

Abstract
Microtubules are fundamental elements of neuronal structure and function. They are dynamic structures formed from protofilament chains of α- and β-tubulin heterodimers. Acetylation of the lysine 40 (K40) residue of α-tubulin protects microtubules from mechanical stresses by imparting structural elasticity. The enzyme responsible for this acetylation event is MEC-17/αTAT1. However, despite its functional importance, the consequences of MEC-17/αTAT1 misregulation on neuronal structure and function are incompletely defined. Using overexpression and loss of function approaches, we have analysed the effects of MEC-17 misregulation on the development and maintenance of synaptic branches in the mechanosensory neurons of Caenorhabditis elegans. We find that synaptic branch extension is delayed, and that synaptogenesis is defective in these animals. Strikingly, by adulthood the synaptic branches specifically and spontaneously degenerate. This phenotype is dependent on the acetyltransferase domain on MEC-17, revealing that correct levels of K40 acetylation are essential for the maintenance of neuronal structure. Finally, we investigate the genetic pathways in which mec-17 functions, uncovering novel interactions with dual leucine-zipper kinase dlk-1 and the focal adhesion gene zyx-1/Zyxin. These interactions link MEC-17 together with factors involved in neuronal and actin remodelling to protect synaptic branches. Together, our results reveal that appropriate levels of α-tubulin K40 acetylation by MEC-17 are crucial for the development and maintenance of neuronal architecture.
Introduction
Maintenance of neuronal structure over the lifetime of an animal is essential for a fully functional and adaptable nervous system. The highly polarized nature of neurons, with axons typically extending magnitudes larger than the size of the cell body, makes them particularly susceptible to mechanical stress associated with normal animal movement. The ability of neurons to resist these stressors is mediated by the cytoskeleton [1]. Major structural and functional components of a neuron’s cytoskeleton are formed by microtubules. These highly conserved cylindrical structures are assembled from chains of α- and β-tubulin heterodimers termed protofilaments [2]. Microtubules are dynamic structures, cycling through phases of polymerisation and depolymerisation [3, 4]. They are essential for neuronal development, structure, and function, providing the platform for intracellular transport and organelle positioning, as well as the scaffolds for signalling molecules [3].
Previous research has provided strong links between microtubule dynamics and maintenance of neuronal structure [5]. In particular, the C. elegans posterior lateral microtubule (PLM) neurons have provided a productive model for defining the cellular mechanisms required for maintaining neuronal branches and synapses. These neurons extend a long axon anteriorly to the mid-body region from which a single branch elongates ventrally to synapse with other neurons in the ventral nerve cord [6]. Chen et al. demonstrated the importance of MEC-12/α-tubulin and MEC-7/β-tubulin for the maintenance of the synaptic branches in these neurons, with loss-of-function alleles inducing post-developmental branch loss [7]. These defects were dependent on the microtubule-associated RhoGEF, RHGF-1, activating the dual leucine-zipper kinase DLK-1 (a mitogen-activated protein kinase (MAP) triple kinase activated by microtubule disruptions) [7, 8]. DLK-1 levels are controlled by the E3 ubiquitin ligase RPM-1/Phr/Pam/Highwire, [9], which is also critical for synaptogenesis in the PLM neurons [10, 11]. Moreover, the microtubule minus end binding and stabilizing protein PTRN-1/Patronin/CAMSAP3 [12] also regulates PLM synaptogenesis in a DLK-1 dependent manner [13]. Overall, these studies highlight the importance of intact and stable microtubules for both synaptogenesis and the maintenance of synaptic branches. However, the role of microtubule post-translational modifications in these processes remains unclear.
Microtubules are subjected to a wide-range of post-translational modifications that affect their organisation, dynamics, and cellular interactions [14, 15]. Unique amongst these modifications, because it occurs inside the microtubule lumen, is acetylation of α-tubulin lysine-40 (K40) [16, 17]. K40 acetylation was long considered a passive marker of stable microtubules. However, the precise functional importance of this modification was finally revealed in 2017: K40 acetylation weakens the interactions between protofilaments, increasing microtubule elasticity and providing resistance against mechanical stress [18, 19]. Due to the frequent mechanical forces applied to cells during normal animal behaviour, K40 acetylation therefore imparts an important protective role for the maintenance of microtubule (and neuronal) integrity. This is particularly true for neurons that are highly polarized cells with neurites frequently extending for magnitudes larger than the size of the soma.
