K4me3) and Pol II, with active enhancers (H3K27ac) and regulatory regions (ATAC-seq), and negatively-correlates with repressive marks (H3K27me3, H3K9me3). (D) DPY-27 (DCC), H3K4me3, and H3K27ac wild type embryo ChIP-seq patterns are plotted across DPY-27 ChIP-seq binding peak summits. The summits were categorized as recruitment element on the X (rex), promoter [+ strand], promoter [− strand], active enhancer, regulatory element, and other (see text for details). Approximately half of DCC containing gene regulatory sites also contain active marks H3K4me3 and H3K27ac. (E) Pattern of DCC, H3K4me3, H3K27az and ATAC-seq signals across X chromosome transcription start sites indicate colocalization of DCC signal with accessible region of the promoters.
The current model for DCC binding to the X chromosomes involves two steps: recruitment and spreading [11]. Recruitment is mediated in a hierarchical manner, where the DCC enters the X at a small number of strong recruitment sites, which are fully distinguished from the autosomes by the presence of multiple copies of a 12-bp recruitment motif within high occupancy transcription factor target (HOT) sites [12]. The stronger and weaker recruitment sites are thought to cooperate over long distances to robustly recruit the DCC to the X [12]. Unlike recruitment, spreading is an X-sequence independent process and can occur on DNA physically attached to the recruitment sites [13]. An estimated 50-100 recruitment sites separated by 0.1-1 Mb distances support binding of the DCC across the ~17Mb X chromosome [12, 14].
Global run on (GRO-seq) [10] and chromatin immunoprecipitation sequencing (ChIP-seq) [8] analyses showed that the DCC is required to reduce RNA Pol II binding at X chromosomal promoters. DCC mediated repression appears to be chromosome-wide, with no large groups of genes escaping from dosage compensation [8, 9]. Previous work has highlighted multiple roles for the DCC in regulation of X chromosome structure; DCC is required for the ~40% compaction of the X compared to autosomes [15], the regulation of subnuclear localization of the X chromosomes [16, 17], and the regulation of topologically associating domains (TAD) on the X [18, 19]. The DCC was also shown to increase and decrease the levels of H4K20me1 and H4K16ac, respectively, on the X chromosomes [20, 21]. Reduction of H4K16ac on the X occurs downstream of H4K20me1 enrichment, and requires the deacetylase SIR-2.1 [21]. H4K20me1 enrichment on the X is mediated by the H4K20me2 demethylase DPY-21, which physically interacts with the condensin core of the DCC [18]. The mechanism by which H4K20me1 enrichment on the X contributes to transcription repression is predicted to be indirect [8]. Previous studies did not address if the DCC regulates X chromosome expression by changing the level or the distribution of histone modifications linked to transcription regulation.
To address this question, we analyzed the distribution of several histone modifications in wild type, DCC mutant and DCC depleted conditions, as well as in X-to-autosome fusion strains in which the DCC ectopically spreads into autosomal sequences [13]. In wild type embryos and L3 larval animals, DCC binding sites coincide with accessible gene regulatory elements marked by ATAC-seq [22]. In DCC mutant (dpy-21 null) or depleted (dpy-27 RNAi) embryos, the levels of repressive histone modifications, including H3K9me3, remain unchanged while levels of active histone modifications, including H3K4me3 and H3K27ac, increase at X chromosomal promoters compared to autosomal ones. Further linking DCC binding to the regulation of active histone marks and gene expression, in X;V fusion chromosomes ectopic spreading of the DCC into autosomal sequence locally reduces both gene expression and H3K4me3 levels. We also found that DCC depletion does not affect binding of PHA-4 transcription factor, the cohesin loader PQN-85 (Scc2 homolog), nor the putative H3K27acetylase CBP-1 as measured by ChIP-seq, thus ruling out a model in which DCC indiscriminately reduces binding of proteins to the X chromosomes. Taken together, our results suggest that the DCC fine-tunes transcription across the X by targeting and modulating the activity of gene regulatory elements by interfering with specific transcriptional coactivators that deposit activating histone modifications on the X.
RESULTS
DCC is preferentially enriched at active gene regulatory elements on the X
To understand the DCC’s effect on histone modifications, we first compared the genomic distribution of the DCC to marks of active and repressed chromatin in embryos and L3 larval stages (Figure 1B). A combination of new and published ChIP-seq data, including those from modENCODE [23], and published accessibility data for ATAC-seq [22] and DNase-seq [24] were used. New data were produced from at least two biological replicates that correlate based on visual examination of genome browser tracks (Supplemental Figure 1). Summary and access information on all data sets is provided in Supplemental File 1.
