

Abstract
The sources of human germline mutations are poorly understood. Part of the difficulty is that mutations occur very rarely, and so direct pedigree-based approaches remain limited in the numbers that they can examine. To address this problem, we consider the spectrum of low frequency variants in a dataset (gnomAD) of 13,860 human X chromosomes and autosomes. X-autosome differences are reflective of germline sex differences, and have been used extensively to learn about male versus female mutational processes; what is less appreciated is that their mutation patterns also reflect chromosome-specific biochemical features that distinguish the X from autosomes. We tease these different features apart by comparing the mutation spectrum in multiple genomic compartments on the autosomes and between the X and autosomes. In so doing, we are able to ascribe specific mutation signatures to replication timing and recombination, and to identify differences in the types of mutations that accrue in males and females. In particular, we identify C>G as a mutagenic signature of male meiotic double strand breaks on the X, which may result from late repair. These results show how biochemical processes of damage and repair interact with sex-specific life history traits to shape germline mutation patterns across both human X and autosomes.
Introduction
Germline mutations, the source of all heritable variation, accrue each generation from accidental changes to the genome during the development of gametes. These mutations reflect a balance of biochemical processes that alter DNA in the germline and those that correctly repair DNA lesions before the next replication (Ségurel, Wyman, and Przeworski 2014). The biochemical machinery that underlies mutagenesis can be conceptualized as a set of genetic loci that modulate the net mutational input in each generation, and variants in these loci as “modifiers of mutation” (Lynch et al. 2016; Harris and Pritchard 2017). Since the activity of distinct biochemical pathways often leaves different signatures in DNA (Alexandrov et al. 2013, 2018; Nik-Zainal et al. 2012, 2016; Roberts et al. 2013; Pleasance et al. 2009), these modifiers influence the distribution of mutation types (the “mutation spectrum”), as well as the total number of mutations inherited by offspring.
The mutational landscape in the germline is also modified by the sex of the parent: in humans, notably, it has long been known that males contribute three times as many mutations on average as females per generation (Crow 2000; Kong et al. 2012). As in other mammals, gametogenesis differs drastically by sex: female germ cells are arrested in meiosis for much of their development whereas male germ cells enter meiosis late in their development (Morelli and Cohen 2005; Fayomi and Orwig 2018; Tang et al. 2016). Male germ cells undergo many more cell divisions than female germ cells; they are also methylated earlier and have higher methylation levels on average throughout ontogenesis (Reik, Dean, and Walter 2001). Due to differences in their cellular biochemistry at different developmental stages, male and female gametes may be subject to different kinds of endogenous and environmental insults, or repair different types of DNA lesions with varying degrees of efficacy. For example, male gametes may accrue oxidative damage due to lack of base excision repair in late spermatogenesis (Smith et al. 2013). Males and females also differ in life history traits such as the timing of puberty and age of reproduction (Fenner 2005), which modulate the exposure of the gamete to the biochemical states associated with particular stages of development and thus alter their mutagenic impact. In that sense, the sex of the parent as well as variants in loci associated with sex-specific biochemistry and life history are also modifiers of mutation. The germline mutation spectrum in each generation is therefore a convolution of signatures of biochemical processes and the effect of sex.
In principle, it is possible to characterize mutational mechanisms by decomposing the mutation spectrum into its component signatures. Such an approach has led to a wealth of insight into the sources of somatic mutations, i.e., mutations that accumulate in somatic tissues during normal development or ageing. Distinct signatures of processes that generate or repair DNA lesions have been identified by analyzing millions of somatic mutations in their immediate sequence context, across tumor samples of diverse etiologies (Pleasance, Stephens, et al. 2009; Nik-Zainal et al. 2016; L. B. Alexandrov et al. 2013; L. Alexandrov et al. 2018). A complementary approach, based on changes in the mutation spectrum with regional variation in genomic features, has further illuminated the influence of local replication timing, transcription, chromatin organization, and epigenetic modifications on somatic mutagenesis (Greenman et al. 2007; Rubin and Green 2009; Pleasance, Cheetham, et al. 2009; Hodgkinson, Chen, and Eyre-Walker 2011; Woo and Li 2012; Schuster-Böckler and Lehner 2012; Liu, De, and Michor 2013; Supek and Lehner 2015; Polak et al. 2015; Blokzijl et al. 2016).
These methods have proved difficult to apply to the germline however, because each offspring inherits only about 70 de novo mutations on average (Jónsson et al. 2017). Thus, the most direct approach to the study of germline mutations, the resequencing of pedigrees (Francioli et al. 2015; Kong et al. 2012; Rahbari et al. 2015; Goldmann et al. 2016; Jónsson et al. 2017), remains limited in its ability to identify determinants of mutation rate variation. For instance, examining 96 possible mutation types considered in a trinucleotide context in ~100,000 de novo mutations, the biggest study to date found only three mutation types for which the proportion transmitted from mothers and fathers differed significantly (Jónsson et al. 2017). Additionally, the mutation patterns from the three largest de novo mutation studies combined show inconsistent patterns of correlation to genomic features, for reasons that remain unclear (Smith, Arndt, and Eyre-Walker 2018).
One way to overcome the limitation of small samples in studies of germline mutation is to use rare polymorphisms as a proxy for de novo mutations. Low frequency variants in large samples are recent enough for effects of direct and indirect selection and biased gene conversion to be minimal; they should therefore recapitulate the de novo mutation spectrum with reasonable fidelity (Carlson et al. 2018; Rahbari et al. 2015; Schaibley et al. 2013). The much higher density of rare variants across the genome can then be used to more robustly investigate associations with genomic features. Using this strategy, a recent study of human autosomal data identified mutation types and contexts significantly associated with a variety of genomic features (Carlson et al. 2018). While the authors suggested putative biochemical sources for three signatures in the germline based on their similarity to patterns that have been reported in tumors, it is unclear to what degree these mechanisms can be directly extrapolated to the germline (Chen et al. 2017; Hodgkinson and Eyre-Walker 2011). Moreover, sex-specific effects on the mutation spectrum were not considered.