The enzyme responsible for K40 acetylation is α-tubulin acetyltransferase 1 (αTAT1) [20, 21]. Loss of the orthologous protein in C. elegans (MEC-17) disrupts microtubule organization, reducing protofilament numbers and affecting the number and length of microtubules [22, 23]. K40 acetylation has been linked with a number of cellular functions, including cell migration and adhesion [24–26], microtubule-based trafficking [27–29], autophagy [30], modulation of kinase signalling [31], as well as neuronal development and function [21, 23, 32–35]. Intriguingly, αTAT1 also has enzyme-independent functions, which include its interaction with microtubules, and in the elongation and maintenance of neuronal structure [23, 36, 37].
Our previous research demonstrated that MEC-17 functions independently from α-tubulin acetylation as a protective factor in the C. elegans mechanosensory neurons [36]. Absence of MEC-17 caused spontaneous, adult-onset axonal degeneration due to severe disruptions in neuronal microtubule structure [22, 23, 36]. To determine if the overexpression of MEC-17 could be used to protect against other genetic or environmental neurodegenerative insults, we generated C. elegans strains that overexpress MEC-17 specifically in the mechanosensory neurons. Surprisingly, far from being a protective factor, we show that overexpression of MEC-17 induces specific loss of the synaptic branches of the PLM neurons. Although these branches are formed largely normally in these animals, we provide evidence that synaptogenesis is also defective. Moreover, we find that disruption of the MEC-17 paralogue gene (ATAT-2) also induces specific loss of the PLM synaptic branches. Finally, we demonstrate that MEC-17 functions together with the dual leucine-zipper kinase DLK-1 and focal adhesion protein ZYX-1/Zyxin to preserve these synapses. Overall our data establish that the correct regulation of α-tubulin K40 acetylation is essential for synaptic development and maintenance of neuronal structure.
Results
MEC-17 and ATAT-2 function to maintain synaptic branches
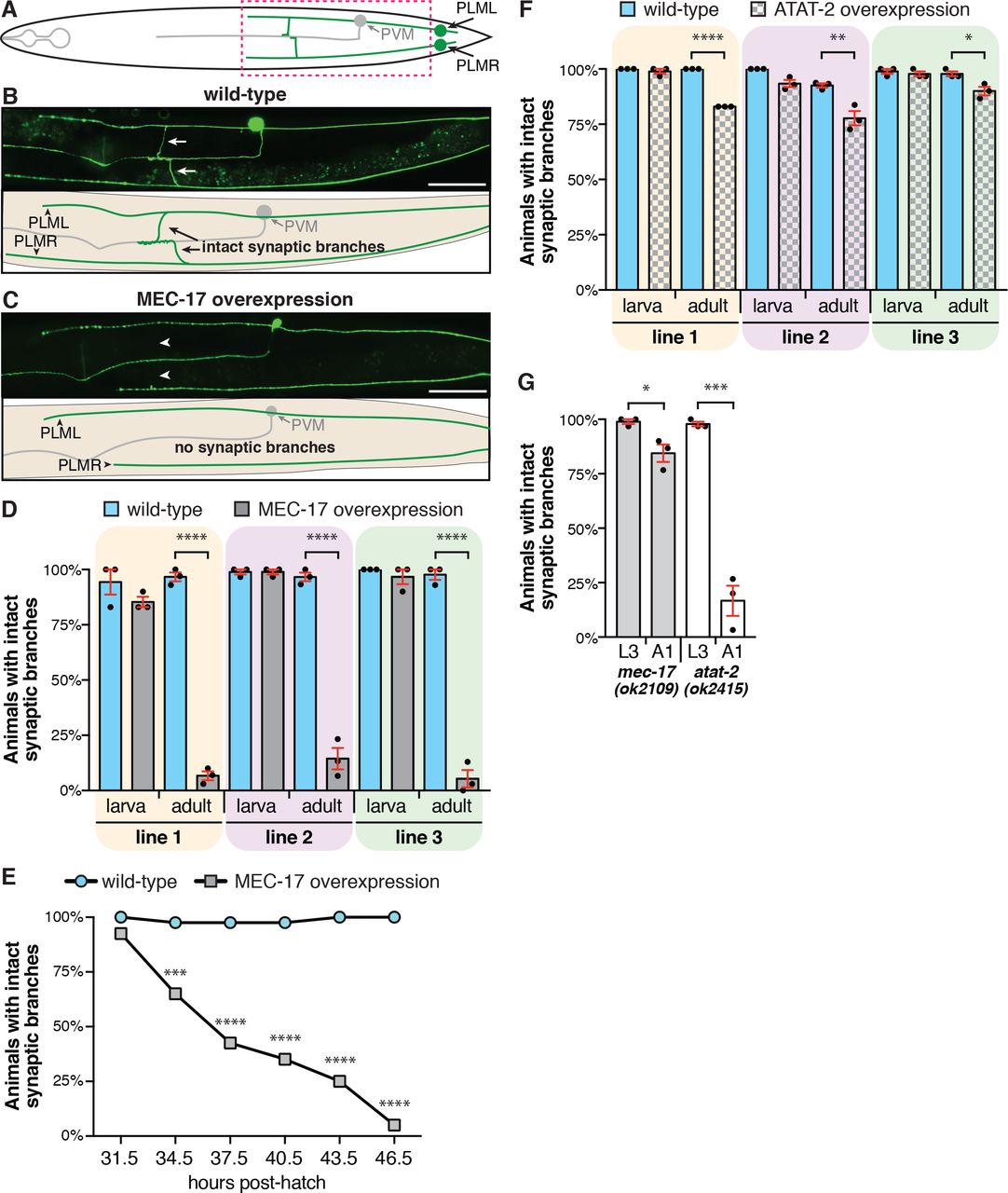
Our previous study revealed that MEC-17 protects the six mechanosensory neurons of C. elegans from spontaneous axonal degeneration [36]. As such, we hypothesised that its overexpression might be beneficial in preventing axonal degeneration induced by other genetic or environmental insults. In order to test this, we generated transgenic strains with MEC-17 overexpressed specifically in the mechanosensory neurons (Pmec-4::mec-17), the only cells in which it is normally expressed. To our surprise and in opposition to our hypothesis, the overexpression of MEC-17 caused the specific loss of the PLM synaptic branches while leaving the main axon shafts intact (Fig. 1A-C). During normal development in