

Abstract
Proper repair of double-strand breaks (DSBs) is key to ensure proper chromosome segregation. In this study, we found that the deletion of the SRS2 gene, which encodes a DNA helicase necessary for the control of homologous recombination, induces aberrant chromosome segregation during budding yeast meiosis. This abnormal chromosome segregation in srs2 cells accompanies the formation of a novel DNA damage induced during late meiotic prophase-I. The damage may contain long stretches of single-stranded DNAs (ssDNAs), which lead to aggregate formation of a ssDNA binding protein, RPA, and a RecA homolog, Rad51, as well as other recombination proteins inside of the nuclei. The Rad51 aggregate formation in the srs2 mutant depends on the initiation of meiotic recombination and occurs in the absence of chromosome segregation. Importantly, as an early recombination intermediate, we detected a thin bridge of Rad51 between two Rad51 foci or among the foci in the srs2 mutant, which is rarely seen in wild type. These might be cytological manifestation of the connection of two DSB ends and multi-invasion. The DNA damage with Rad51 aggregates in the srs2 mutant is passed through anaphase-I and -II, suggesting the absence of DNA damage-induced cell-cycle arrest after the pachytene stage. We propose that Srs2 helicase resolves early protein-DNA recombination intermediates to suppress the formation of aberrant lethal DNA damage during late prophase-I.
Introduction
In sexually reproducing organisms, meiosis, a specialized form of cell division, produces haploid gametes from diploid germ cells. Following DNA replication, reciprocal recombination takes place to connect the homologous chromosomes and to generate genetic diversity of gametes. With arm cohesion, the connection between the chromosomes, which is cytologically visualized as chiasma, is essential for faithful chromosome segregation during meiosis I by antagonizing the pulling force by spindle microtubules to create tension (Petronczki et al. 2003).
Meiotic recombination is initiated by the generation of DNA double-strand breaks (DSBs) by a meiosis-specific topoisomerase-like protein, Spo11, at recombination hotspots (Keeney et al. 1997). Subsequently, the end of DSBs is quickly resected to produce 3’-overhanging single-stranded DNAs (ssDNAs). Replication protein A (RPA) binds to the ssDNAs, followed by the loading of Rad51, a homolog of bacterial RecA (Shinohara et al. 1992), with the assistance of auxiliary proteins, such as Rad52, Rad55-Rad57 and Pys3-Csm2-Shu1-Shu2 (a.k.a. Shu) (New et al. 1998; Sasanuma et al. 2013b; Shinohara and Ogawa 1998; Sung 1997). Rad51 filaments on ssDNA are active protein machinery for DNA homology search and strand exchange (Ogawa et al. 1993; Sung 1994). Rad51 filament activity is helped by Rad54, which belongs to the SNF2/SWI2 DNA helicase family (Shinohara et al. 1997b).
Whereas Rad51 is sufficient for the homolog search in recombination during mitosis, meiosis requires a meiosis-specific RecA homolog, Dmc1, for the recombination (Bishop et al. 1992). Dmc1 is essential for homology search/strand exchange in inter-homolog recombination during meiosis while Rad51 plays an auxiliary role by assisting Dmc1 assembly (Bishop 1994; Cloud et al. 2012; Shinohara et al. 1997a). Indeed, the Rad51 activity for inter-sister recombination during meiosis is suppressed by the action of a meiosis-specific Rad51 inhibitor, Hed1 (Tsubouchi and Roeder 2004). Like Rad51, Dmc1 forms a nucleo-protein filament on ssDNAs to catalyze the strand invasion of the DNA into its homologous duplex DNA for the formation of an intermediate, D(displacement)-loop (Hong et al. 2001).
In D-loop, DNA synthesis occurs from 3’-end of invading strand as a primer. When the synthesized DNA strand is ejected from the D-loop (Allers and Lichten 2001; Hunter and Kleckner 2001), the ejected synthesized ssDNA is able to anneal with the complementary ssDNA in the other end of the DSB. Annealing induces the second DNA synthesis to complete the recombination by producing non-crossovers. This pathway is called synthesis-dependent strand annealing (SDSA) (Allers and Lichten 2001). On the other hand, when the newly synthesized DNA is stably bound to the D-loop, ongoing DNA synthesis can extend a D-loop with a large displaced ssDNA, which is able to anneal with ssDNA on the opposite DSB ends. Additional processing of the intermediates leads to the formation of double-Holliday junction (dHJ) (Schwacha and Kleckner 1994). dHJs are specifically resolved into crossovers. Importantly, meiotic recombination is tightly coupled with chromosome morphogenesis such as the formation of the synaptonemal complex (SC), a meiosis-specific zipper-like chromosome structure, which juxtaposes homologous chromosomes in near vicinity (Cahoon and Hawley 2016).
Srs2 is a 3’-to-5’ SF1 helicase related to bacterial UvrD helicase (Rong et al. 1991). Srs2 protein has some distinct functional domains: 3’-5’ DNA helicase domain, Rad51-interaction domain, and also SUMO- and PCNA-binding domains in the C-terminus (Marini and Krejci 2010). Genetic analyses showed positive and negative roles of Srs2 in the recombination (Marini and Krejci 2010). Biochemical studies have demonstrated that purified Srs2 protein can dislodge Rad51 filament on ssDNAs and dramatically inhibits Rad51-joint molecules via direct interaction with Rad51 in vitro (Krejci et al. 2003; Veaute et al. 2003). This biochemical activity of Srs2 supports the idea of Srs2 function as an anti-recombinase. The Rad51-dismantling activity of Srs2 is confirmed by in vivo analysis (Sasanuma et al. 2013a).
Deletion of SRS2 gene shows different kinds of genetic interaction with mutants deficient in DNA transaction. The srs2Δ is synthetic lethal with a mutation of the SGS1, encoding a RecQ-type DNA helicase. By forming a complex with Top3 and Rmi1, Sgs1 is known to dissolve the dHJ structure into noncrossovers (Cejka et al. 2010; Wu and Hickson 2003). Moreover, the srs2Δ is synthetic lethal with the deletion of the RAD54 (Klein 2001), suggesting the role of Srs2 in a late stage of the recombination such as the post-invasion step in the recombination. This lethality is thought to be caused by a fatal defect in the resolution of toxic intermediates in the recombination process. This is supported by the fact that the deletion of RAD51 can suppress the lethality of srs2Δ sgs1Δ and srs2Δ rad54Δ mutants (Gangloff et al. 2000; Schild 1995).
During mitosis, crossovers should be suppressed when DNA damage is spontaneously introduced, because the crossover between homologous chromosomes and sister chromatids results in the loss of heterozygosity. In contrast, as described above, meiotic recombination must give rise to at least one essential crossover per chromosome, which is fostered by a group of proteins called ZMM (Zip-, Msh-, Mer) (Shinohara et al. 2008). Previous genetic studies showed a role of Srs2 in meiosis (Palladino and Klein 1992; Sasanuma et al. 2013b). However, the molecular defects associated with srs2 deletion in meiosis have not been described in detail. Therefore, it remains elusive how Srs2 regulates meiotic recombination.