Insight into sex-specific effects can be gained by contrasting polymorphism levels on the sex chromosomes and autosomes, since autosomes reflect mutational processes in the male and female germlines equally, while the X chromosome disproportionately reflects the female germline, and the Y chromosome exclusively reflects the male germline. This approach to studying sex differences has been used extensively; notably, its application to divergence data provided the first systematic evidence for a higher contribution of males to mutation in humans and other mammals (Shimmin, Chang, and Li 1993; Makova and Li 2002) Yet no significant influence of sex on the mutation spectrum was inferred in a recent comparison of ~3000 rare variants on the X and Y chromosomes (Rahbari et al. 2015). Despite their importance, therefore, the genesis of germline mutations remains poorly understood to date, and the role of sex-specific modifiers particularly enigmatic.
To fill this gap, we compare the spectrum of rare polymorphisms across subsets (“compartments”) of the genome using genome-wide SNPs in the gnomAD dataset (Lek et al. 2016). We focus on specific genomic compartments on the X chromosome and autosomes with unique combinations of biochemical and sex-specific properties, enabling us to tease apart biochemical and sex-specific influences on the germline mutation spectrum. With over 120 million SNPs to analyze across the genome, this approach can detect even subtle differences in mutational patterns between genomic compartments.
Results and Discussion
We use whole genome SNP data from 15,496 individuals made available by the Genome Aggregation Database (gnomAD), which includes 9,256 Europeans and 4,368 African or African-American individuals (Lek et al. 2016). We limit our analysis to the 6,930 female individuals in the dataset to sample X-chromosomes and autosomes in equal numbers. We then compare the diversity levels of different mutation types in pairs of genomic compartments (Fig. 1a). In these data, there are ~120 million SNPs, of which 53% of the variants are singletons, 11% doubletons, and about 10% of variants are at frequency 1% or greater (Fig. 1b).

(a) A schematic of genomic compartments on the X chromosome and autosomes. Three compartments on the X chromosome are depicted: the pseudoautosomal region (PAR), regions of the X that escape inactivation (E), regions of the X that undergo inactivation (I). Also depicted are two hypothetical autosomal compartments with distinct biochemical properties (a1 and a2). Analyses include pairwise comparisons of mutational patterns between autosomal compartments and between the X chromosome and autosomes. (b) The frequency spectrum of variants in the 13,860 chromosomes analyzed (AC = Allele count; AF = Allele frequency).
As in other recent studies, we extract the single base pair flanking sequence on each side of the variant position using the hg19 reference to obtain mutations in their trinucleotide context, and combine mutations in reverse complement classes (for example, the ACG>ATG and CGT>CAT classes are collapsed into the former) to obtain 96 mutation types. Unless otherwise noted, we treat the major allele as the ancestral state at a site; however, we obtain similar results using the ancestral allele and context from the 1000G reconstruction of the ancestral human genome sequence (1000 Genomes Project Consortium et al. 2015) (Supplementary Fig. 5). We include multi-allelic sites (~6% of the data) by counting the multiple derived alleles separately as if they had occurred at separate bi-allelic sites with the same major allele (Supplementary Methods). To obtain the diversity for each mutation type within a genomic compartment, we divide the number of segregating sites of a particular type by the number of mutational opportunities, i.e., sites where a single change could have given rise to that mutation type; this approach accounts for base composition within a compartment.
To compare mutation types across two genomic compartments, we normalize the diversity for each mutation type by the total diversity within each compartment. In this way, we control for the effect of population genetic processes that affect diversity across compartments but do so evenly across all mutation types, and isolate differences in the mutation spectrum; this step is particularly important for comparisons between the X chromosome and autosomes. For each of 96 mutation types, we test if the observed relative diversity in the two compartments differs from what would be expected by chance. To this end, we designate one of the two compartments as the “test” and the other as the “reference” compartment. Our null expectation is that the number of mutations of a particular type in the test compartment is binomially distributed with a mean value proportional to the observed diversity for that type in the reference compartment, adjusted for overall differences in diversity between the two compartments (Supplementary Methods). Mutation types are considered significantly different in their frequencies between the two compartments if the two-tailed p-value from the binomial test is below the Bonferroni-corrected 5% significance threshold.
Biochemical properties vary along the genome, both on autosomes and the X chromosome. In turn, sex-specific influences from the germline are the same across autosomes, but differ between the X chromosome and autosomes. We therefore first compare autosomal compartments with distinct biochemical features to illuminate biochemical influences on the mutation spectrum. Then, by comparing compartments across the X chromosome and autosomes and accounting for average biochemical differences between them, we disentangle sex-specific and biochemical influences on the mutation spectrum.
Replication timing and its covariates influence the germline mutation spectrum
We consider autosomal compartments that differ with regard to specific biochemical properties in the germline. In cases where these data are unavailable for germline tissue and we are limited to somatic cell lines, we focus on biochemical features that have broadly similar distributions across tissue types. Replication timing is consistently an important predictor of local mutation rates (Stamatoyannopoulos et al. 2009; Smith, Arndt, and Eyre-Walker 2018; Chen et al. 2017) in both the soma and the germline, and broad-scale replication timing maps are relatively concordant across tissues (Hiratani et al. 2010; Ryba et al. 2010) (Supplementary Figure 1). The observed mutagenic effect of late replication has been hypothesized to be due to a decline in the efficacy of mismatch repair with delayed replication, less time for repair, or the accumulation of damage-prone single-stranded DNA at stalled replication forks (Stamatoyannopoulos et al. 2009; Supek and Lehner 2015).