wild-type C. elegans, these synaptic branches extend during the first larval stage (L1) and are preserved across the animals’ lifespan [11]. Analysis of larval stage (larval stage 3, L3) animals overexpressing MEC-17 revealed that the synaptic branches were present, however, by early adulthood (one day old adults, A1) the vast majority of synaptic branches in these animals were lost (Fig. 1D and S2A). We consistently observed thinning of the PLM branches in animals with MEC-17 overexpression, as well as remnants of the branch emanating from the main axon shaft, the pre-synaptic side or both (Fig. S1). These data suggest that the branch is physically breaking in these animals, rather than undergoing a retraction process.

(A) Schematic representation of a ventral view of the PLM (green) and PVM neurons (gray). Dashed box highlights the regions imaged in panel B and C. (B) Image and schematic showing the intact PLM synaptic branches of a wild-type animal carrying the zdIs5(Pmec-4::GFP) transgene. Arrows point to the synaptic branches of PLML and PLMR. (C) Image and schematic of an animal with overexpression of MEC-17 in which the synaptic branches of both PLML and PLMR are disrupted. Arrowheads point the expected position of the lost branches. Scale bars represent 50 μm. (D) Quantification of the number of animals with an intact synaptic branch at L3 (larva) or A1 (adult) stages. Three independent transgenic strains are shown (gray bars) compared to their non-transgenic siblings (wild-type, blue bars). Bars show mean ± SE; symbols show the mean of three-independent experiments, each with n ≥ 30 (total n ≥ 90). (E) Quantification of the number of animals with an intact synaptic branch across the L4 stage of development in animals carrying the same transgene as line 3 in panel D (cjnEx036(Pmec-4::mec-17); gray squares) compared to their non-transgenic siblings (wild-type, blue circles). Different groups of animals were analysed for each time-point; n ≥ 36 for each time-point. (F) Quantification of the number of animals with an intact synaptic branch at L3 (larva) or A1 (adult) stages. Three independent transgenic strains with overexpression of ATAT-2 (gray bars) or their non-transgenic siblings (wild-type, blue bars) are shown. Bars show mean ± SE; symbols show the mean of three-independent experiments, each with n ≥ 28 (total n ≥ 89). (G) Quantification of the number of animals with intact synaptic branches in mec-17(ok2109) and atat-2(ok2415) mutant background at both L3 and A1 stages. Bars show mean ± SE; symbols show the mean of three-independent experiments, each with n ≥ 29 (total n ≥ 90). P values * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001 from one-way ANOVA with Tukey’s post-hoc tests (panels D, F), Fisher’s exact test (panel E) or unpaired t-tests (panel G).