In this study, we analyzed the role of Srs2 helicase in meiotic recombination, particularly looking at dynamics of its interacting partner, Rad51. We found that, in the absence of Srs2, abnormal DNA damage associated with Rad51 aggregation accumulates during late prophase-I, after the completion of meiotic recombination. The formation of this DNA damage in the srs2 requires meiotic DSB formation, but is independent of chromosome segregation. We also detected thin line-staining of Rad51 connecting between two adjacent Rad51 foci in early prophase in the absence of Srs2, which is rarely seen in the wild type. We propose that Srs2 protects chromosomes in late meiotic prophase-I from accumulation of abnormal DNA damage by properly coupling the completion of meiotic recombination with chromosome morphogenesis.
Materials and Methods
Yeast strains and medium conditions
All yeast strains used in this article are isogenic derivatives of SK1 and listed in Table S1. pCLB2-SGS1 and RAD54-RFB strains were a gift by Dr. Neil Hunter and Dr. Andreas Hochwagen, respectively. Mediums and culture conditions regarding meiosis are described in (Sasanuma et al. 2008).
Antibodies and chemicals
The primary anti-sera were used as following concentrations; anti-Rad51 (guinea pig, 1/500), anti-Dmc1 (rabbit, 1/500), anti-Rad52 (rabbit, 1/300), anti-Rfa2 (rabbit, 1/500), anti-Zip1 (rabbit, 1/500), anti-Red1 (rabbit, 1/500), Anit-Hed1 (rabbit, 1/200) and anti-Mei5 (rabbit, 1/500) for cytology. Anti-Hed1 serum from rabbit was prepared for denatured Hed1 protein purified from E. coli. Anit-Nop1(mouse) is from Encor Biotech (MCA28-F2). α-tubulin is monoclonal antibody of rat that can recognize alpha subunit (AbD Serotec/BioRad, MCA77G). The second antibodies for staining were Alexa-fluor 488 (Goat) and 594 (Goat) IgG used at a 1/2000 dilution (Molecular Probes).
Rapamycin (LC-Laboratories, R-5000) and benomyl (methyl 1-[butylcarbamoyl]-2-benzimidazolecarbamate; Sigma Aldrich, PCode 1002355429) were dissolved in DMSO at a concentration of 1 mM and 30 mg/ml, respectively.
Immuno-staining
Chromosome spreads were prepared using the Lipsol method as described previously (Shinohara et al. 2000; Shinohara et al. 2003). Immnostaining was conducted as described (Shinohara et al. 2000). Stained samples were observed using an epi-fluorescence microscope (BX51; Olympus, Japan) with a 100X objective (NA1.3). Images were captured by CCD camera (CoolSNAP; Roper, USA), and afterwards processed using IP lab and/or iVision (Sillicon, USA), and Photoshop (Adobe, USA) software tools.
SIM imaging
The structured illumination microscopy was carried out using super resolution-structured illumination (SR-SIM) microscope (Elyra S.1 [Zeiss], Plan-Apochromat 63x/1.4 NA objective lens, EM-CCD camera [iXon 885; Andor Technology], and ZEN Blue 2010D software [Zeiss]) at Friedrich Miescher Institute for Biomedical Research, Switzerland. Image processing was performed with Zen software (Zeiss, Germany), NIH image J and Photoshop.
Whole cell staining
Cells were fixed with 1/10 volume of 37% formaldehyde (Wako) and treated with 10 μg/ml Zymolyase 100T (Seikagaku) for 1.5 h. Cells were placed to the poly L-lysine (Sigma) coated slides and then fixed with cold 100% methanol, cold 100% acetone and cold 1X PBS. Slides were used for immuno-staning.
Western Blotting
Western blotting was performed for cell lysates extracted by TCA method. After being harvested and washed twice with 20% TCA, cells were roughly disrupted by Yasui Kikai (Yasui Kikai Co Ltd, Japan). Protein precipitation recovered by centrifuge at 1600 g for 5min was suspended in SDS-PAGE sample buffer adjusting to pH8.8 and then boiled for 95°C, 2min.
Southern Blotting
Southern blotting analysis was performed with the same procedure as in (Storlazzi et al. 1995). Genomic DNA prepared was digested with both MluI and XhoI (for crossover/non-crossover, upper panels) and PstI (for meiotic DSB, lower panels). Probes for Southern blotting were Probe “155” for crossover/non-crossover and Probe 291 for DSB detection as described in (Storlazzi et al. 1995). Image gauge software (Fujifilm Co. Ltd., Japan) was used for quantification for bands of R1, R3 and DSB I.
Pulsed-field gel electrophoresis
For pulsed-field gel electrophoresis (PFGE), chromosomal DNA was prepared in agarose plugs as described in (Bani Ismail et al. 2014) and run at 14 °C in a CHEF DR-III apparatus (BioRad) using the field 6V/cm at a 120° angle. Switching times followed a ramp from 15.1 to 25.1 seconds. Durations of electrophoresis were 41 h for chromosome III.
Statistics
Means ± S.D values are shown. Graphs were prepared using and Microsoft Excel and GraphPad Prism 7. Datasets were compared using the Mann-Whitney U-test. χ2-test was used for proportion.
Results
SRS2 deletion markedly decreased spore viability
As reported previously (Palladino and Klein 1992; Sasanuma et al. 2013a), the srs2 deletion mutant exhibits reduced spore viability of 36.8%, indicating a critical role of this helicase for meiosis (Fig. S1A). This marked reduction of the spore viability is somehow unexpected given a negative role of this helicase in recombination.
We also confirmed the kinetics of meiotic progression in srs2Δ strains by DAPI staining. In the wild-type strain, meiosis I started at 5 h after incubation with sporulation medium (SPM) and was sequentially followed by meiosis II. Finally, ~90% of the wild-type cells completed MII at around 8 h (Fig. 1A). In the srs2Δ mutant, the appearance of cells undergoing MI was delayed by ~2 h and ~75% of cells finished MII at 14 h (Fig. 1A). A similar delay was observed for a srs2 mutant in a different strain background previously (Palladino and Klein 1992). This indicates a defect during prophase-I in the srs2 mutant. In srs2 cells after sporulation; e.g. 12 h, we often detected fragmented DAPI bodies in a cell/spore (Fig. 1B), indicating a defect in chromosome segregation during the mutant meiosis.

A. Meiosis I was analyzed by DAPI staining of the wild type (open circles; NKY1303/1543) and srs2 (filled circles; HSY310/315) cells. The number of DAPI bodies per nucleus was counted in a minimum of 150 DAPI positive cells at each time point. B. DAPI image of wild-type and srs2 cells at 12 h. C. Immunostaining analysis of Rad51 (green) and Dmc1 (red) on chromosome spreads in wild type (NKY1303/1543) and srs2 (HSY310/315) cells. Representative images with or without DAPI (blue) dye at 4, 6 and 8 h for wild type and the srs2 are shown. D. Kinetics of Rad51 focus-positive cells in various yeast strains. A spread with the Rad51 foci is defined as a cell with more than five foci. Spreads containing Rad51 aggregates were also counted. A minimum of 100 cells were analyzed at each time point. Graphs show kinetics of one representative experiment for the wild type cells (top; NKY1303/1543), and srs2 cells (bottom; HSY310/315). Circles and triangles show spreads with Rad51 foci and aggregate, respectively. E. F. Immunostaining analysis of a component of RPA, Rfa2 (green) with Rad51 (red) in wild type (NKY1303/1543; top) and srs2 (HSY310/315; bottom) cells at 4 h (E) and at 8 h (F). In F, a dashed square is enlarged in the right. G. Kinetics of Rfa2 foci-positive cells in wild type (NKY1303/1543; top) and srs2 (HSY310/315; bottom) cells as shown in (B). Circles and triangles show spreads with Rfa2 foci and aggregates, respectively.