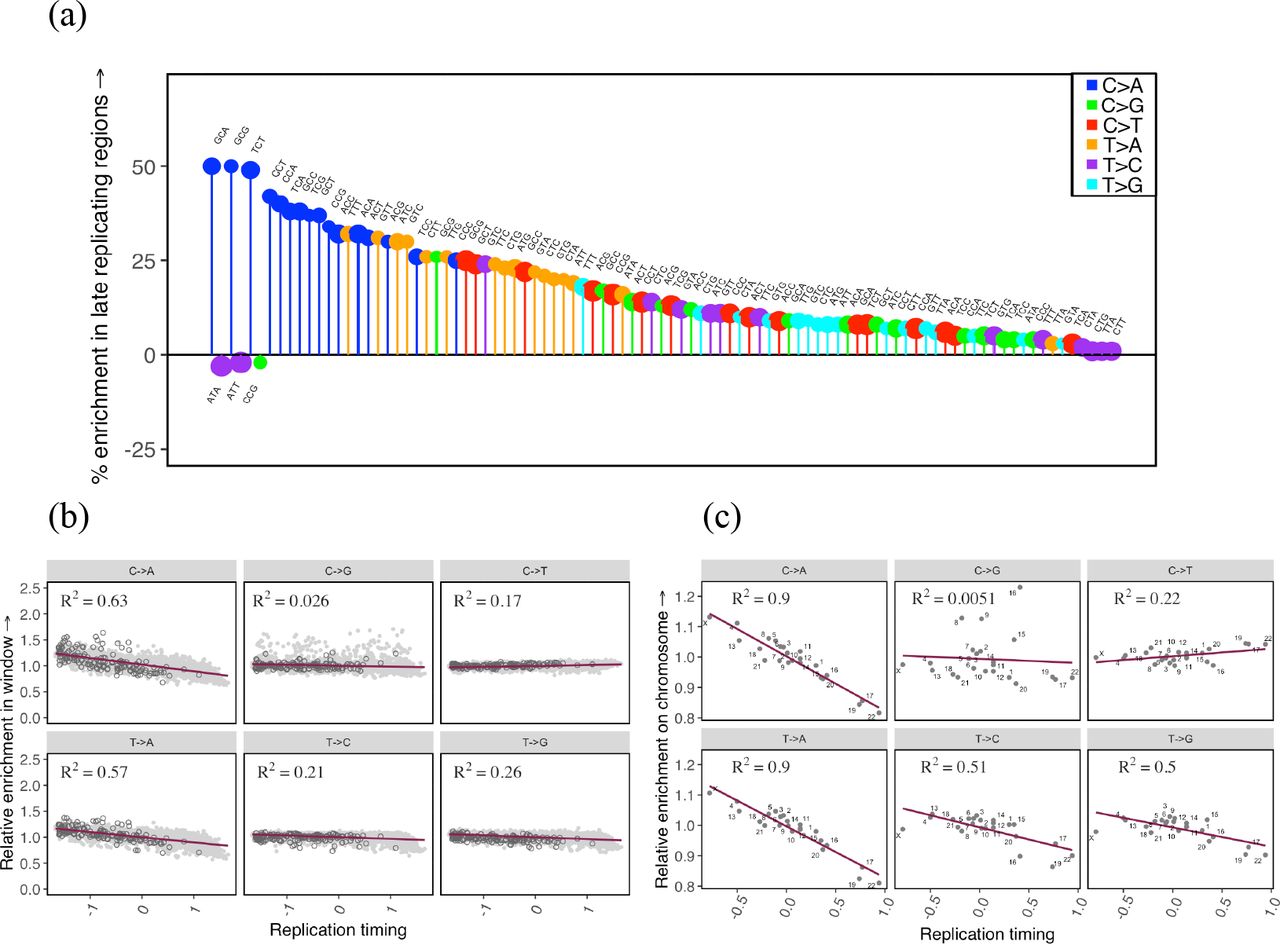
To assess if replication timing affects the germline mutation spectrum, we compare autosomal regions that differ in their replication timing using available data from LCL and H9-ESC cell lines (Hiratani et al. 2010; Koren et al. 2012). As expected, almost all mutation types are significantly enriched in late replicating regions relative to early replicating regions (Fig. 2a, Supplementary Fig. 2). In particular, we observe a substantial enrichment of C>A and T>A mutations in late replicating regions, a pattern also observed by Carlson et al., 2018 in a different sample of rare variants. Moreover, the mean replication timing in 1 Mb windows across the genome explains ~60% of the variation in C>A and T>A enrichment in those windows relative to the autosomal average and between 2% and 26% for all other mutation types (p ≪ 10−5), suggesting that these two mutation types are particularly sensitive to replication timing (Fig. 2b, Supplementary Fig. 2, Supplementary Methods).

The effect of replication timing on the mutation spectrum at different scales. (a) Comparison of the spectrum of 96 mutation types in late replicating autosomal regions relative to early replicating autosomal regions. Only significant differences are shown, ranked in order of effect size from left to right. The size of the circle reflects the number of mutations of that type. Late replicating regions are defined as having a replication timing score <= −0.5 and early >= 0.5; positive scores indicate earlier than average replication. (b) For each of six mutational classes, the enrichment in 1Mb windows relative to all other autosomal windows combined, ordered by the mean replication timing. Positive replication timing scores indicate earlier than average replication. Enrichment in 1 Mb windows on the X chromosome relative to all autosomal windows combined have been overlaid in dark grey for reference. (c) For each of six mutational classes, enrichment of individual autosomes and X relative to all other autosomes combined, ordered by the mean replication timing.