Genome browser analysis of DCC binding revealed a correlation with active chromatin (Figure 1B). DCC ChIP-seq enrichment coincides with ATAC-seq peaks at promoters and enhancers containing RNA Pol II, H3K4me3 and H3K27ac (Figure 1B) [22]. Conversely, marks of repressive chromatin, including H3K27me3 and H3K9me3 do not coincide with DCC binding (Figure 1B). Comparison of additional DCC subunits DPY-30 and SDC-3, histone modifications, and proteins including the transcription factor PHA-4, cohesin loader subunit PQN-85 (Scc2 homolog), putative H3K27 acetylase CBP-1 (p300 homolog), and the mediator subunit MDT-15 (Med15 homolog) support the conclusion that DCC binding coincides with gene regulatory sites (Supplemental Figure 2). To further compare the distribution of active and repressive marks with DCC localization, we plotted the Spearman rank correlation of average ChIP-seq enrichment within 1 kb contiguous windows across the X chromosome (Figure 1C). DCC subunit DPY-27 binding correlates best with H3K4me3, RNA Pol II, MDT-15, and CBP-1, proteins enriched at active gene regulatory elements.
The complex pattern of DCC ChIP-seq profile suggest different modes of binding
The DCC has a complex pattern of binding as measured by ChIP-seq, including somewhat uniform baseline enrichment across the X, peaks of different heights at promoters, enhancers and within genes, and strong enrichment at the recruitment sites [12, 25]. To further analyze sites of DCC enrichment, we focused on the top 50% of peaks sorted by their ChIP-seq score at the summit, mostly eliminating peak calls due to baseline DCC binding (Supplemental Figure 3). Next, we categorized the DCC peaks as those located at recruitment sites [12], promoters (within 250 bp of a GRO-seq or 500 bp of a Wormbase defined transcription start site (TSS) [10]), active enhancers (overlapping a H3K27ac peak that is not a promoter), gene regulatory elements (overlapping an ATAC-seq or DNase-seq peak and not promoter or active enhancer), and unknown categories. We then plotted DCC, H3K4me3 and H3K27ac enrichment patterns across the DCC summits in each category (Figure 1D). This analysis revealed that a majority of DCC binding peaks occur at active promoters and enhancers, and that the strength of DCC binding correlates with the activity of the gene regulatory site, as measured by H3K4me3 and H3K27ac enrichment. DCC binding at promoters coincides with chromatin accessibility, and surrounding H3K27ac and H3K4me3 enrichment at the +1 nucleosome (Figure 1E).
DCC binding at promoters partially correlates with their transcriptional activity
Unlike the majority of sites, approximately one third of DCC binding sites show little H3K4me3 and H3K27ac enrichment (Figure 1D), suggesting that DCC binding is not restricted to elements marked by these modifications. To understand the different modes of DCC binding, we next scrutinized the level of correlation between DCC binding and transcription. Previous ChIP-chip analysis of DCC binding in embryos and L4/young adults showed a correlation between binding and gene expression [13]. Similarly, DCC and RNA Pol II binding (as measured by ChIP-seq using 8WG16 antibody recognizing the unmodified C terminal of AMA-1 (large subunit)) show DCC enrichment changes at genes differentially bound by RNA Pol II in embryos and L3s (Figure 2A). Supporting the conclusion that DCC correlates with active transcription, DCC binding at promoters is higher at genes that are being transcribed compared to silent genes and genes whose mRNAs were maternally deposited in embryos (Figure 2B).