The srs2Δ mutant showed a defect in meiotic DSB repair
We analyzed meiotic recombination defects the srs2 deletion mutant in more detail. First, we checked the repair of meiotic DSBs in the mutant by Southern blotting. DSB formation was monitored at the HIS4::LEU2 locus, an artificial meiotic recombination hotspot in chromosome III (Fig. S1B)(Cao et al. 1990). In wild type, DSB frequencies reached its maximum value at 3 hours of meiosis (~10% of total signals) and then decreased gradually (Fig. S1C, D). The srs2Δ accumulates DSB at higher levels (~20%) with more hyper-resection than wild type and delays the disappearance by ~ 2h. (Fig. S1D), indicating that Srs2 is required for efficient meiotic DSB repair. We also checked the formation of two recombinant species, crossover (CO) and non-crossover (NCO) at the same locus. The srs2Δ reduces both CO and NCO to 52% and 64% of the wild-type levels (at 6 h; Fig. S1C, D), respectively. These show that Srs2 is necessary for efficient formation of meiotic recombinants. This is consistent with previous return-to-growth experiment showing delayed recombinant prototroph formation in the srs2Δ mutant (Palladino and Klein 1992).
During meiotic prophase, homologous chromosomes are tightly coupled with the formation of the synaptonemal complex (SC), a zipper-like chromosome structure linking two homologous chromosomes. Zip1 is a component of the central region of SC, which serves as a marker for synapsis (Sym et al. 1993). A defect in meiotic recombination results in defective SC formation. We checked the SC formation in the srs2Δ mutant by immuno-staining analysis of Zip1 on chromosome spreads as well as a meiosis-specific cohesin component, Rec8 (Fig. S1E). We classified three categories according to Zip1 staining; Dotty Zip1 (Class I), partially extended (Class II) and fully-elongated (Class III), which roughly correspond with leptotene, zygotene and pachytene stages, respectively. In wild type, ~66% of nuclei contained full-elongate Zip1 lines at 4 h and Zip1 signal gradually disappeared from chromosomes. In srs2Δ strains, although Zip1 focus-positive nuclei exceeded 80% at 4 h, the proportion of cells with fully-elongated Zip1 was significantly reduced to 13 and 26% at 4 and 5 h, respectively (Fig. S1F). Consistent with this, the proportion of polycomplexes (PCs), which are an aggregate of Zip1, dramatically increased; ~60% of the srs2Δ nuclei contained PCs at 4 h (Fig. S1G). SCs disassembled more slowly in the mutant than wild type, consistent with delayed meiotic DSB repair (Fig. S1F).
The srs2Δ mutant accumulated aggregates of Rad51 during late meiotic-prophase I
Immuno-staining analysis of chromosome spreads can detect recombination proteins such as Rad51 and Dmc1 on the spreads as a focus, which marks a site of ongoing recombination (Bishop 1994). Previous study indicated that the number of Rad51 foci on chromosome spreads in the srs2Δ mutant at 4 h incubation of SPM is slightly reduced compared to those in wild-type (Sasanuma et al. 2013b). We performed kinetic analysis of Rad51 and Dmc1 focus formation. In wild-type cells, dotty signals of both Rad51 and Dmc1 peaked at 4 h of meiosis (Figs. 1C and S2A). The appearance of Rad51 foci in cells lacking Srs2 is slightly delayed, and the disappearance of the foci is delayed relative to wild-type cells (Fig. 1D), consistent with delayed DSB repair in the mutant.
Interestingly, after disappearance of Rad51 foci, we observed reappearance of Rad51 staining with a unique structure after 5 h incubation in the srs2Δ mutant (Figs. 1C and S2A). This staining shows clustering of beads-in-line of Rad51 foci, in which 1-5 bright aggregates of Rad51 are connected with each other through thin threads containing Rad51 as well as much simple big aggregation of Rad51 (referred to as Rad51 aggregates) (Fig. 1C). The formation of Rad51 aggregates reach a plateau at 6 h, slightly decreases thereafter, but some cells at 10 or 12 h contained Rad51 aggregates (Fig. 1D), when most of srs2 mutant cells finished MII (Fig. 1A). At 6 and 12 h, 56 and 40 percent of cells contained aggregates of Rad51, respectively (Fig. 1D, bottom).
Interestingly, this aggregate staining is specific to Rad51, not seen to Dmc1 (Figs. 1C and S2A). Western blots show that Dmc1 and its mediator Mei5 (Hayase et al. 2004) are still present at MI and MII (Fig. S2B). On the other hand, like Rad51 foci, we do see the aggregates of Rad52, a mediator of Rad51 (Shinohara and Ogawa 1998), on chromosomes only in the srs2Δ mutant, but not in wild type cells at late times (Fig. S2C, D). We also found that a Rad51 -inhibitor protein, Hed1 (Tsubouchi and Roeder 2006), formed an aggregate with Rad51 with co-localization (Fig. S2E, F). The kinetics of appearance of Rad52 and Hed1 aggregates in the srs2Δ mutant are similar to those of Rad51 (Fig. S2D, F).
In order to know the nature of the late Rad51 foci/aggregates, we also studied the localization of RPA (Rfa2, a middle subunit of RPA) at late prophase I of the srs2Δ mutant. Immuno-staining showed that, in addition to early RPA foci (Fig. 1E, F), like Rad51-aggregates, aggregate staining of Rfa2 re-appeared at late times of the srs2 meiosis; e.g. 6-10 h (Fig. 1G). Closer examination reveals that RPA also exhibits a long-line like staining (Fig. 1F). The kinetics of Rfa2 aggregates in the srs2 mutant is very similar to that of Rad51 (Fig. 1D, G). Some RPA lines and aggregates co-localized with Rad51 lines and aggregates (Fig. 1F). This suggests that the formation of ssDNAs during late prophase-I in srs2 cells.
One possibility is that Rad51 aggregates bind to DNA damage in ribosomal DNA (rDNA) region, whose segregation defect is often observed in the recombination defective mutants (Li et al. 2014). We co-stained Rad51 with anti-Nop1, a marker for an rDNA region (Schimmang et al. 1989). As shown in Fig. S3A, a single Nop1 signal does not co-localize with late Rad51 aggregates as well as early Rad51 foci in the srs2 mutant. This excludes the possibility that late Rad51 aggregates are induced by abnormal recombination in the rDNA repeat.