Because replication timing is correlated with multiple genomic features, including higher order chromatin structure, epigenetic modifications, and in particular, DNA methylation at CpG sites, some of the observed patterns could be reflective of these processes rather than replication per se. To assess the marginal impact of CpG methylation on the effect of replication timing, we consider early and late replicating regions within and outside CpG islands, which are regions of CpG hypomethylation across tissue types (Deaton and Bird 2011; Wu et al. 2010). We find that at both CpG sites inside and outside islands, C>A mutations are enriched in late replicating regions (Supplementary Fig. 3), suggesting that this signal is not due to differences in methylation. Moreover, we also observe this pattern at non-CpG sites (Supplementary Fig. 3).
The association of C>A mutations with replication timing does not necessarily imply that they are “replicative” in origin, i.e., due to errors directly introduced by the replication machinery while copying intact DNA, as they could also reflect greater unrepaired damage in later replicating regions (Supek and Lehner 2015). In particular, since C>A mutations are a known consequence of oxidative damage in somatic tissues (Alexandrov et al. 2018; David, O’Shea, and Kundu 2007; Neeley and Essigmann 2006; De Iuliis et al. 2009; Alexandrov et al. 2013), it is plausible that these mutations accumulate in regions of late replication due to greater damage to exposed single-stranded DNA, or poorer repair in these regions.
Considering other factors shown to influence mutation patterns, we recover a known signature of CpG methylation: transitions at CpG sites (C>T mutations in the ACG, CCG, GCG, and TCG trinucleotide contexts), which are thought to be due to the spontaneous deamination of methyl-cytosine to thymidine, are highly depleted in the hypomethylated CpG islands compared to the rest of the genome (Supplementary Fig. 4a). Similarly, we detect an increase in C>G mutations in a subset of autosomal regions previously shown to be enriched for this signature (Supplementary Fig. 4b). This C>G signature is thought to reflect inaccurate repair of spontaneous damage-induced double-strand breaks in the germline (Jónsson et al. 2017; Gao et al. 2018).
Importantly, the impact of these biochemical features on mutation does not average out across chromosomes. Comparing individual autosomes to all other autosomes reveals ubiquitous variation in the mutation spectrum at the chromosome-level (Supplementary Fig. 4c). In particular, individual chromosomes that replicate later on average show greater enrichment of C>A and T>A mutation types: differences in mean replication timing for individual autosomes explain ~90% of the variation in C>A and T>A enrichment at the chromosome level (p ≪ 10−5), while they explain less than 50% for other mutation types (Fig. 2c). These results demonstrate that replication timing, and potentially other genomic features such as methylation and propensity for accidental double strand break damage, lead to chromosome-level differences in diversity, hinting at some plausible sources for observed but unexplained chromosome-level differences in average divergence (Hodgkinson and Eyre-Walker 2011).
Sex differences in the mutation spectrum are subtle but likely ubiquitous
Next, we assess the impact of sex on the germline mutation spectrum by comparing mutational patterns on the X chromosome and autosomes. The X chromosome is disproportionately exposed to mutational processes in the female germline; in other words, each X chromosome spends more time in females than in males, while each autosome spends the same amount of time in males and females. Thus, mutation types that arise more commonly in the female germline are expected to be enriched (and mutation types that arise more commonly in the male germline depleted) on the X chromosome relative to autosomes. We account for population-level properties that may affect the mutation spectrum differently on the X and autosomes (Supplementary Methods). Having done so, we find most mutation types to be differentially enriched on the X and autosomes (Fig. 3a).

Comparison of the mutation spectrum on the X chromosome and autosomes. The pseudoautosomal region (PAR) and CpG sites are excluded from this analysis. Only significant differences are shown, ranked in order of effect size from left to right. The size of the circle reflects the number of mutations of that type. (a) Enrichment of mutation types on the X chromosome relative to autosomes. (b) Enrichment of mutation types in the genic compartment of the X chromosome that constitutively escapes inactivation, relative to genic regions on autosomes. Hollow circles represent mutation types enriched or depleted in both the escaped and inactive compartments of the X relative to autosomes. (c) Enrichment of mutation types in the genic X chromosome compartment that undergoes X-inactivation, relative to genic regions on autosomes. Hollow circles represent mutation types enriched or depleted in both the escaped and inactive compartments of the X relative to autosomes. The larger number of significant differences in (c) compared to (b) likely reflects at least in part the approximately five-fold greater amount of data in the inactive versus the active genic regions of the X chromosome.
Importantly, however, these X-autosome differences do not only reflect differences in the types of mutations in male and female germlines; given the substantial effect of biochemical features on mutational patterns across autosomes, they can also stem from differences in the distribution of these biochemical features on the X chromosome and autosomes. For instance, in de novo mutation studies (Jónsson et al. 2017; Goldmann et al. 2016), C>A mutations are found to arise more often in males, suggesting that they should be depleted on the X. Instead, they are found enriched on the X chromosome relative to autosomes (Fig X). A possible explanation is that the X accrues excess C>A mutations because it replicates late in the germline. C>A mutations are known to be associated with oxidative damage (Alexandrov et al. 2018; David, O’Shea, and Kundu 2007; Neeley and Essigmann 2006; De Iuliis et al. 2009; Alexandrov et al. 2013), which remains unrepaired in sperm (Smith et al. 2013), and is likely repaired at or before the first cell division in the zygote (Harland et al., 2017; Huang et al., 2014; Ju et al., 2017). Late replication of the X chromosome at this stage, perhaps due to the inactive status of the paternally inherited X in female embryos (Reik and Ferguson-Smith 2005), could then indeed be expected to result in an enrichment of C>A mutations on the X despite a primarily male source of damage. This example underscores that accounting for X-specific effects of biochemical features is important for uncovering true sex differences in X-autosome comparisons.