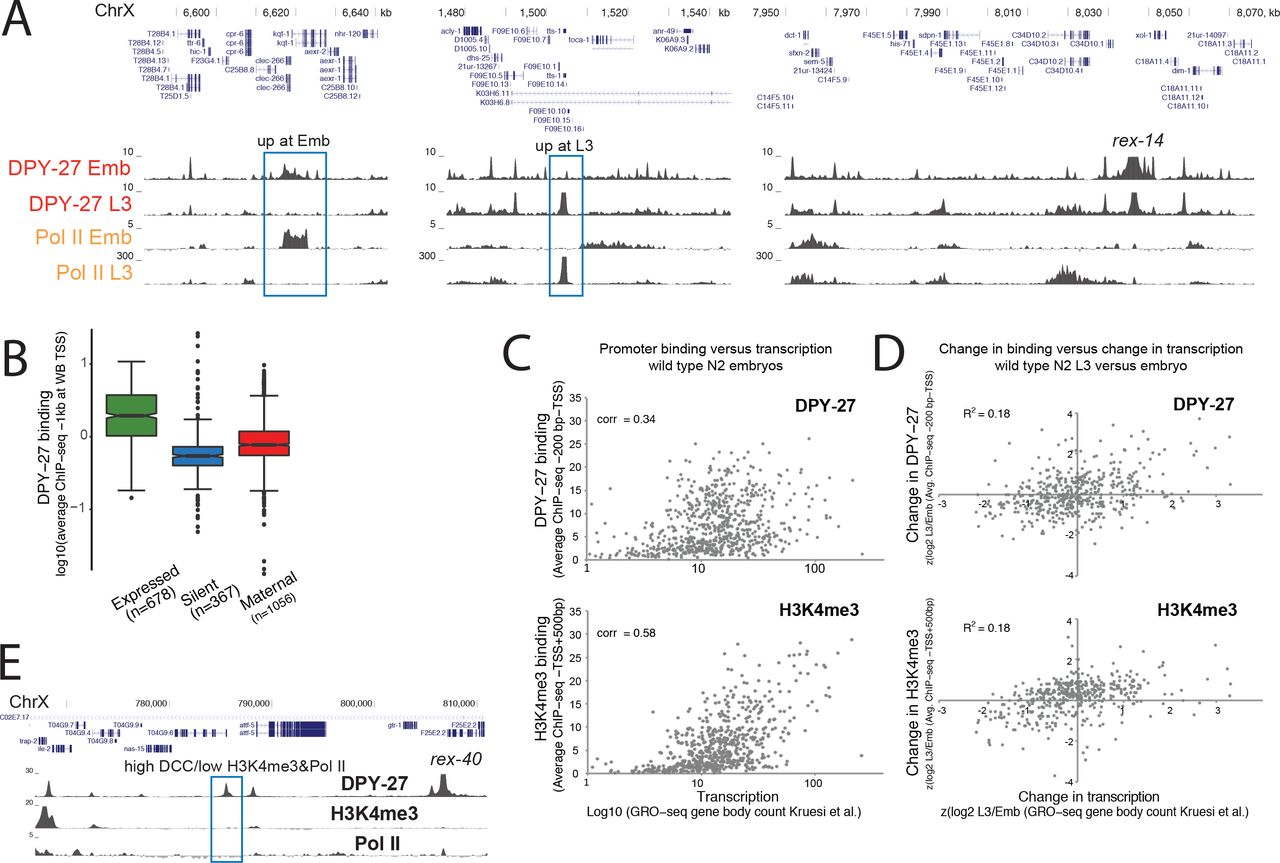
(A) DPY-27 ChIP-seq binding across example X chromosomal regions with differential transcription in embryos versus L3s. (B) DPY-27 binding at 1kb around the TSS of expressed genes (N2 embryos FPKM >1 [8] and detected in GRO-seq [10]), silent genes (FPKM = 0 and not detected in GRO-seq), and maternally loaded genes (FPKM >1 and not detected in GRO-seq) on the X chromosomes. (C) Average DPY-27 and H3K4me3 ChIP-seq scores at promoters positively correlate with transcription as measured by GRO-seq counts at corresponding gene bodies. (D) Standardized ratio of binding (y axis) is compared to transcription (x axis) in L3 versus embryos. Change in DPY-27 and H3K4me3 partially correlate with the change in transcription at individual promoters. (E) A 40 kb region around a recruitment site shows a strong recruitment site, and additional DCC binding peaks (blue rectangle), which show low Pol II and H3K4me3, suggesting that DCC enrichment and transcriptional activity at promoters can be uncoupled
The level of positive correlation between DCC binding and transcription (as measured by GRO-seq [10], Spearman rank correlation of 0.34) is less than that observed between H3K4me3 and transcription (0.58), suggesting that the DCC and H4K3me3 differ in relation to RNA Pol II binding (Figure 2C). To evaluate if the levels of DCC and H3K4me3 are tuned to transcription at individual promoters, we plotted the change in DCC or H3K4me3 levels versus change in transcription between embryos and L3s (Figure 2D). While there is a slight positive correlation, both DCC and H3K4me3 do not perfectly follow transcription changes at individual promoters. Furthermore, at and near recruitment sites, we found sites that with high DCC and low H3K4me3 and RNA Pol II (Figure 2E). These results suggest that while DCC binding generally correlates with transcriptional activity, the two are not strictly coupled.
DCC modulates the levels of active histone modifications on the X
To determine DCC’s effect on gene regulatory elements, we analyzed the level of several histone modifications associated with active and repressed chromatin upon DCC knockdown (dpy-27 RNAi) and mutation (dpy-21(e428) V) in embryos. Since DCC represses transcription by approximately 2-fold, we expected and observed subtle changes. To quantify such subtle changes, we used the autosomes as an internal control for ChIP efficiency and calculated the standardized ratio of ChIP enrichment in mutant versus wild type. This approach detected previously described X-specific changes upon DCC mutation and knockdown, including reduction of H4K20me1 [20, 21] (Figure 3A) and increase of RNA Pol II [8, 26] (Figure 3B). The levels of histone H3 and negative control IgG at promoters do not show a significant change in dpy-21 mutant embryos (Figure 3C), ruling out a comparable effect on nucleosome occupancy. The same analysis approach revealed a DCC-dependent reduction in the X chromosomal levels of H3K4me3 (Figure 3D), H3K27ac (Figure 3E), H4pan-ac, and H4K16ac, but not H3ac (Figure 3F), suggesting that the DCC reduces specific active histone modifications at X chromosomal promoters.