In order to know the relationship of Rad51 aggregate formation with chromosome segregation, we performed whole cell immuno-staining for Rad51 and Dmc1. At early time points, both wild-type and srs2Δ mutant cells showed punctate staining for both Rad51 and Dmc1 with some background diffuse staining in a nucleus (Fig. 2A). Rad51-positive nuclei appear at 2 h, peaks at 4 h, and then disappear in wild-type cells while the positive nuclei peaks at 5 h in the srs2 cells (Fig. 2C). Consistent with results for chromosome spreads (Fig. 1C), the srs2Δ cells start to show a big aggregate of Rad51, but not of Dmc1 in nuclei from 5 h and this staining reached to plateau at 8 h (Fig. 2C). Rad51 aggregates in a nucleus often contained thin lines and the number of the aggregate varies up to 2-5 per a nucleus. Importantly, we could also detect Rad51 aggregates in srs2Δ cells with two and four big DAPI bodies in a cell, which correspond with cells finishing MI and MII, respectively (Fig. 2B, D). This suggests that DNA damage associated with Rad51 aggregates does not induce delay or arrest of the progression of meiosis. To see the DNA damage checkpoint activation at late meiosis of the srs2 cells, we analyzed the phosphorylation status of Hop1, which is a substrate of Mec1/ATR and Tel1/ATM as a marker of the activation (Carballo et al. 2008), and found that the srs2 cells accumulated more phosphorylated Hop1 at 4 h compared to wild type and showed residual phosphorylation during late time points (Fig. 2E).

A, B. Whole cell immunostaining analysis of Rad51 (green) and Dmc1 (red) in wild type (NKY1303/1543) and srs2 (HSY310/315) cells at 4 and 8 h in meiosis. In (B), cells with Rad51 in prophase-I (top), after MI (middle), and after MII (bottom) are shown. C. Kinetics of Rad51 foci-positive cells in wild-type and srs2 cells. A foci-positive cell is defined as a cell with more than five foci (closed circles, wild type, NKY1303/1543; open circles, srs2 cells, HSY310/315). A minimum of 100 cells were analysed at each time point. Graphs show the mean values with S.D. from three independent experiments. D. Kinetics of srs2 cells containing Rad51 aggregates prior to MI (blue; one DAPI body in a cell) after MI (green; two DAPI bodies in a cell) and MII (red; three or more DAPI bodies in a cell) are shown. Mean values with S.D. from three independent experiments are shown. E. Western blotting analysis of Hop1 phosphorylation during meiosis. Cell lysates at different time points in meiosis in wild type (NKY1303/1543) and srs2 (HSY310/315) cells were probed with anti-Hop and anti-tubulin antibodies. Phosphorylated Hop1 (shown as “Hop1-P”) shows slower mobility relative to un-phosphorylated Hop1.
Rad51-aggregate formation in the srs2 mutant depends on Spo11
To know the nature of Rad51 aggregates in the srs2Δ mutant, we looked for genetic requirement of the aggregate formation in the mutant. Rad51–aggregate formation in srs2Δ cells is dependent on DSB formation, since a catalytic-dead spo11 mutation, spo11-Y135F (Keeney et al. 1997), almost abolishes both early focus and aggregate of Rad51 staining in the srs2Δ mutant (Fig. S3B). It is likely that early DSB-related events in the srs2Δ cells may trigger Rad51 aggregates during late meiosis.
In mitosis, the sgs1 mutation is synthetic lethal with the srs2 mutation, indicating a redundant role of these two helicases (Gangloff et al. 2000). Sgs1 helicase, together with Top3 and Rmi1, is known to prevent the formation of the untangled chromosomes. The absence of Sgs1 results in abnormal meiosis divisions due to accumulation of un-resolve recombination products involving multi-chromatids (Jessop and Lichten 2008; Jessop et al. 2006; Oh et al. 2007; Oh et al. 2008; Tang et al. 2015). In mammals, the lack of Sgs1 ortholog, BLM helicase, induces anaphase bridges, which are associated with DNA damage generated during S-phase (Biebricher et al. 2013; Chan et al. 2007). We examined the late Rad51 aggregate formation in a meiotic-null allele of sgs1, sgs1-mn (CLB2p-SGS1) (Oh et al. 2007). The sgs1-mn forms early Rad51 foci with delayed disappearance in prophase of MI, but, unlike the srs2, the mutant does not form late Rad51 aggregates (Fig. S3C, D), suggesting that unresolved recombination intermediates formed in the absence of the Sgs1 do not trigger Rad51 aggregates formation.
The effect of Rad54 depletion on the kinetics of Rad51 aggregates in the srs2 mutant
We postulated that some Rad51 aggregates turned over during meiosis and could expect to stall its dynamics by blocking late stage of the recombination reaction. We focused on Rad54, which functions at post-assembly stage of Rad51 (Shinohara et al. 1997b), and tried to examine the effect of RAD54 deletion on Rad51 aggregates. However, it is reported that the rad54 deletion is synthetically lethal with the srs2 deletion (Klein 2001; Palladino and Klein 1992; Schild 1995). To circumvent this, we used Rad54-anchor away system, which specifically depletes nuclear Rad54 fused with RFB by the addition of the drug rapamycin (Haruki et al. 2008; Subramanian et al. 2016). The srs2 RAD54-RFB cells grow normally in the absence of rapamycin while the srs2 RAD54-RFB cells grow poorly on the plate containing the drug, confirming synthetic lethality of the rad54 and srs2 (Fig. 3A). In order to know the functional relationship between Rad54 and Srs2 during late meiosis, first, we added rapamycin at 4 h to RAD54-RFB and srs2 RAD54-RFB cells and analyzed both spore viability and Rad51 foci. The srs2 RAD54-RFB decreased spore viability to 64% in the absence of the drug. As reported (Shinohara et al. 1997b), RAD54-RFB cells decreased spore viability to 48% in the presence of the drug. Addition of the rapamycin also reduced the spore viability of the srs2 RAD54-RFB to 24%, indicating the additive effect of the srs2 deletion and RAD54 depletion on spore viability (Fig. 3B). RAD54 depletion does not affect delayed MI progression in the srs2 deletion (Fig. 3C). As in wild-type cells, RAD54-RFB cells showed normal assembly and disassembly of Rad51 foci in the absence of the drug (Rapa−; Fig. 3D, E). However, we found that, from 5 h, one hour after the addition of the drug (Rapa+), a new class of Rad51 staining appeared. This class contains 5-10 brighter foci of Rad51, called “Rad51 clump”, which is distinct from the typical Rad51 foci and aggregates (Fig. 3D). This Rad51 clamp peaks at 6 h and then disappears (Fig. 3E), indicating the role of Rad54 in the post Rad51-assembly stage. In the absence of rapamycin, the srs2 RAD54-RFB mutant shows the similar kinetics for both Rad51 foci and aggregates to the srs2Δ mutant. By the addition of the rapamycin at 4 h, like RAD54-RFB cells, the srs2 RAD54-RFB mutant formed Rad51 clamp from 5 h and showed the similar kinetics to that in the RAD54-RFB (Rapa+). In addition, Rad51 aggregates appeared at 5 h and accumulated during further incubation. Rad51 aggregate kinetics in the absence of RAD54 (Rapa+) is delayed relative to its presence (Rapa−) (Fig. 3E). This result indicates that Rad51 clumps formed without Rad54 are independent of Srs2. And also, Rad51 aggregate kinetics in the srs2 cell is independent of Rad54 function.