One well-characterized idiosyncratic feature of the X is X-inactivation, which is associated with sex-specific changes in methylation, transcriptional activity, and notably, replication timing: because the inactive X chromosome exhibits a significant lag in replication, on average the X replicates later than autosomes (Koren et al. 2014). Though X-inactivation is a short-lived process in the germline—limited to early embryogenesis in females, and brief meiotic and post-meiotic periods in males (Nguyen and Disteche 2005; Heard and Disteche 2006; Payer, Lee, and Namekawa 2011; Sangrithi and Turner 2018)—it could nevertheless lead to observable differences in the mutation spectrum between different regions of the X. The “active” compartment of the X chromosome, i.e., the approximately 15% of the transcribed X that constitutively escapes inactivation across tissues (Tukiainen et al. 2017; Carrel and Willard 2005) may therefore differ in its mutation spectrum from the rest of the X. Comparing autosomes, X inactive and X active regions, we find T>C mutations at GTC sites and C>T types at ACT sites enriched in both active and inactive regions of the X relative to autosomes and T>G mutations at ATG sites depleted both in the active and inactive regions of the X relative to autosomes (Fig. 3b and 3c, Supplementary Figure 10). Since these cases cannot be attributed to X-inactivation and are enriched (or depleted) concordantly on compartments of the X chromosome that differ in their replication timing, methylation levels and other features, they are strong candidates for true sex differences in mutation. Given that the genic compartment known to escape inactivation across tissues is a small fraction of the X chromosome, there are likely many more subtle ones that we miss.
A complementary approach to minimizing the effect of X-specific features on the mutation spectrum of the X chromosome is to consider regions of the X chromosome that are comparable to autosomes in their average replication timing. The replication timing on the X chromosome across multiple human cell lines depends on whether one of the X chromosomes is inactivated (Supplementary Figure 1; Supplementary Methods) (Allegrucci and Young 2007; Vallot et al. 2015; Patel et al. 2017; Ryba et al. 2010; Hiratani et al. 2010; Tang et al. 2016). This observation suggests that controlling for replication timing differences between the X chromosome and autosomes may also control for the effects of other correlated features, including X-inactivation. Using this approach, all three mutation types that we highlight as putative sex differences based on their differential enrichment in the active compartment of the X relative to autosomes are also observed as significant differences between the X chromosome and autosomes (Fig. 4a, Supplementary Fig. 12). That we find the same types with this complementary approach provides further evidence that they are true sex differences.

The mutation spectrum on the X and autosomes matched for average replication timing. The pseudoautosomal region (PAR) and CpG sites are excluded from this analysis. (a) Comparison of the mutation spectrum on the X chromosome and autosomes matched for average replication timing. Only significant differences are shown, ranked in order of effect size from left to right. Hollow markers represent mutation types concordantly enriched or depleted in the active regions of the X chromosome compared to autosomes. Mutation types also reported as significant sex differences by Jonsson et al., 2017 are marked with crosses. (b) The X-autosome spectrum for six mutation classes, controlling for mean replication timing (in red), compared to known male female differences from Jonsson et al., 2017 (in black). Solid points are statistically significant differences.
We also detect a number of additional differentially enriched types between X and autosomes after controlling for replication timing differences (Fig. 4a); many of these types are concordantly enriched in early and late replicating regions of the X relative to autosomes (Supplementary Fig. 12). Assuming that a majority of X-specific effects are accounted for when we control for replication timing, these types can also be considered putative sex differences. In that respect, it is noteworthy that C>A mutations are enriched in inactive or late replicating regions of the X, but depleted in the active or early replicating regions of the X, when compared to autosomes (Supplementary Figs. 10, 12). This pattern is what we would expect from the combined influences of a male bias and an effect of replication timing, as we suggested earlier for these types.
We further assess these putative sex-specific signatures by comparing them to results from the largest human pedigree study of de novo mutations to date (Jónsson et al. 2017). Among the six broad mutational classes, Jónsson et al. find C>T mutations significantly enriched in maternal, and C>A, C>G, and T>G mutations relatively enriched in paternal de novo mutations. The mutational patterns we observe on the X chromosome and autosomes after controlling for differences in replication timing are consistent with these effects: we find C>T mutations significantly enriched, and C>A, C>G, and T>G classes significantly depleted on the X chromosome relative to autosomes (Fig. 4b; Supplementary Fig. 11). Jónsson et al. also find three mutation types in their trinucleotide context (TCC>TTC, ACC>AAC, ATT>AGT) as significant sex differences: of these we find two as significant X-autosome differences. As expected, the maternally enriched TCC>TTC type is relatively enriched on the X chromosome, and the paternally enriched ACC>AAC type is relatively enriched on autosomes (Fig. 4a). We do not observe the third type as differentially enriched on the X and autosomes, possibly because there are genomic features specific to the X that mask its enrichment in females.
In turn, the types that we identify as putative sex differences from the comparison of X active, X inactive and autosomes are not reported as significant sex differences in Jónsson et al. (2017). The reason may be that most of them reflect subtle X-autosome differences, with X-enrichment or depletion in the range of 5-10%. Translating these enrichments into a difference between males and females requires a full population genetic model, including assumptions about demography and life history (Amster and Sella 2017). Nonetheless, such subtle X-autosome differences likely correspond to small sex differences that current de novo studies are underpowered to detect.
Components of the meiotic recombination machinery are sex-specific modifiers of the mutation spectrum
In the preceding section, we suggest a plausible mechanism through which sex-specific properties of the germline and the biochemical properties of X-inactivation and late replication jointly influence the distribution of C>A mutations. Here we highlight another mutation type, C>G, which is distributed in a sex-specific manner, but is largely insensitive to replication timing. In this case we are able to leverage the sex-specific properties of recombination on the X chromosome to gain biological insight into the likely source of this mutation and the factors shaping its distribution along the genome.