Change in level of histone modifications upon DCC defect are plotted. Average ChIP enrichment within 1 kb windows centered at the GRO-seq defined transcription start sites were calculated in wild type (N2), DCC mutant (dpy-21) and DCC depleted (dpy-27 RNAi) embryos. Change in the level of each histone modification was measured by standardizing log2 ratio of experimental to control ChIP-seq scores. Difference in values from each chromosome were tested against all the other autosomes using a two-tailed Student’s t-test and resulting p-values that were less than ≤ 0.001 were marked with an asterisk. This analysis captured the expected changes in H4K20me1 (A) and RNA Pol II (B) at X chromosomal promoters upon DCC defect. (C) Neither H3 nor IgG negative control ChIP-seq data showed a comparable difference in the dpy-21 mutant, suggesting that the measured effects through this analysis are specific. (D-F) Analysis of different histone modifications associated with active transcription. (G) ChIP-seq enrichment for H3K4me3 and H3K27ac in wild type, dpy-21 mutant and dpy-27 RNAi knock down embryos was plotted across the TSS sites on the X and chromosome I. The level of enrichment is ordered in a descending manner using maximum coverage in wild type. (H) Genome browser view of ChIP-seq profiles in wild type, mutant and knockdown embryos over a 250kb representative region of the X chromosome. The similar order of enrichment in (G) and the similar patterns in (H), and correlation between wild type and mutant ChIP-seq signal (see text) suggest that the DCC regulates the level of histone marks rather than their distribution. (I) Similar analysis as in (A), but ChIP-seq scores in wild type and mutant were calculated over 200 bp window centering at the summit of peaks in wild type embryos. (J) Similar analysis as in (A), but ChIP-seq scores in wild type and mutant were calculated over top 1% of 1kb windows ordered by average ChIP-seq scores in wild type embryos.
The increase in the levels of active histone modifications upon DCC depletion was due to reduction of these histone modifications at their canonical locations rather than a different distribution of ChIP-seq binding patterns (Figure 3H). H3K4me3 and H3K27ac enrichment across the transcription start sites in wild type, dpy-21 mutant and dpy-27 RNAi conditions show small differences, but generally, sites with high enrichment in the wild type are still highly enriched in the mutant conditions (Figure 3G). Supporting the same conclusion, Spearman rank correlation values between wild type and mutant H3K4me3 and H3K27ac enrichment within 1 kb contiguous windows were similar on the autosomes and the X (H3K4me3 N2-CB428 on X: 0.59 on autosomes:0.57-0.65 ; H3K4me3 control-dpy-27 RNAi on X:0.55 on autosomes:0.52-0.65; H3K27ac N2-CB428 on X:0.79 on autosomes:0.79-0.85; H3K27ac N2- dpy-27 RNAi on X:0.85 on autosomes:0.84-0.89), indicating a lack of X-specific change in the distribution of histone modifications upon DCC defect. Collectively, these results suggest that the DCC defect did not create or eliminate new sites of enrichment on the X but increased the level of H3K4me3 and H3K27ac at their canonical locations.
Since the distribution of modifications remains similar in the DCC knockdown embryos, we focused on the changes at their canonical sites (ChIP-seq peaks in wild type). H3K4me3 and H3K27ac are enriched near the transcription start sites (Figure 3G) and show distinct patterns of enrichment (Figure 1E). The levels of H3K3me3 and H3K27ac within 200 bp of their canonical binding summits increase specifically on the X upon DCC defect (Figure 3I). Peak calling on lower and broader ChIP-seq patterns observed for H4pan-ac, H4K16ac, and H3ac was difficult, therefore to analyze their binding, we took the top 1% of 1 kb windows based on wild type ChIP-enrichment. In the dpy-21 mutant, the level of H4pan-ac increases specifically on the X, but H4K16ac and H3ac do not (Figure 3J). Greater variability in the dpy-21 mutant compared to dpy-27 RNAi may be due to additional dpy-21 activity outside dosage compensation [8, 18]. For H4K16ac, an X-specific effect at the TSS, but not at top 1% H4K16ac sites suggests spatial specificity for DCC–mediated reduction of H4K16ac.
DCC-mediated change in histone modifications and gene expression
Our results indicated that the DCC reduces activating histone modifications at X chromosomal promoters. Since DCC also reduces RNA Pol II binding to promoters [8, 10], we asked if DCC-dependent decrease in Pol II binding correlates with decrease in histone modifications at individual promoters. RNA Pol II and DCC binding positively correlate with the levels of active histone modifications at promoters in both wild type and dpy-21 mutant embryos (Supplemental Figure 4A). At individual promoters on the X, change in RNA Pol II binding upon DCC defect did not correlate as well with the change in active histone modifications (Supplemental Figure 4B). Nevertheless, RNA Pol II and H3K4me3 ChIP-seq ratios in dpy-21 mutant versus wild type positively correlated (0.21 for AMA-1 antibody). Interestingly, differential binding of DPY-27 and H3K27ac in dpy-21 versus wild type negatively correlated specifically on the X (spearman rank correlation of −0.32 on X and 0.04 on autosomes), supporting the idea that the DCC reduces H3K27ac on the X chromosomes.