To temporally define branch loss, we analysed animals at three-hour time points across the final larval stage (L4) of development, which extends from 31.5-33.5 to 44-46 hours post-hatch [38]. At the earliest time point (31.5 h), MEC-17 overexpression caused no detectable defect (Fig. 1E). At each subsequent time point, we observed a progressive loss of the synaptic branches, with less than 50% of animals presenting intact branches at 37.5 h and only 5% at the final 46.5 h time point. This final level of defect is consistent with that observed in adults (5%, Fig. 1D, line 3), demonstrating that the overexpression of MEC-17 leads to the destruction of the PLM synaptic branches specifically during the L4 stage of development.
MEC-17 functions redundantly with the ATAT-2 protein in the acetylation of α-tubulin [20, 21]. To determine if correct regulation of ATAT-2 is also important for maintenance of the PLM synaptic branches, we generated strains with overexpression of ATAT-2 specifically in these neurons (Pmec-4::atat-2). Similar to the phenotypes observed with MEC-17 overexpression, we observed no defect at L3 stage, but a significant proportion of animals with overexpression of ATAT-2 displayed disrupted synaptic branches in adulthood (Fig. 1F and S2B). However, whereas MEC-17 overexpression led to synaptic defects in more that 85% of animals (Fig. 1D), ATAT-2 overexpression had a more modest effect, with a 10-22% decrease observed across three independent transgenic lines (Fig. 1F).
Interestingly, loss of function mutations in either mec-17 or atat-2 phenocopied the overexpression studies, with the PLM synaptic branch lost in a significant proportion of adult animals (Fig. 1G and S2C, D). Though in contrast to the overexpression studies, loss of atat-2 caused the stronger phenotype, with only 17% of adults presenting intact synaptic branches, compared to 84% of adults lacking mec-17. This difference in penetrance may be driven by the stronger defect in mechanosensation in mec-17 animals [21, 23], which results in reduced locomotion and as a result, their neurons might encounter less movement-induced mechanical stress.
Misregulation of MEC-17 disrupts synaptic branch development
We next investigated whether MEC-17 also functions during development of the PLM synaptic branches. Whilst the main axon shaft of PLM develops during embryogenesis [39], the synaptic branch extends during the first larval period (L1) [11]. To quantify any temporal changes in branch development, we analysed animals at 4, 6 and 8 hours post-hatching for both the presence of a branch and for a complete branch (one extending into the ventral nerve cord). Compared to their non-transgenic siblings, animals with overexpression of MEC-17 displayed a significant delay in the development of the PLM branch (Fig. 2A, compare bars with yellow shading to those with green shading). Deletion of mec-17 caused a similar, but more significant delay in branch formation (Fig. 2A, compare bars with gray shading to those with brown shading). This defect was most pronounced at the 8-hour time point, at which 97% of wild-type animals had developed a branch (with 80% complete), whereas only 58% of mec-17(ok2109) animals had a branch (and only 38% were complete).

(A) Quantification of the number of animals displaying a branch from the main PLM axon shaft (present) and those with a branch extending into the ventral nerve cord (complete). Both PLML and PLMR analysed at 4, 6 and 8 h post-hatch; n ≥ 30. P values * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001 from Chi-square tests; asterisks above the bars compare ‘present’ branches; asterisks within the bars compare ‘complete’ branches. (B) Quantification of the PLM branch position 8 h post-hatch. Position calculated relative to the PLM soma and axon terminus. Circles represent individual axons analysed; n ≥ 38; mean ± SE shown in red; no significant differences observed from one-way ANOVA with Tukey’s post-hoc test. (C) Maximum projection confocal images of the PLM synaptic branches in a non-transgenic animal. The top panel displays the neurons labelled with tagRFP, the middle image shows the pre-synaptic sites labelled with SNB-1, the bottom image is an overlay with co-localization displayed in white, and the bottom panel is a schematic of the overlay. Arrowheads point to synaptic expansions/accumulations; scale bars represent 5 μm. (D) Representative images of an animal with MEC-17 overexpression displayed as per panel (C). This animal has two intact synaptic branches, but no SNB-1 accumulation at the synaptic sites. Arrow points to accumulation of SNB-1::GFP at the branch point. (E) Quantification of the size of the SNB-1 pre-synaptic area in non-transgenic versus transgenic animals with MEC-17 overexpression. (F) Relative fluorescence levels of SNB-1::GFP in non-transgenic versus transgenic animals with MEC-17 overexpression. For panels E and F, Circles represent individual axons analysed; n ≥ 20; mean ± SE shown in red. P values * < 0.05, ** < 0.01 from unpaired t-tests.