A. Mitotic plate assay to confirm synthetic lethality of srs2 RAD54-anchor on YPAD plates containing 1 μM rapamycin. B. Spore viability of RAD54-RFB (H7790/7791) and RAD54-RFB srs2 (HYS71/82) cells in the absence (−) and the presence (+) of rapamycin. C. Kinetics of MI entry in RAD54-RFB (H7790/7791) and RAD54-RFB srs2 (HYS71/82) cells in the absence (−) and the presence (+) of rapamycin. Rapamycin was added at 4 h at a concentration of 1 μM. Graphs show the mean values with S.D. from three independent experiments. D. Immunostaining analysis of Rad51 (green) and Dmc1 (red) on chromosome spreads in RAD54-RFB (H7790/7791) and RAD54-RFB srs2 (HYS71/82) cells in the absence and the presence of rapamycin. Rapamycin (1 μM) was added at 4 h in meiosis. Representative images of Rad51 staining with or without the addition of rapamycin are shown. E. Kinetics of Rad51 foci-positive cells in RAD54-RFB (top green graph; H7790/7791) and RAD54-RFB srs2 (bottom red graph; HYS71/82) cells in the absence (−) or the presence (+) of rapamycin. Rad51 foci (circles), -clumps (square), and –aggregates (triangles); open symbols (without rapamycin) and closed symbols (addition of the rapamycin at 4 h).
Rad51 aggregate in the srs2 mutant is dependent of pachytene exit but is independent of the onset of meiosis I
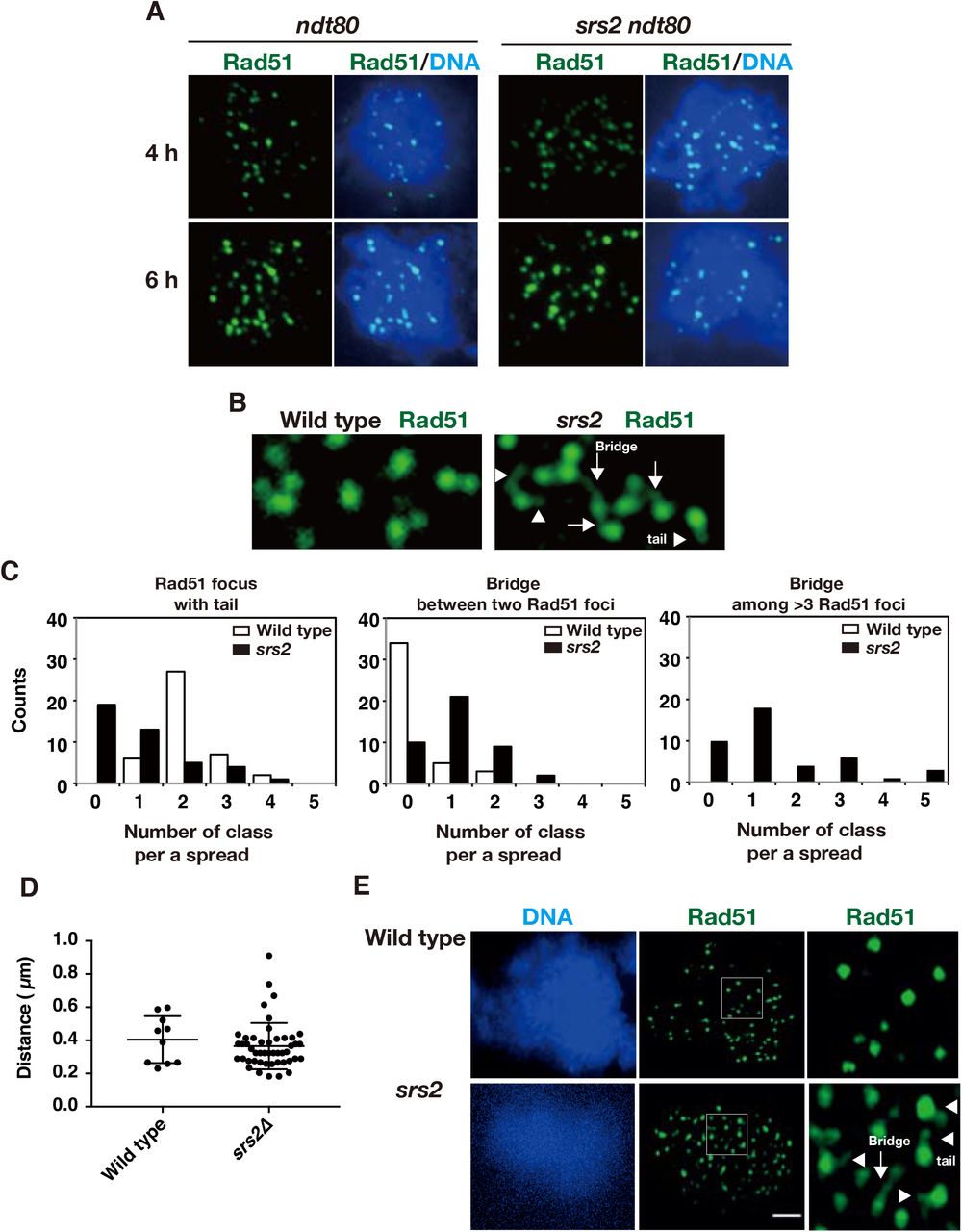
Rad51 aggregates in the srs2Δ mutant are formed at late times during prophase-I. To know the relationship between the focus formation and the progression of meiosis, we first analyzed the Rad51 aggregate formation in the srs2Δ with the ndt80 mutation, which induces pachytene arrest due to the inability to express genes necessary for exit from mid pachytene stage (Xu et al. 1995). Staining of chromosome spreads in ndt80 cells reveal accumulation of cells with Rad51 foci (Figs. 4A and S3E), which is induced by persistent DSB formation during pachytene arrest by the ndt80 (Carballo et al. 2013). At later times, the ndt80 mutant showed the reduced number of Rad51 foci compared to early time points (Fig. S3E). However, Rad51 foci seemed to turn over less efficiently in the ndt80 mutant (Fig. S3E, F). Little Rad51 aggregate formation was seen in srs2 ndt80 cells arrested at mid-pachytene both on chromosome spreads and in whole cells (Figs. 4A and S3E). This indicates that the formation of Rad51 aggregates in the srs2 mutant depends on Ndt80, thus after the exit of mid-pachytene stage.