DCC binding does not affect repressive histone modifications
To test if the DCC affects histone modifications indiscriminately, we performed ChIP-seq analysis of H3K4me1, H3K27me1, H3K27me2 and H3K9me3 (Figure 4A). H3K4me1, H3K27me1 and H3K27me2 did not yield strong signals, precluding clear conclusions (Supplemental Figure 1). Nevertheless, changes in these modifications were neither X-specific nor consistent between dpy-27 RNAi and dpy-21 mutant (Supplemental Figure 5A). We observed no difference in H3K9me3 distribution on the X chromosomes between control and dpy-27 RNAi treated embryos (Figure 4A). H3K9me3 levels showed higher variability in control and dpy-27 RNAi conditions, but the difference is not restricted to the X chromosome suggesting that the DCC does not specifically regulate H3K9me3 (Figure 4B). We also considered the opposite, and tested if H3K9me3 affects DCC localization by using a strain in which H3K9 methylation is eliminated [27]. In the absence of H3K9me3, RNA Pol II binding pattern is similar to that of wild type (Figure 4C), consistent with the lack of overt effect on growth in laboratory conditions [27]. The reason for a general reduction in ChIP scores in the mutant is unclear. Regardless, RNA Pol II binding is not specifically different on the X (Figure 4D), consistent with a lack of effect for H3K9me3 on DCC binding and dosage compensation. Similarly, SDC-3 (DCC subunit required for DPY-27 recruitment to the X), and CAPG-1 (HEAT domain subunit of condensin DC) binding profiles are similar between the wild type and mutant (Figure 4C). Furthermore, CAPG-1 peaks in the H3K9me3 mutant largely overlap with those of the wild type (Figure 4E), and genomic sites with high H3K9me3 enrichment do not coincide with new SDC-3 sites (Figure 4F). These results suggest that DCC binding does not regulate and is not regulated by heterochromatin mark H3K9me3 on the X.

(A) ChIP-seq profile of H3K9me3 in control and DPY27 RNAi along a representative region of chrX exemplifies no significant change in H3K9me3 upon DCC knockdown. (B) Change in H3K9me3 ChIP-seq scores at the top 1% most enriched 1kb windows in the control and dpy-27 RNAi compared to N2 embryos. (C) ChIP-seq profiles of DPY-27, DCC subunits (SDC-3, CAPG-1), Pol II, and H3K9me3 in wild type and the H3K9me3 null mutant (GW638, met-2, set-25) across a representative region of the X. DCC and Pol II binding profiles remained similar in the met-2, set-25 mutant, including in regions enriched in H3K9me3 in wild type (blue rectangle). (D) Pol II binding in H3K9me3 null mutant compared to N2 wild type suggest no specific effect of met-2, set-25 on dosage compensation. Average ChIP enrichment within 1 kb windows centered at the GRO-seq defined transcription start sites was calculated, and change was measured by standardized log2 ratio of the experimental to the control condition. None of the chromosomes showed a significant difference in Pol II binding (p≤ 0.001) compared to the rest, based on a two-tailed Student’s t-Test. (E) ChIP-seq peak overlap of DCC subunit CAPG-1 between wild type and H3K9me3 null mutant. (F) ChIP-seq peak overlap between SDC-3 and top 1% H3K9me3 enriched 1kb windows. E & F suggest that lack of H3K9me3 in met-2, set-25 does not lead to improper SDC-3 binding to H3K9me3 containing regions in the wild type
DCC spreading into autosomal loci in X;A fusion chromosomes represses gene expression
To determine if DCC spreading reduces gene expression and histone modification levels locally, we analyzed strains containing X-to-autosome fusion (X;A) chromosomes. Previous work showed that the DCC spreads into the autosomal regions of X;V, X:II, and X:I fusion chromosomes, and that spreading is linear and reduces with distance from the X [13]. Ectopic DCC binding leads to increased H4K20me1 in the autosomal region of spreading [20]. An earlier microarray analysis in embryos did not detect a significant difference in gene expression in the fusion strains [13]. Subsequent experiments suggested that dosage compensation starts in embryogenesis but is not complete until larval stages [8], therefore we repeated the experiment using mRNA-seq in larvae.
DPY-27 ChIP-seq analysis verified that the DCC binds to the autosomal region of spreading in the X;V fusion chromosomes in larvae (Figure 5A). To test if gene expression specifically changed in the region of spreading, first we took three 0.5 Mb windows at the middle, left- and right-most end of each chromosome, and plotted the ratio of mRNA-seq levels for genes within each window. Average gene expression is significantly and specifically reduced at the side of X fusion, which is the right most end of chromosome V in the X;V strain, and the left-most end of chromosome II in the X;II strain (Figure 5B). The level of repression reduces with distance from the fusion site (Figure 5C), and is proportional to the level of spreading demonstrated for each fusion [13]. These results indicate that DCC spreading into the autosomal regions of the X;V and X;II fusion strains results in repression.