The PLM synaptic branches are formed in a stereotypical position along the main axon shaft. This is controlled by Wnt-Frizzled and Planar Cell Polarity signalling pathways that function to spatially restrict the focal accumulation of filamentous (F-) actin [40]. To examine whether MEC-17 is important for developmental patterning of the branch, we calculated the relative position of the branch to the cell body and axon terminus. As shown in Figure 2B, the branch consistently extended from the main axon shaft at around two-thirds of its length, and this was unchanged by either the overexpression of MEC-17 or its loss-of-function.
To more closely analyse the synapses in animals overexpressing MEC-17, we visualized the accumulation of the RAB-3 and SNB-1 proteins in the PLM neurons. The synaptic vesicle-associated small guanosine triphosphatase RAB-3 encodes the orthologue of the RAS GTPase rab3 [41], while SNB-1 encodes synaptobrevin, a synaptic vesicle protein required for synaptic transmission [42] that is orthologous to the vesicle-associated membrane proteins (VAMP1 and VAMP2) in humans. Both RAB-3 and SNB-1 strongly localize to presynaptic termini in discrete punctate patterns for each PLM neuron [36, 41, 43]. Confocal microscopy analysis of L3 stage animals expressing tagged versions of SNB-1 and RAB-3 revealed that the overexpression of MEC-17 significantly affected accumulation of these molecules at pre-synaptic sites (Fig. 2C, D, and S3). SNB-1::GFP accumulated in 100% (20/20) of synaptic sites in non-transgenic animals, but in only 65% (11/17) of synapses in animals overexpressing MEC-17 (P = 0.00282 from unpaired t-test). Similarly, the majority of non-transgenic animals displayed the expected accumulation of mCherry::RAB-3 at pre-synaptic sites (95%, 18/19), but the same was true for less than half of animals with MEC-17 overexpression (47%, 7/15, P = 0.000954 from unpaired t-test). Notably and as can be seen in Figure 2D (yellow arrows), we frequently observed SNB-1::GFP accumulating at the branch point in animals with MEC-17 overexpression (39%, 7/18). This was never observed in the non-transgenic controls (0/20). This may suggest the presence of a defect in axonal transport that specifically affects trafficking along the synaptic branch. In addition, the relative area of the pre-synaptic accumulation in PLM was also significantly reduced (Fig. 2E), as was the relative intensity of the SNB-1::GFP marker (Fig. 2F), suggesting that the synaptic connections of these neurons are abnormally formed when MEC-17 is overexpressed.
These results demonstrate that correct regulation of mec-17 is important for temporal control of synaptic branch development, as well as for the correct trafficking of pre-synaptic components. Thus, a precise balance of acetylated versus non-acetylated α-tubulin appears to be important for these processes. In contrast, MEC-17 does not appear to participate in the developmental patterning of the PLM synaptic branch, suggesting that it does not impact on the localized stabilization of F-actin. Overall, these data establish an important role for MEC-17 in the normal development of the PLM synapses.
MEC-17 functions through the acetylation of α-tubulin to maintain synaptic branches
MEC-17 possesses both acetylation-dependent and -independent functions. To determine how MEC-17 functions to maintain the PLM synaptic branches, we overexpressed a mutated version of the protein (D144N) lacking enzymatic activity [21, 23, 36]. Across five independent transgenic lines, this mutant version failed to phenocopy the overexpression of wild-type MEC-17, with the vast majority of animals maintaining intact synaptic branches into adulthood (Fig. 3A and S2E). Thus, we demonstrate a novel role for the enzymatic function of MEC-17 in maintaining the PLM synaptic branch.

(A) Quantification of the number of animals with an intact synaptic branch at A1 stage. Five independent transgenic strains are shown (gray striped bars) compared to their non-transgenic siblings (wild-type, blue bars). Bars show mean ± SE; symbols show the mean of three-independent experiments, each with n ≥ 29 (total n ≥ 89). (B) The proportion of animals displaying intact synaptic branches in mec-17(e1607) mutants containing no rescue transgene (black bars) compared to transgenic strains expressing wild-type MEC-12 (magenta bars), MEC-12(K40Q) (chequered magenta bars), or MEC-12(K40R) (striped magenta bars). For each independent experiment n ≥ 27 (total n ≥ 87). (C) Quantification of animals with intact synaptic branches in mec-17(ok2109) and mec-12(e1607) single and double mutant animals. For each independent experiment, n ≥ 29 (total n ≥ 89). (D, E) The percentage of control (non-transgenic) and MEC-17 overexpressing animals (transgenic) with intact synaptic branches when treated with 1% DMSO (white bars) or increasing concentration of colchicine (D) or paclitaxel (E). For three independent experiment n ≥ 27 (total n ≥ 88). P values ** < 0.01, *** < 0.001, **** < 0.0001 from one-way ANOVA with Tukey’s post-hoc test.