First, we find the C>G mutational class to be enriched as a whole on autosomes relative to the X chromosome, suggesting that mutations of this type are relatively more common in the male germline compared to the female germline (Fig. 3a). This notion is supported by results from Jónsson et al., 2017, who find the C>G mutational class to be enriched in males relative to females. This study also showed that clustered C>G de novo mutations are concentrated in particular autosomal regions, and increase substantially with maternal age. Maternal age at reproduction determines the duration of oocyte arrest, since females are born with their entire complement of oocytes, which remain in dictyate arrest until ovulation. The authors therefore speculated that the C>G clusters could be due to the more frequent spontaneous occurrence of damage-induced double strand breaks (DSBs) in some genomic regions and in older oocytes. In this view, C>G mutations arise from the repair of spontaneous double-strand break damage in both males and females, with such damage being more age-dependent in females but more common overall in males.
Accidental damage is not the only source of double strand breaks in the germline, however, as during meiosis, double strand breaks are deliberately induced along the genome, through targeting of PRDM9-binding motifs (Myers et al. 2010; Diagouraga et al. 2018). These DSBs are repaired through the homologous recombination pathway: a small minority are resolved through crossovers (COs), which involve exchanges of large segments between homologous chromosomes, and the rest are thought to be repaired through non-crossover gene conversion events (NCOGCs), though a small minority may involve non-homologous end joining and other mechanisms (Baudat, Imai, and de Massy 2013; Li et al. 2018). Because meiotic double-strand breaks are required to initiate homolog search and pairing, they are made all over autosomes and on sex chromosomes both inside and outside the pseudoautosomal region (PAR), in male and female germ cells (Kauppi et al. 2011; Pratto et al. 2014; Lu and Yu 2015), even though male germ cells do not have a sex-chromosome homolog outside the PAR. Specifically in male meiosis, however, the number of DSBs and the timing of DSB induction and repair differs systematically between compartments on the sex chromosomes and autosomes; for instance, PAR1 experiences an exceptionally high rate of meiotic DSBs (Kauppi et al. 2011; Kauppi, Jasin, and Keeney 2012), and DSBs are repaired late on sex chromosomes relative to autosomes (Kauppi et al. 2011; Kauppi, Jasin, and Keeney 2012; Pratto et al. 2014; Lu and Yu 2015).
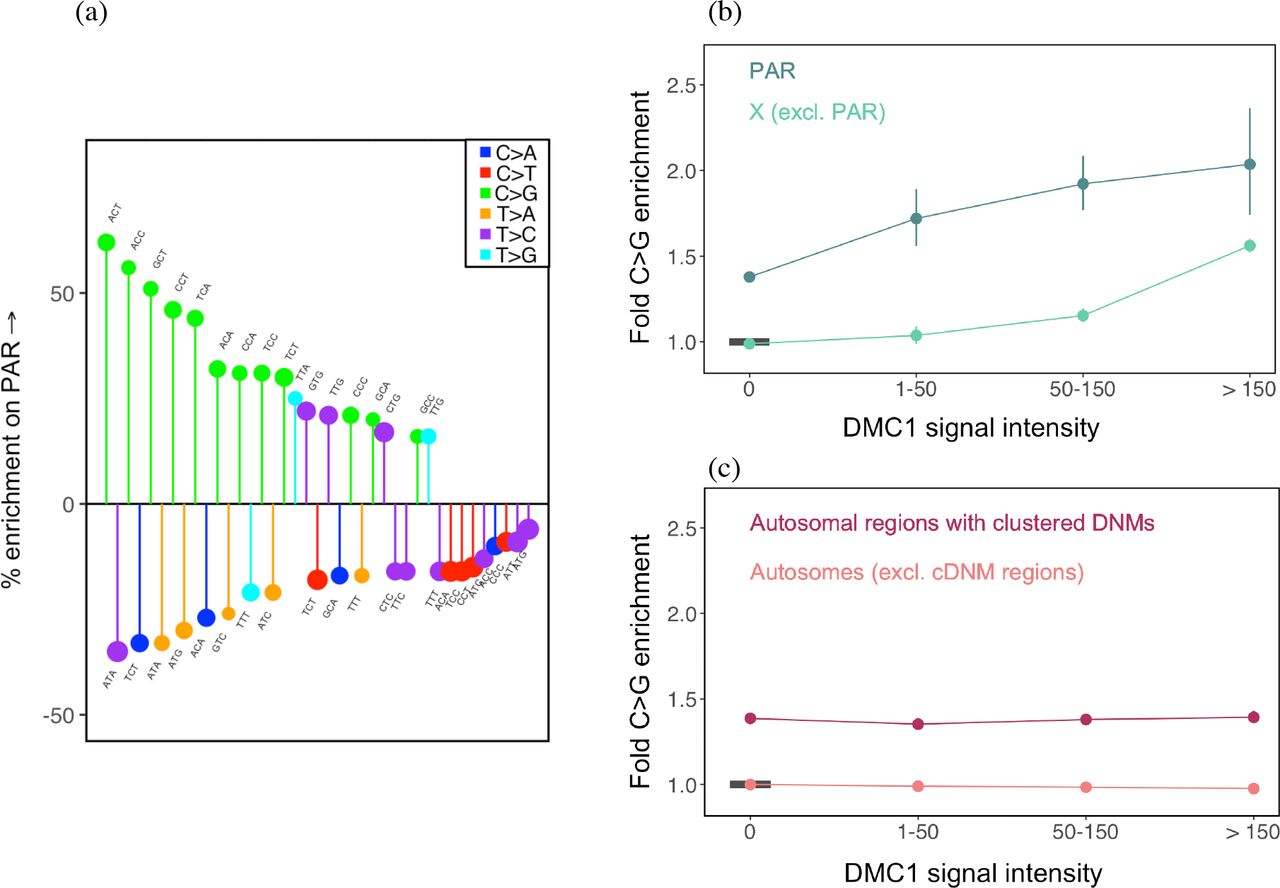
To explore whether these meiotic DSBs also lead to C>G mutations, we first consider the pseudoautosomal region 1 (PAR1), which experiences an obligate crossover in males, but normal levels of recombination in females (Kauppi et al. 2011; Kauppi, Jasin, and Keeney 2012; Lange et al. 2016; Hinch et al. 2014). PAR1 is a 2.6 Mb region on the X chromosome that does not undergo X-inactivation (Mangs and Morris 2007); since two copies are carried by both males and females, it is exposed to the male and female germlines to the same degree as autosomes. Comparing the mutational patterns, we find that C>G mutation types are systematically enriched on the PAR1 relative to autosomes (Fig. 5a), indicating that repair of double-strand breaks made deliberately during meiotic recombination in males is associated with C>G enrichment. Thus, meiotic recombination is mutagenic at least with regard to this one type of mutation.