(A) DPY-27 (DCC) ChIP-seq profile in the wild type and X;V fusion chromosome containing strains in L3 larvae. The spreading profile of the DCC in the autosomal region of the fusion chromosome is similar to that on the X, as indicated by Pol II and ATAC-seq signal in wild type. (B) mRNA-seq analyses in strains containing wild type X, V and II, X;V and X;II fusion chromosomes. DESeq log2 expression ratios were calculated and plotted for genes located within middle, left and right-most 500kb windows of chromosomes II, V, and X. The schematics above the boxplots show which chromosome arms are fused. For X;II, the right end of X was fused to the left end of chr II, and for X;V the right end of X was fused to the right end of V [49]. * p-value ≤ 0.001 (two-tailed Fisher test). (C) Similar to (B), but expression ratios are plotted for genes within 1 Mb windows stepping out from the fusion site. The amount of repression decreases as a function of distance from the fusion border, following the pattern of DCC spreading [13]. (D) Standardized log2 ratio of H3K4me3 ChIP-seq scores within 200bp centering at wild type H3K4me3 peak summits located at the middle, left and right-most 1Mb windows of chromosomes II, V, and X. H3K4me3 significantly decreases in the DCC spreading region (two-tailed Student’s t-Test). (E) Average DPY-27 ChIP-seq scores for 1 kb GRO-seq defined TSS regions are plotted in grey. Change in expression and histone modifications were calculated by a moving average analysis using a 200 kb window with a 20 kb step size. For each 200 kb window, ChIP-seq and mRNA-seq ratios in X;V/wt were compared to the rest of the chromosome and a p-value statistic was generated through t-test. In this analysis rather than asking if there is a significant change for each gene (as in DEseq), we ask whether the values in each window are significantly higher or lower than the values observed for the rest of the windows along the chromosome. Windows with a p-value ≤ 0.01 are plotted.
DCC spreading into autosomal loci leads to H3K4me3 reduction
In the X;V fusion strain, DCC spreads further [13] and causes stronger repression (Figure 5C). Thus, we assayed the change in H3K4me3 and H3K27ac levels in the X;V fusion chromosomes. Since the expected change is small, we focused the analysis on where the signal is highest by taking a standardized ratio of ChIP-seq enrichment in X;V versus wild type at 200 bp around canonical peak summits. Despite high variability, there is a significant reduction in average H3K4me3 levels within the spreading domain of the X;V fusion chromosomes compared to wild type (Figure 5D). For H3K27ac, there is higher variability across chromosomes, yet a slight reduction within the autosomal region of spreading is also observed (Supplemental Figure 6A).
To be able to analyze DCC spreading with respect to the subtle changes in gene expression and histone modification levels, we used a sliding window analysis with 200 kb windows and 20 kb steps. For each window, we performed a student’s t-test asking whether the ChIP-seq or mRNA-seq ratio within each window is significantly increased or decreased compared to the rest of the windows across chrV. Windows with p values less than 0.01 were plotted under the DPY-27 ChIP-seq enrichment in the X;V fusion chromosome (Figure 5E). Although noisy, the level of gene expression, H3K4me3 and H3K27ac were significantly reduced in windows close to the fused end of chrV (Figure 5E). Lack of a similar pattern on other chromosomes (Supplemental Figure 6B) supports the conclusion that DCC spreading into the autosomal region of the fused chromosome reduces active histone modifications and represses transcription.
DCC depletion does not significantly alter binding of PHA-4, CBP-1 and PQN-85
To test if the DCC represses transcription by reducing binding of all proteins to the X chromosomes, we performed ChIP-seq analysis of the transcription factor PHA-4, the putative H3K27 acetylase CBP-1 (p300 homolog), and the cohesin loader subunit PQN-85 (Scc2 homolog) (Figure 6A). We found no X-specific difference in the binding of these proteins as measured by ChIP-seq upon dpy-27 RNAi knockdown (Figure 6B), suggesting that the DCC does not indiscriminately displace proteins from the X.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) ChIP-seq profiles of DPY-27 (condensin DC subunit), PHA-4 (FOXA transcription factor), PQN-85 (S. cerevisiae Scc2p homolog), and CBP-1 (putative H3K27 acetyltransferase), in representative regions on chr X and III. Protein binding patterns were largely similar in wild type and DCC depleted conditions. (B) Analysis as in figure 3I, plotting change in protein binding across 200 bp wild type peak summits. CBP-1, PQN-85, and PHA-4 level on the X chromosomes did not change significantly upon DCC knockdown. (C) Summary of DCC binding and regulation of histone modifications on the X chromosomes. DCC binding sites coincide with gene regulatory elements marked by accessible chromatin on the X. Majority of these elements also contain histone modifications associated with active transcription. Remaining include recruitment elements and sites that do not contain the analyzed histone modifications. DCC tunes the level of specific histone modifications (shown in boxes denoted by up and down arrows) and not their distribution on the X.