To further analyse the role of α-tubulin acetylation, we studied animals carrying different versions of MEC-12, the sole C. elegans α-tubulin protein that contains a K40 acetylation site. Specifically, we compared the proportion of intact PLM synaptic branches in animals carrying a missense mutation in the GTP domain of MEC-12 [mec-12(e1607)] with those carrying a single-copy insertion of wild-type mec-12, an acetylation mimic [mec-12(K40Q)], or a version that cannot be acetylated [mec-12(K40R)] [20]. Consistent with the findings of Chen et al. [7], mutation of mec-12 caused a severe synaptic branch defect, with only 6% animals maintaining both their synaptic branches into adulthood (Fig. 3B and S2F). This defect could be partially rescued with expression of either wild-type MEC-12 or with the K40R version (Fig. 3B and S2F). Expression of MEC-12(K40Q), which mimics acetylation at every possible K40 site, failed to provide any rescue and slightly worsened the defect compared to the mec-12(e1607) animals alone (Fig. 3B and S2F). These data provide further support for a crucial role of α-tubulin K40 acetylation in the maintenance of the PLM synaptic branches.
We next investigated the interaction between mec-12 and mec-17 in the maintenance of the PLM synaptic branches. Surprisingly, animals carrying mutations in both mec-12 and mec-17 partially suppressed the mec-12 single mutant phenotype (Fig. 3C and S2G). This suggests that the mec-12 mutant phenotype is dependent on mec-17. Together, our analyses demonstrate that MEC-17 functions through its acetyltransferase domain to preserve PLM synaptic branches. Curiously, and similarly to what has been proposed for the DLK-1 MAP protein [7], our genetic studies with mec-12 and mec-17 suggest that MEC-17 may function as a microtubule damage signal that is required for the activation of neuronal remodelling pathways.
Overexpression of MEC-17 disrupts microtubule stability and structure
In order to gain insight into how the overexpression of MEC-17 impacts microtubule stability, we treated animals with microtubule stabilizing (paclitaxel) and de-stabilizing (colchicine) drugs [36, 44]. Firstly, to assess the impact of destabilization, animals were grown on low concentrations of colchicine, which promotes microtubule depolymerization by binding to free tubulin [45]. Colchicine induced a dose-dependent loss of the synaptic branches in wild-type (non-transgenic) animals (Fig. 3D and S4A). No change in the penetrance of defects was observed in MEC-17 overexpressing animals (Fig. 3D and S4A). This indicates that destabilization of the microtubule network by promoting depolymerization causes the loss of the PLM synaptic branches. Next, to evaluate the effect of microtubule hyper-stabilization, animals were grown on low concentrations of paclitaxel, which suppresses microtubule dynamics by binding to microtubule bundles [45]. Treatment with paclitaxel led to a modest, but significant rescue of the synaptic branch defect in animals with MEC-17 overexpression, while non-transgenic control animals were unaffected (Fig. 3E). Interestingly, this rescue was more pronounced when we compared the proportions of defects in these animals (S4B). The percentage of animals lacking both PLM synaptic branches was reduced in a dose-dependent manner from 75% in the controls, to 46% in those treated with the highest concentration of paclitaxel (3 μM). These data imply that the overexpression of MEC-17 leads to destabilization of the microtubule network and highlight the importance of normal microtubule dynamics for maintaining synaptic branches.
MEC-17 functions together with DLK-1 and ZYX-1 in the maintenance of synaptic branches
Previous research has established roles for the E3 ubiquitin ligase RPM-1 [10, 11], the MAPK DLK-1 [7], the microtubule minus end-binding protein PTRN-1 [13, 46], and the focal adhesion protein ZYX-1 [47] in the development and/or maintenance of the PLM synaptic branches. To analyse potential interaction between MEC-17 and these pathways, we studied animals carrying mutations in either rpm-1, dlk-1, zyx-1, or ptrn-1 together with the overexpression or mutation of MEC-17. Analysis of single mutant animals revealed that only mutation of dlk-1 and zyx-1 caused a progressive loss of the PLM synaptic branches (Fig. 4 and S5, S6). Mutations in rpm-1 caused a reduction in the proportion of animals with intact synaptic branches in larval stages that was not worsened with age (Fig. S5A and S6A), while the synaptic branches were present at wild-type levels in ptrn-1 mutants (Fig. S5C and S6C). However, the synaptic branch loss in animals with MEC-17 overexpression was exacerbated by mutations in rpm-1, dlk-1, ptrn-1 and zyx-1 (Fig. S5 and S6). In each case, the proportion of animals with intact branches at L3 stage was significantly reduced in these animals. This suggests that both rpm-1 and ptrn-1 may have largely redundant roles in the maintenance of the PLM synaptic branches that can be revealed with misregulation of MEC-17. Furthermore, our results suggest that enhanced K40 acetylation compounds the deficits induced by rpm-1, dlk-1, ptrn-1 and zyx-1 mutations.