A. Immuno-staining analysis of Rad51 in ndt80 (HSY596/597) and srs2 ndt80 (LPY058/059) cells. B. Rad51-bridges in srs2 cells at 4h. Rad51 tail and bridge are shown in arrowheads and arrows, respectively. C. Rad51-tail or bridge is classified into three classes; Rad51 focus with tail (left), Rad51 bridge between two foci (middle), Rad51 bridge among three or more foci (right). On each spread, the number of each class per a spread was counted, and then a count of the spreads in each class is shown. 42 spreads of wild-type (NKY1303/1543) and srs2 (HSY310/315) cells were analyzed and counted. D. The length of the Rad51 bridge between two Rad51 foci was measured and plotted. Three horizontal lines from the top indicate the 75, 50 (median), and 25 percentiles, respectively. P=0.39; Mann-Whitney U test. E. SR-SIM microscopic observation of Rad51 (green) in wild-type (NKY1303/1543) and srs2 (HSY310/315) cells. Representative image DAPI (blue; left) dye and Rad51 (green, middle) is shown. White insets in middle images are shown in a magnified view at right. The bar indicates 2 μm.
When the kinetics of Rad51 aggregate formation in the srs2 mutant was compared to kinetics of meiosis I entry, Rad51 aggregate in the srs2 mutant appear 1 h earlier than the entry into meiosis I (Fig. 1D). To confirm this, we blocked the microtubule dynamics by treating cells with a benomyl, a microtubule depolymerization drug. As shown previously (Hochwagen et al. 2005), the addition of benomyl to yeast meiosis at 4 h prior to the formation of the aggregates, largely suppressed the entry of meiosis I, thus the onset of anaphase-I, in both wild-type and srs2 cells (Fig. 5A). The treatment with benomyl does not affect Rad51-focus kinetics in both wild-type and srs2 mutant (Fig. 5A, C). Moreover, the srs2 cells formed Rad51 aggregates in the presence of benomyl with similar kinetics in its absence (mock treatment with DMSO) (Fig. 5C). This indicates that Rad51-aggregate formation in the srs2 mutant occurs in the absence of microtubule dynamics, thus chromosome segregation, suggesting that Rad51 aggregate formation in srs2 mutants is associated with an event during late prophase-I, not with events during the metaphase-I or anaphase-I.

A. Immunostaining analysis of Rad51 (green) in wild type (NKY1303/1543) and srs2 (HSY310/315) cells in the presence of benomyl. The benomyl was added at 4 h at a concentration of 120 μg/ml. B. Kinetics of MI entry in wild type (green) and srs2 (red) cells in the absence (open symbols) or the presence (closed symbols) of benomyl. C. Kinetics of Rad51 foci and Rad51 aggregates in wild type (top, green) and srs2 (bottom, red) cells in the absence (open) or the presence (closed) of benomyl. Circles, Rad51 foci; triangles, Rad51 aggregates. D. Immuno-staining analysis of Rad51 and Red1 in CDC20-mn (YFY74/77) and srs2 CDC20-mn (YFY80/83) cells. The chromosome spreads at 5 and 7 h were immuno-stained against Rad51 (green) as well as chromosome protein Red1 (red). E. Kinetics of Rad51 aggregate-positive cells in Red1-postive and -negative spreads. Rad51-focus and Rad51-aggregate positive spreads were classified into Red1-negative (open bars) and Red1-positive (closed bars) at each time point. At each time point, more than 50 spreads were counted.
Rad51 aggregates in the srs2 mutant appear when SC is disassembled
In order to confirm that Rad51-aggregate formation in the srs2 is independent of the onset of anaphase I, we used a meiosis-specific null mutant of the CDC20, which encodes an activator of Anaphase promoting complex/cyclosome (APC/C), the cdc20-mn (CLB2p-CDC20). As reported previously (Lee and Amon 2003), the cdc20-mn shows an arrest at the onset of anaphase I. In the cdc20-mn, Rad51 foci appear and disappear like in wild-type control. As expected from the results with benomyl, the Rad51-aggregate formation occurs after the disappearance of Rad51 foci in the srs2 cdc20-mn double mutant as in the srs2 mutant (Fig. 5D, E). This supports the notion that Rad51-aggregate formation in srs2 mutant is independent of the entry into anaphase-I, thus chromosome segregation.
The relationship between the formation of Rad51 aggregates and late meiotic prophase I such as SC disassembly was compared by immuno-staining of Rad51 with Zip1 (Fig. S4A). After the pachytene exit, the central region of SCs is dismantled as seen in the loss of Zip1 -line signals from chromosomes (Sym et al. 1993). The srs2 cells containing Rad51 aggregates were almost negative for Zip1 lines (Fig. S4A).
We also performed the staining of Red1, which is a component of chromosome axes (Smith and Roeder 1997). Most cells with Rad51 foci at 3-5 h are almost positive for Red1 staining in both cdc20-mn and srs2 cdc20-mn cells (Fig. 5D, E). In contrast, srs2 cdc20-mn cells with Rad51 aggregates were negative for Red1 signal. These indicate that Rad51 aggregate formation in the srs2 occurs after or during disassembly of Red1-axes. This is confirmed in the background of wild type too (Fig. S4B, C).
We confirmed this by staining of Rec8, a kleisin subunit of cohesin (Klein et al. 1999). At late time points such as 6 h, Rec8 showed dotty staining compared to 5 h (Challa et al. 2019), when most of Rec8 show line staining. Rec8 line positive spreads contained Rad51 foci (Fig. S4D, E). In srs2 cells with or without cdc20-mn, Rad51 aggregates are predominantly seen in cells with Rec8-dots (Fig. S4D, E).
The srs2 mutant accumulated bridge staining of Rad51 between two recombination foci during early prophase-I
During our staining analysis, we noticed that the srs2 cells show very unique thin line staining of Rad51 during early prophase such as 4 h (Fig. 4B). The thin Rad51-line in srs2 cells is connected from one Rad51 focus to the other focus/foci, which we refer to as “Rad51 bridge”. At least one clear Rad51 bridge between two Rad51 foci were observed at ~40% frequency of srs2 spreads at 4 h (middle graph of Fig. 4C). A few Rad51 bridges were seen in wild type. We also found the Rad51-bridge staining among more than three Rad51 foci in srs2 cells, but not in wild type (right graph of Fig. 4C). Careful examination of Rad51 foci in the wild type often detected a Rad51 focus with “single tail (or whisker)” (left graph of Figure 4C). The number of Rad51 tail from a single Rad51 focus is almost one. There is few focus with more than 2 tails. When measured the length of the bridge between two foci, we found both wild-type and the srs2 cells show similar distribution of the lengths (Fig. 4D). These results indicate that Srs2 suppresses the formation of Rad51 bridges. Indeed, the srs2 cells increased the frequency of the Rad51 bridges and more connections among more than two foci relative to the wild type (Fig. 4C).
We then used super-resolution microscopy to analyze Rad51 localization on meiotic chromosomes at high resolution. A structural illumination microscope (SIM) was used to determine Rad51 localization in wild type and srs2 cells at 4h (Fig 4E). As shown above, in the srs2 mutant, we detected both Rad51 bridges and tails more than in wild type. The wild type the srs2 mutant shows Rad51 foci with tail/bridge at a frequency of 15.4±4.2% (n=18) and 54.3±8.5% (n=20), respectively.