DISCUSSION
Here, our analysis of DCC distribution with respect to various chromatin marks reflects multiple modes of binding, including a baseline distribution, strong enrichment at the recruitment sites, and peaks of DCC enrichment at gene regulatory elements, partially correlating with active chromatin. Our results suggest that the DCC is required for reducing the level of histone modifications that are associated with active transcription, including H3K4me3 and H3K27ac, but does not regulate or regulated by heterochromatin-associated histone modification H3K9me3. GRO-seq and ChIP-seq analysis of RNA Pol II in DCC mutants showed that the DCC reduces RNA Pol II binding to X chromosomal promoters [8, 10, 26]. Collectively, our study suggests a model in which the DCC represses transcription by specifically targeting and modulating the activity of gene regulatory elements by reducing the level of activating histone modifications (Figure 6C).
The first question this model raises is how the DCC targets active gene regulatory elements? Accumulation at active promoters and enhancers is a conserved feature of condensins in C. elegans, D. melanogaster, chicken, mouse and human cells [5]. Condensins bind chromosomes by entrapping and/or encircling DNA through multiple interactions mediated by its ring structure [28, 29]. One mechanism by which condensins may target gene regulatory elements is through binding to accessible DNA in vivo, which tends to coincide with active promoters and enhancers. Another possibility is through specific recruitment by transcription factors. In yeast and mammals, condensins are recruited to tRNA gene promoters and extra TFIIIC sites by interacting with TFIIIC [14, 30–34], TBP [35] and sequence specific transcription factors [36]. In C. elegans, the strong DCC recruitment elements are HOT sites that are bound by multiple transcription factors [12]. Binding to accessible DNA and recruitment by specific transcription factors are not mutually exclusive mechanisms [37]. Indeed, condensin DC binding through both DNA accessibility and specific recruiter proteins may result in the complicated pattern of DCC distribution that we observe in vivo.
The second question that our model raises is how specific histone modifications are regulated by the DCC? Our work suggests that the DCC does not indiscriminately reduce binding of proteins to the X. It is possible that the DCC regulates binding of specific histone modifying complexes. For instance, physical interaction of the DCC with a subunit of a chromatin-modifying complex may serve as a barrier to complex binding or function. Supporting this idea, DPY-30, an essential subunit of the MLL/COMPASS complex physically interacts with the DCC [26]. Intriguingly, a recent proteomic analysis found that mitotic chromosomes disproportionately lose chromatin-modifying complexes associated with euchromatin and not heterochromatin [38]. Furthermore, the level of displacement differed for different histone acetylases [38]. It is possible that reduced acetylation is connected to mitotic transcriptional repression, which is thought to be important for chromosome segregation [39]. Therefore, DCC mediated transcriptional repression may have evolved from a conserved condensin role in regulating specific chromatin modifying complexes in the formation of mitotic chromosomes.
The third question is how do histone modifications regulate RNA Pol II binding to X chromosomal promoters? H3K4me3 and H3K27ac are particularly instructive in models that predict gene expression from histone modifications [23, 40, 41]. We also observed a strong correlation between RNA Pol II, H3K4me3 and H3K27ac. At individual promoters, the differential binding of RNA Pol II and histone modifications upon DCC defect were less correlated, perhaps due to insufficient sensitivity of the ChIP-seq assay in C. elegans embryos and/or a complex quantitative relationship between Pol II recruitment and histone modifications at a given promoter [42]. While it remains unclear how much H3K4me3 activates transcription directly [43], it has been shown that H3K4me3 interacts with specific transcriptional activators [43], and ectopic recruitment of H4K4me3 activates and maintains transcription [44]. H3K27ac also regulates transcription, presumably by controlling transcription factor binding and RNA Pol II release from promoters [45]. In C. elegans a small proportion of genes show promoter pausing [46], thus H3K27ac may regulate dynamics of activator binding upstream of RNA Pol II recruitment to promoters.
Evolution of diverse dosage compensation strategies reveals how different transcriptional regulatory mechanisms can be co-opted to regulate large domains within the genome. DCC belongs to the deeply conserved SMC family of complexes that are involved in genome organization and gene regulation across species [2, 47, 48]. Here, we show that the DCC targets gene regulatory elements and modulates the level of active histone modifications rather than their distribution, suggesting that C. elegans dosage compensation evolved to control transcriptional output without interfering with the underlying transcriptional program. A similar condensin-mediated tuning of histone modifications on mitotic chromosomes may be important for proper inheritance of transcriptional programs after cell division. Future work on how the DCC modulates histone modifications and transcriptional activity of gene regulatory elements will be important to cross-reveal mechanisms by which condensin-mediated organization of mitotic chromosomes affect gene regulation across cell division [4].