Panels provide ventral view examples of the phenotypes observed in animals carrying the zdIs5(Pmec-4::GFP) transgene and overexpression of MEC-17: (A) branch thinning; (B) remnants on the main axon shaft; (C) remnants on the pre-synaptic side; (D) remnants on both sides. White arrows point to intact branches; yellow arrows to thin branches; arrowheads to branch remnants; boxes highlight the enlarged images shown below each larger image. Scale bars represent 50 μm.

All panels display the proportions on animals with either both PLM synapses intact (gray), PLML disrupted (orange), PLMR disrupted (purple), or both disrupted (red). Data is replotted from that shown in Figures 2 and 3, with the combined mean proportions from three independent experiments shown (total n ≥ 89). (A) The three MEC-17 overexpression lines. (B) Lines with overexpression of ATAT-2. (C) Defects in the mec-17(ok2109) line. (D) The atat-2(ok2415) mutant line. (E) Five-independent MEC-17(D144N) overexpression lines. (F) Four different mec-12 lines analysed: QH4769 [mec-12(e1607)], QH4802 [mec-12(e1607) + MEC-12 rescue], QH4804 [mec-12(e1607) + MEC-12(K40Q) rescue], QH4806 [mec-12(e1607) + MEC-12(K40R) rescue]. (G) Analysis of single mec-12(e1607) and mec-17(ok2109) mutants, as well as mec-12(e1607); mec-17(ok2109) double mutants.

(A) False-coloured maximum projection confocal images of the PLM synaptic branches in a non-transgenic animal. The top panel displays the neurons labelled with GFP, the middle image shows the pre-synaptic sites labelled with RAB-3, the bottom image is an overlay with co-localization displayed in white. Arrowheads point to synaptic expansions/accumulations; scale bars represent 5 μm. (B) Images as per panel A, in a representative animal with overexpression of MEC-17. Note the absence of RAB-3 accumulation in one of the PLM synapses, which also appears dysmorphic in the top panel.

Proportions of A1-stage animals with either both PLM synapses intact (gray), PLML disrupted (orange), PLMR disrupted (purple), or both disrupted (red) for BXN232 [MEC-17 overexpression] animals treated with (A) 1% DMSO or colchicine, and (B) 1% DMSO or paclitaxel. Data is replotted from that shown in Figure 3, with the combined mean proportions from three independent experiments shown (total n ≥ 88).

(A) Quantification of the proportion of animals with intact synaptic branches in animals with MEC-17 overexpression and/or the rpm-1(ok364) or rpm-1 (ju41) mutations. Bars show mean ± SE; symbols show the mean of three-independent experiments, each with n ≥ 27 (total n ≥ 86). (B) Percentage of intact synaptic branches in animals carrying MEC-17 overexpression and/or mutations in dlk-1. For each independent experiment, n ≥ 27 (total n ≥ 87). (C) The percentage of animals with intact synaptic branches when overexpressing MEC-17 and/or carrying the ptrn-1(tm5597) mutation. For each independent experiment, n ≥ 30 (total n ≥ 90). (D) Analysis of synaptic branch defects in animals carrying MEC-17 overexpression and/or mutations in zyx-1. For each independent experiment, n ≥ 30 (total n ≥ 90). P values * < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001 from one-way ANOVA with Tukey’s post-hoc test.

(A) Quantification of animals with intact synaptic branches in mec-17(ok2109) and rpm-1(ok364) single and double mutants. Bars show mean ± SE; symbols show the mean of three-independent experiments, each with n ≥ 29 (total n ≥ 88). (B) Analysis mec-17(ok2109) and dlk-1(km12) single and double mutant animals. For each independent experiment, n ≥ 28 (total n ≥ 87). (C) The percentage of animals with intact synaptic branches in mec-17(ok2109) and ptrn-1(tm5597) single and double mutant animals. For each independent experiment, n ≥ 28 (total n ≥ 88). (D) Analysis mec-17(ok2109) and zyx-1(gk190) single and double mutant animals. For each independent experiment, n ≥ 29 (total n ≥ 90). P values ns > 0.05, * < 0.05, ** < 0.01, **** < 0.0001 from one-way ANOVA with Tukey’s post-hoc test.