The average length of the bridge is ~0.4 μm (Fig. 4D). If the bridge is postulated to consist of a single Rad51 filament on the ssDNA, which is extended 2-fold relative to the B-form DNA (Ogawa et al. 1993), we can calculate the bridge contains ~600 nt (400 nm/2X3.3/10.5). This might be a range of reasonable estimate ssDNA length at a single DSB site with ~900 nt (Mimitou et al. 2017; Zakharyevich et al. 2010). Rad51 bridge described here might be similar to the staining of “ultra fine bridge” seen in anaphase of damage mammalian cells (Chan and Hickson 2011) (see Discussion). The formation of Rad51 thin bridges in early prophase-I of the srs2 cells suggests entanglement of recombination intermediates.
Little chromosome breaks are formed in the srs2 mutant during late prophase-I
In order to detect chromosomal breaks, we tried to analyze chromosome status by pulse field gel electrophoresis (PFGE). At 3, 4 h time points in both wild-type and srs2 mutant cells, chromosomal band becomes a smear due to the introduction of DSBs (Fig. S5). While the smear pattern disappeared at 5 h in wild type, the srs2 mutant showed persistent smear bands by 5 h and then disappeared. Interestingly, we again detected smear bands at 10 and 12 h when most of the srs2 diploid makes spores, indicating the formation of DSBs in the srs2 spores. The smear bands were barely observed in wild type spore. More importantly, the srs2 cells did not show breaks at 5-8 h, when Rad51 aggregates are induced.
Discussion
Rad51 bridges and Rad51 aggregates in the srs2 mutant
The srs2Δ mutant shows decreased levels of CO and NCO relative to the wild-type, indicating a positive role of Srs2 in meiotic recombination (pro-recombination role). This weak defect in the recombination is consistent with delayed DSB repair (delayed disassembly of Rad51 foci) as well as defective SC formation in the mutant. Our studies also showed that the srs2 mutant is partially defective in a step after the DSB processing. However, this “weak” defect in the recombination cannot explain reduced spore viability of the mutant, since the mutants with 50% reduction of CO show high spore viability; e.g. spo11, xrs2, msh4/5 hypomorphic mutants (Martini et al. 2006; Nishant et al. 2010; Shima et al. 2005). Consistent with low spore viability of the mutant, we and others detected abnormal chromosome segregation in srs2 meiosis, suggesting the presence of DNA abnormality in the mutant.
In this study, we described “unusual” DNA damage formed in the absence of Srs2 helicase during meiosis. This damage is marked with the association of the recombination protein, Rad51, with a large quantity, which we refer to as “Rad51 aggregate”. The Rad51 aggregate is not a protein aggregate since it contains another recombination protein, Rad52, as well as RPA, but not meiosis-specific recombination proteins such as Dmc1. The presence of RPA strongly suggests the presence of ssDNAs. Indeed, thin line-like staining of Rad51 and RPA emanating from the aggregate are often observed.
The formation of Rad51 aggregates in srs2Δ mutant requires Spo11 catalytic activity, thus DSB formation. On the other hand, kinetic analysis revealed that Rad51 aggregates in srs2Δ mutant appear in late prophase-I after the disappearance of Spo11-dependent Rad51 foci associated with meiotic recombination. Rad51 aggregates appear just after the disappearance of “normal” Rad51 foci. This suggests that the formation of Rad51 aggregates occur after the completion of DSB repair such as Rad51-mediated strand invasion. Consistent with this, the ndt80 mutation, which induces an arrest at mid-pachytene stage, blocks the aggregate formation in the srs2Δ mutant. The ndt80 mutant accumulates dHJ as a product of completion of Rad51-dependent strand invasion (Allers and Lichten 2001), and also shows persistent formation of Spo11-dependent meiotic DSBs (Carballo et al. 2013). Therefore, persistent DSBs and dHJs are unlikely to be directly linked with Rad51 aggregate formation.
Mutant analysis shows the formation of Rad51 aggregates in the srs2Δ requires pachytene-exit, but occurs prior to the transition of metaphase-I to anaphase-I, chromosome segregation. Indeed, Rad51 aggregate formation occurs even when chromosome segregation was inhibited by the treatment with a microtubule polymerization inhibitor and the CDC20 depletion, which delays and blocks the onset of anaphase-I. These indicate that the aggregate formation is induced around the disassembly of meiotic chromosome structure; e.g. diplotene or diakinesis.
One possibility to explain Rad51 aggregate formation in the srs2 mutant is that, after the exit of Ndt80-execution point, there might be unrepaired DSBs, which could be repaired by Rad51-dependent pathway (but not Dmc1-pathway) during late prophase-I. The srs2 mutant might be specifically defective in this DSB repair after the pachytene exit. In this pathway, Srs2 may be essential for Rad51 removal, which may lead to the accumulation of unrepaired ssDNAs. However, this is unlikely since even DSB ends formed during pachytene are bound by Dmc1 as well as Rad51. However, the Rad51 aggregates in the srs2 mutant do not contain Dmc1 even when Dmc1 protein is present in a cell.
Alternatively, Rad51 aggregates and/or its associated DNA damage are formed in two-step process. Frist, DSB repair in the absence of Srs2 may result in the formation of aberrant recombination products/intermediates such as entangled duplexes DNAs (see Fig. 6B). Second, this aberrant product/intermediate might be converted into DNA damage with Rad51 aggregates in late prophase-I. Consistent with this two-step model, we found a novel structure called Rad51-bridge (or whisker), thin lines of Rad51 which connect Rad51 foci. This bridge is seen at early prophase I of the srs2 mutant more frequently than in wild type.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A. A model of Rad51 focus comprised of the Rad51 filament. Rad51 filament is accommodated into a three-dimensional structure. B. A recombination pathway with Rad51 focus and filaments is described. In left, the second end capture by a displaced strand from D-loop, which is mediated by the Rad51 filament may trigger the disassembly of Rad51 filament (focus) on the second end, which is promoted by Srs2. Srs2 also prevents the formation of a multi-invasion intermediate. These abnormal recombination intermediates are processed into a final product, which may contain a pathological damage that may lead to the formation of a long ssDNAs with Rad51 aggregates during late prophase-I.
The presence of Rad51-bridge and -whisker from Rad51 focus suggests that Rad51 focus is not a simple Rad51 filament, rather may contain a three-dimensional configuration of Rad51 filament (Fig. 6A). Rad51-bridge line staining is reminiscent of anaphase bridge or ultra-fine bridge of chromosomes in mammalian cells (Chan et al. 2007). The formation of anaphase bridges in mammalian cells is a two-step process. Although these bridges are formed during M phase with onset of anaphase, the initiation event leading to the bridge formation occurs during S-phase. These bridges are induced by the treatment of the cell with DNA replication inhibitor(s) or in the absence of DNA repair protein such as BLM helicase. The bridges are decorated with repair proteins such as BLM and RPA, but not Rad51.