MATERIALS AND METHODS
Worm strains and growth
Mixed developmental stage embryos (wild type N2) were isolated from gravid adults by bleaching. Mutant strains used in this study were CB428 (dpy-21(e428) V), OP37 (wgIs37 [pha-4::TY1::EGFP::3xFLAG + unc-119(+)]), YPT41 (X;II) and YPT47 (a.k.a. 15eh#1, X;V) [49], and GW638 (met-2(n4256) set-25(n5021) III) [27]. For ChIP samples, embryos or larvae were incubated in 2% formaldehyde for 30 minutes. Synchronized L3 worms were isolated by growing starved L1s for 24 hours at 22°C. L1-L3 worms were isolated from asynchronous plates by passing larvae with a 20-micron filter, where the embryos and larvae with expanded germline are not capable of flowing through. Large scale RNAi knockdown for ChIP and RNA-seq analyses was performed as described previously [14]. Briefly, bacteria with RNAi inducing plasmids were grown in liquid, and concentrated 130-fold to seed 4 6×10 cm plates. Synchronized N2 L1s were plated on RNAi plates and grown at 20°C for four days to obtain gravid adults. Knockdown was verified by western blot analysis of DPY-27 compared to control (vector only) RNAi.
Antibodies and chromatin immunoprecipitation
Information on antibodies used in this study is given in Supplemental File 1. Two new antibodies were used. MDT-15 antibody was validated by western blot analysis upon RNAi knockdown and immunoprecipitation (Supplemental Figure 7). CBP-1 antibody did not show a measurable signal on western blot hybridization and immunofluorescence assays but showed the expected ChIP-seq pattern overlapping with H3K27ac (Supplemental Figure 1). Experiments were from at least two biological replicates with matching input samples as reference (Supplemental File 1). ChIP-seq [14] and mRNA-seq [50] experiments were performed as previously described.
ChIP-seq data processing
Single-end sequencing was performed by Illumina Genome Analyzer IIx, HiSeq-2000, HiSeq-2500, HiSeq-4000 or NextSeq 500. The raw and processed data are provided at Gene Expression Omnibus database (GEO, http://www.ncbi.nlm.nih.gov/geo) under accession number GSE122639. ChIP data processing and peak finding was performed as described previously [14]. Briefly, 50-75 bp single-end reads were aligned to the C. elegans genome version WS220 using bowtie version 1.2.0 [51], allowing two mismatches in the seed, returning the best alignment, and restricting multiple alignments to four sites in the genome. Mapped reads from ChIP and input were used to call peaks and obtain read coverage per base using MACS version 1.4.3 [52] with default parameters. ChIP scores per base were obtained by normalizing to the median coverage and subtracting the input coverage. To obtain summits for binding profiles that are a combination of focused and broad patterns, large peaks were split using PeakSplitter version 1.0 [53], with a minimum height cut-off of 4 and a separation float of 0.86. The replicate, number of reads, and access information for the data sets are provided in Supplemental File 1.
ChIP-seq data analysis
Data were visualized using UCSC genome browser, ce10 (http://genome.ucsc.edu/). Heatmaps of ChIP enrichment across WS220 TSS and GRO-seq defined TSS sites [10] were produced using Deeptools [54] with default parameters in Galaxy (doi:10.1093/nar/gkw343). Change in ChIP binding scores across TSS and peak summits were calculated by standardizing average ChIP scores within a 1 kb window centering at the TSS or 200 bp window centered at the summit through calculating = (log2(mut/wt)-mean(log2(mut/wt)) / stdev (log2(mut/wt)). Box plots were produced in R using ggplot2 (http://ggplot2.org). The whiskers extend from the hinge to the largest value no further than +/−1.5 IQR (distance between the first and third quartiles) from the hinge. Outliers are not plotted. The notch shows the 95% confidence interval of the median (median +/− 1.58*IQR/sqrt(n)). Data analysis scripts are available at the Ercan lab github: https://github.com/ercanlab/street_et_al_2019/.
mRNA-seq data processing and analysis
Single-end sequencing was performed by Illumina HiSeq-2000. mRNA-seq data processing was performed as described previously [12]. Briefly, 50 bp single-end reads were aligned to the C. elegans genome version WS220 using Tophat version 2.1.1 [55], using default parameters. Count data was calculated using HTSeq version 0.6.1 [56] and normalized using the R package DESeq2 [57]. The resulting mRNA levels and expression ratios are provided in Supplemental File1.
ACKNOWLEDGEMENTS
We thank Susan Gasser for providing the GW638 strain, Dominic Balcon and Jacob Carmichael for help with growing worms and NYU-CGSB, UNC, and MDC Berlin High Throughput Sequencing Facilities for sequencing. We thank Gyorgyi Csankovszki for providing H4K16 antibody. Research reported in this publication was supported by NIGMS of the National Institutes of Health under award number R01GM107293. Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
REFERENCES