Distribution of C>G mutations in genomic compartments relative to autosomes. CpG sites are excluded from these analyses. (a) The mutation spectrum on PAR1 relative to autosomes. Only significant differences are shown, ranked in order of effect size from left to right. (b) Enrichment of C>G mutations in DMC1 hotspots of varying intensity on the X chromosome, relative to autosomes outside DMC1 hotspots and excluding autosomal regions rich in clustered de novo mutations. The reference is denoted by the horizontal black bar. For each estimate, the 95% confidence interval from a binomial test is represented by the vertical bars (and is sometimes too small to be apparent). (c) Enrichment of C>G mutations in DMC1 hotspots of varying intensity on autosomes. For each estimate, the 95% confidence interval from a binomial test is represented by the vertical bars. The horizontal black bar shows the reference, namely autosomes outside DMC1 hotspots and excluding regions rich in clustered de novo mutations (identified by Jonsson et al., 2017).
To further characterize the source of the C>G enrichment associated with meiotic recombination in males, we use DMC1 ChIP-Seq data from human spermatocytes (Pratto et al. 2014). The DMC1 signal reflects intermediates in the homologous recombination pathway; high levels of DMC1-binding can reflect either an increased frequency of double strand breaks (hotspots of greater intensity) or a greater duration of intermediates, i.e., a longer time to repair (Kauppi et al. 2011; Pratto et al. 2014). Using these data, we find that there is clear C>G enrichment not only in PAR1, but also in hotspots on the X chromosome outside PAR1; moreover, the enrichment increases with the strength of the DMC1 signal (Fig. 5b, Supplementary Fig. 13a). In contrast to the X, we do not find an appreciable enrichment of C>G mutations in hotspots of similar average intensities on autosomes once we exclude the regions of clustered de novo C>G mutations reported by Jónsson et al., 2017 (Fig. 5c; Supplementary Fig. 13b). We note that consistent with the patterns we observe in autosomal hotspots, Pratto et al., 2014 found an enrichment of C>T and T>C types around male autosomal hotspots; they also observed C>G enrichment to a small degree but the source of these types was not discussed further by the authors, and could potentially be due to overlap with regions of clustered de novo C>G mutations (Jónsson et al., 2017).
That we observe C>G enrichment in hotspots on the X chromosome but not those on autosomes leads us to speculate that the predominant source of this C>G signature is the delay in repair of DSBs on the X chromosome relative to autosomes in male meiosis. One possibility is that the enrichment of C>G mutations stems from repair using the sister chromatid rather than the homolog. While there are mechanisms to ensure that meiotic DSBs are preferentially repaired using the homolog, this “homolog enforcement” is thought to be lifted late in meiosis (Lao and Hunter 2010; Lu and Yu 2015); DSBs still not repaired by this stage may be repaired using the sister chromatid (Li et al. 2018). In this scenario, in males, the late repair of meiotic DSBs using the sister chromatid on the X chromosome leads to an enrichment of C>G mutations specifically in recombination hotspots on the X chromosome (and not in autosomal hotspots). In females, since the two X chromosomes recombine like autosomes, meiotic DSBs are not expected to lead to excess C>G mutations on the X chromosome or autosomes. While we do not have a DMC1 map in females to test this directly, the lack of a C>G signal in female recombination hotspots identified in pedigree studies (Supplementary Fig. 13c) suggests that this conjecture holds, at least on autosomes. The source of the C>G signature noted by Jónsson et al. in autosomes could also be late repair using the sister chromatid instead of the homolog; indeed, if these areas reflect damage, as the authors surmise, they may only be repaired after homolog enforcement is lifted later in meiosis. In summary, we hypothesize that C>G mutations in female gametes arise due to some property of inter-sister repair of spontaneous DSBs. In male gametes, C>G mutations could arise from inter-sister repair of both meiotic and spontaneous damage-induced DSBs, but the effect of meiotic DSBs is limited to the X chromosome.
Because they arise from subtly different manifestations of the same biochemical processes in males and female germlines, C>G mutations exemplify true sex differences in mutation. In that sense, components of the recombination machinery that are involved in late repair of double-strand breaks are sex-specific modifiers of mutation.