Rad51 aggregate formation in srs2 meiosis is clearly different from the anaphase bridge in the following two aspects. First, the formation is not required for chromosome segregation. Second, the initiation event should be Spo11-dependent DSB formation in early prophase-I (meiotic G2 phase). If the two-step model as described above is true, the conversion of the aberrant product/intermediate into the aggregate in srs2Δ mutant should occur after mid-pachytene exit. Given drastic chromosome morphogenesis such as chromosome compaction and disassembly of the meiosis-specific chromosome structure, SC, occur during late meiotic prophase-I (Challa et al. 2019), these events might induce the aberrant DNA damage with Rad51 aggregates.
Given that Rad51-bridge in the srs2 mutant is formed between Rad51 foci, Srs2 might play a role of this kind of Rad51-associated DNA entanglement between the two DSB sites (Fig. 6B). One likely intermediate is multiple invasion (Piazza et al. 2017), which are formed by Rad51-mediated strand invasion into multiple loci. Therefore, Srs2 might play a role in resolution of multiple-invasion by controlling Rad51 filament dynamics using its Rad51-dismantling activity.
Bishop and his colleagues show a pair of Rad51 foci during early meiotic pro-phase I are formed in the two ends of a single DSB site (Brown et al. 2015). Thus, it is likely that the Rad51 bridge we observed is formed between a pair of Rad51 foci on the two DSB ends. If so, one likely possibility is that the bridge is a ssDNA between two DSB ends. One way to connect the two DSB ends is bridged by the annealing of ejected ssDNA from the D-loop after the DNA synthesis (Fig. 6B). Since the bridge is mainly seen in the absence of Srs2, we propose that Rad51 dismantling activity of Srs2 promotes the removal of Rad51 from the rejected ssDNA. Moreover, it is likely that Srs2 also remove Rad51 in the other end of the DSB during the second end-capture. This idea could explain the formation of the bridge between two Rad51 foci in the srs2, but not in wild type. In wild-type cells, Srs2 seems to remove Rad51 assembly from the intermediates for the second end capture. Importantly, genetic analysis of mitotic recombination in the srs2 mutant suggest the role of Srs2 to facilitate the annealing of the newly synthesized strand to second resected ends by removing Rad51 from the second end (Elango et al. 2017; Ira et al. 2003; Liu et al. 2017; Mitchel et al. 2013).
We still cannot figure out recombination products formed in the absence of Srs2, which trigger the formation of the Rad51 aggregate. 2D gel analysis has shown that there is few accumulation of abnormal recombination intermediate such as multiple dHJs (Lichten/Goldman, accompanying paper). Thus, multiple dHJs is unlikely. Rather, there might be an entanglement of DNA strands after the completion of the meiotic recombination (Fig. 6B). This intermediate seems to be related to a lethal recombination intermediate formed in the srs2 mutant during mitosis.
In either scenario, our analysis reveals a novel pathway to protect meiotic cells in late prophase-I from the formation of aberrant DNA damage induced by Spo11. This repair pathway heavily depends on Srs2 function. For this function, Srs2 is almost essential for meiosis. We would like to point out that Rad51 aggregates in the srs2 mutant is related to lethal recombination intermediates in mitotic cells with SRS2 deletion, which is postulated to form through two-step model.
Rad51 aggregate-associated DNA damage seems unrepaired during meiosis. During late prophase-I, there should be sister chromatid or other recombination partners to repair the damage, this might be due to the presence of Rad51-inhibitor Hed1, which clearly suppresses Rad51-mediated DNA repair.
The result that CHEF-Southern for chromosome III did not detect any DNA fragmentation at times of Rad51-aggregates in the srs2Δ mutant; e.g. 6 and 8 h, implies Rad51 aggregate-associated DNA damage does not contain DSBs. One simple interpretation is that Rad51 aggregates are on either the ssDNA gaps or unwound duplex DNAs.
No activation of DNA damage checkpoint during late prophase I, meiosis I and meiosis II
In the absence of Srs2, DNA damage with Rad51 aggregates is formed and passed into MI and MII with activation of DNA damage checkpoint, which leads more catastrophic events such as chromosome fragmentation with DSB formation in spores. This may explain quite a big reduction of spore viability of the srs2 diploid with reasonable levels of meiotic recombination.
The absence of DNA damage-induced delay in late meiosis-I in the srs2 cells is quite different from cell cycle delay induced by the recombination (pachytene) checkpoint during early prophase-I (MacQueen and Hochwagen 2011; Tsubouchi et al. 2018). In the recombination checkpoint, DSBs and associated ssDNA activate sensor kinases Tel1(ATM) and Mec1(ATR), respectively. During meiosis, activated Mec1 and Tel1 induce the activation of a meiosis-specific kinase, Mek1, Rad53 homolog, by phosphorylating its partner protein Hop1. High Mek1 activity down-regulates Ndt80 activity, thus, blocking the exit of mid-pachytene stage. During meiosis, the activation of mitotic DNA damage downstream kinases, Rad53 and Chk1, is blocked through an unknown mechanism. At late times in the srs2 mutant, we did not see prolonged phosphorylation of Hop1, thus little activation of Mec1 (and Tel1). This strongly suggests that DNA damage with Rad51 aggregates in the srs2 is masked by the checkpoint activation or there is no such mechanism in late G2 phase of meiotic cells. Alternatively, although not exclusive with the above, Srs2 may function in the activation of the checkpoint during this phase.
Role of Rad54 in late recombination
Upon Rad54 depletion after the assembly of Rad51 on the ssDNA during meiosis, we found a novel staining of Rad51 called Rad51 clump, which is different from typical Rad51 foci in wild-type and aggregates in the srs2 cells. The presence of Rad51 clumps supports the idea of a role of Rad54 after the assembly of Rad51 filaments. Moreover, the Rad54-Rad51, not with Dmc1, may function in the repair of DSBs in late prophase-I. Previous cytological analysis of the rad54 deletion does not show the formation of Rad51 clump (Shinohara et al. 2000; Shinohara et al. 1997b). One possibility is that Rad54 depletion may remove Rad54-associated proteins also from the nuclei. As a result, we do see clear defect in Rad51 dynamics upon Rad54 depletion in late prophase-I.
In the accompanying paper, Goldman and his colleagues described the formation of Rad51 aggregates during srs2 meiosis.
Author’s contribution
H.S., M.S., and A.S. designed the experiments. H.S., H.S.M.S., Y.F., and L.P. performed all experiments. M.S. provided reagents. H.S., H.S.M.S., and A.S. analyzed the data. A.S. prepared manuscripts with help by H.S., H.S.M.S. and M.S.
Funding
This work was supported by JSPS KAKENHI Grant Number; 22125001, 22125002, 15H05973 and 16H04742 to A.S.; 21770005 to H.S.; 15H05973, M.S. H.S.M.S. was supported by Institute for Protein Research.
Acknowledgements
We are grateful for Drs. Alstair Goldman and Michael Lichten for sharing unpublished results prior to publication. We thank Dr. Neil Hunter (UC, Davis) for pCLB2-SGS1 yeast and Dr. Andreas Hochwagen (New York University) for the anchor-away yeast strains. We thank Ms. H. Matsumoto, C. Watanabe, and H. Wakabayashi for excellent technical assistance.
References