Sex-specific life history traits play a role in the evolution of the mutation spectrum
The case of DSB-repair in germ cells is illustrative of a biochemical process of mutagenesis that differs between males and females. Even if there is no such clear-cut difference, however, much of the biochemical machinery that influences mutation must in theory have subtle sex-specific effects, simply because sex-specific life history traits modulate exposure to biochemical influences differently in males and females. This heterogeneous set of variants associated with sex-specific life history traits, and with biochemical processes in the germline that differ in the two sexes directly or through their interactions with sex-specific life history traits, shapes the mutation spectrum in each generation: changes in the allele frequency of these variants over time should then change the mutation spectrum.
For example, the sex-specific biochemical process of DSB-repair that leads to C>G mutations in the germline clearly interacts with life history traits: the proportion of C>G mutations transmitted in a single generation increases with the age of the mother (Jónsson et al. 2017). It follows that a sustained increase in average maternal age at reproduction could lead to an overall increase in the proportion of C>G mutations relative to other mutation types, thus altering the mutation spectrum. Consistent with an increase in maternal age in recent human evolution, C>G mutations are relatively enriched in rare variants relative to more common variants (Gao, Moorjani et al., unpublished).
Changing allele frequencies of sex-specific modifiers could alter not only the overall mutation spectrum, but also the mutation spectrum on the X chromosome relative to autosomes. As demonstrated for mechanisms of DSB-repair in the germline, some processes that alter the mutation spectrum at the level of the germline also have an auxiliary effect on the mutation spectrum of the X chromosome. Consistent with this notion, we observe a few mutation types for which the level of enrichment on the X chromosome relative to autosomes differs between rare and common variants (Fig. 6; where rare variants are defined as those that occur 1-5 times, and common variants as those that are at a frequency of 1% or greater in the sample). As one example, C>G mutations in a CCC context are significantly enriched on the X chromosome in rare variants to a greater degree than in more common variants, a pattern that could reflect an increase in the proportion of meiotic DSBs in males, or perhaps an effect of increasing maternal age at reproduction towards the present. We note that interpreting X-Autosome differences over time is complicated by changing effective population sizes of the X chromosome and autosomes (Amster and Sella 2017) and by the effects of GC-biased gene conversion on some older variants. Nevertheless, a role for evolving life history traits and other sex-specific modifiers in the evolution of the mutation spectrum is to be expected.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The enrichment of mutation types on the X chromosome, relative to autosomes, for rare variants (allele counts 1-5) versus common variants (variants at frequency 1% or greater) in the sample. Diamonds represent mutation types that differ by 5% in their mean fold enrichment between the two comparisons. This analysis excludes CpG sites and the pseudoautosomal region. The black diagonal represents the x=y line. For each estimate, the 95% confidence interval from a binomial test for Xenrichment among common and rare variants, respectively, is represented by the grey horizontal and vertical bars.
Implications
By comparing the mutation spectrum across different compartments of the genome, we identify putative signatures of sex differences in the germline and plausible biochemical sources of mutagenesis. Notably, we show that replication timing affects the mutation spectrum along the genome and find a mutagenic effect of meiotic recombination that is both sex-specific and X-specific, revealing an appreciable effect of double-strand breaks, both accidental and deliberate, on the mutation spectrum.
Interestingly, our analysis suggests that signatures of sex differences in the germline are likely abundant, but their contributions to the mutation spectrum are subtle relative to those of biochemical processes shared in the two sexes. This finding is hard to reconcile with the idea that male mutations are mostly replication-driven whereas female mutations reflect a large contribution of spontaneous damage, as then we might expect substantially different types of mutations inherited from mothers and fathers. Instead, consistent with a greater role of spontaneous damage and its repair in both male and female germlines (Gao et al. 2018), our results are most readily explained if male and female mutational mechanisms are overall highly similar, underpinned by the shared mechanisms associated with replication, transcription, methylation, and recombination, and other sources of damage. Subtle differences in the mutation spectrum between males and females could then be expected to arise due to sex-specific rates of damage and repair at different stages in germline development, modulated by sex-specific life history traits.
In this respect, we note that a number of recent studies have shown that the mutation spectrum changes slightly across populations (Harris 2015; Harris and Pritchard 2017; Mathieson and Reich 2017; Narasimhan et al. 2017). These findings have been attributed to biochemical modifiers of mutation that alter the relative rates of different mutation types by influencing the biochemical process of error/repair over time. We show that life history traits and other sex-specific modifiers could potentially result in the same kinds of changes in the mutation spectrum and the mutation rate over time. Moreover, sex-specific age of reproduction explains much of the observed mutation rate variation among ~1500 individuals at present (Jónsson et al. 2017). Variants that contribute to sex-specific life history (Perry et al. 2014; Barban et al. 2016) may therefore be a useful starting point to identify genetic sources of inter-individual variation in the mutation rate in humans.
Beyond these insights into mutagenesis, our analysis makes clear that X-autosome comparisons of mutation patterns cannot be taken as directly reflective of germline sex differences. Though historically comparisons of the sex chromosomes to autosomes have been taken to reflect only the effects of sex, mutation patterns on the X chromosome in fact reflect a convolution of X chromosome specific effects and sex. In particular, taking this point into consideration may help to explain why estimates of the male bias in mutation for CpG sites from phylogenetic studies that used X-autosome comparisons were much lower (Taylor et al. 2006) than those obtained directly from male-female differences in de novo mutation data (Kong et al. 2012; Jónsson et al. 2017).
Acknowledgements
We thank Guy Amster, Ziyue Gao, Priya Moorjani, Itsik Pe’er, Jonathan Pritchard, Guy Sella, Arbel Harpak, Felix Wu, and additional members of the Przeworski lab for helpful discussions and/or comments on a draft version of the manuscript and Priya Moorjani, Konrad Karczewski, and Kelley Harris for assistance with gnomAD and SGDP data sets. This work was supported by R01 GM122975 to M.P.
References