To determine if mec-17 functions in the same genetic pathway as rpm-1, dlk-1, ptrn-1, and/or zyx-1 we quantified synaptic branch defects in single and double mutant animals. Intriguingly, loss of mec-17 in rpm-1 mutant animals rescued the synaptic branch defects observed at L3 stage, but the double mutants were not significantly different from the single rpm-1 mutants in adulthood (Fig. 4A and S6A). This may indicate that different pathways are required during development and adulthood to maintain the PLM synaptic branches. At the L3 stage, it appears that rpm-1 is dependent on mec-17. This is supported by the large increase in defects observed with the overexpression of MEC-17 in rpm-1 mutant animals at L3 stage (Fig. S5A and S6A). This is also consistent with the findings of Borgen et al., who demonstrated that loss of mec-17 can suppress PLM axon termination defects observed in rpm-1 mutants [48]. In adulthood however, our data suggest that these two genes function in the same pathway to maintain the PLM synaptic branches.
Loss of dlk-1 could not suppress the defects observed in mec-17 mutant or overexpressing animals (Fig. 4B, S5B and S6B). Previously reported synaptic branch defects in mec-12, mec-7, rpm-1 and ptrn-1 have demonstrated that these defects are dependent upon DLK-1 signalling [7, 10, 13]. DLK-1 is also required for axonal overextension phenotypes associated with the loss of mec-17 function [13, 23]. In contrast, the penetrance of synaptic branch loss in dlk-1; mec-17 double mutant animals was not significantly different from the single mec-17 mutant animals (Fig. 4B and S6B). This indicates that these two genes function in the same genetic pathway to maintain the synaptic branches.
As shown in Figure 4C and S6C, mutation of ptrn-1 could significantly suppress the synaptic branch defect observed in mec-17 mutant animals. PTRN-1 has been shown to inhibit microtubule minus-end disassembly [12], while MEC-17 protects microtubules against mechanical stress through K40 acetylation. Thus, it would be expected that loss of both these proteins would promote microtubule instability. It is therefore not clear why the loss of both mec-17 and ptrn-1 would reduce the penetrance of synaptic branch loss, but it may point towards the importance of interactions between microtubules and actin filaments, for which PTRN-1 has been implicated [49] (see discussion section).
The phenotype observed in animals carrying mutations in mec-17 together with one of two different zyx-1 alleles was not worse than the single zyx-1 mutants (Fig. 4D and S6D). Thus, mec-17 functions in the same genetic pathway as zyx-1 to maintain the PLM synaptic branches. Of note, the two alleles of zyx-1 analysed produced large differences in the penetrance of defects (Fig. 4D and S6D). The stronger gk190 allele of zyx-1 deletes a 777 bp region towards the 3′ end of the gene, removing the protein-binding zinc finger LIM domains and affecting all seven annotated isoforms [50]. In contrast, the weaker ok834 allele produces a 637 deletion + 7 bp insertion within an intronic region that does not affect isoforms b and d [50]. Luo et al. previously demonstrated that the LIM domains are necessary and sufficient for ZYX-1 function in synapse development [47]. Thus, our data supports these findings, and establish a genetic interaction between zyx-1 and mec-17.
Overall, our study reveals the importance of correct levels of K40 acetylation for the maintenance of neuronal architecture. Aberrant regulation of the acetyltransferase enzyme caused defects in synaptic development and led to the dramatic breakdown of synaptic branches. Our genetic analysis uncovered a novel interaction between mec-17 and zyx-1, a gene previously associated with actin remodelling under mechanical stress conditions [51–54]. This result therefore implies that interaction between microtubules and actin networks is important for the preservation of neuronal structure over an animals’ lifetime.
Discussion
Previous research has provided significant links between microtubule dynamics and synaptogenesis. For example, the formation of synaptic boutons in Drosophila melanogaster has been shown to require a transition from dynamic microtubules in growth cones to stable loop-like structures that promote synaptic formation [55–57]. Interestingly, mutation of mec-12/α-tubulin or mec-7/β-tubulin in C. elegans results in smaller presynaptic varicosities [7], suggesting that similar mechanisms may also be important in the nematode. Our data adds further support to the importance of microtubule dynamics in this process, revealing that correct levels of MEC-17 are required for temporal control of synaptic branch formation, and for the correct localization of pre-synaptic components. Moreover, the misregulation of α-tubulin acetylation levels had dramatic consequences for the maintenance of the synaptic branches. As such, our study demonstrates the importance of flexible microtubules for the formation and maintenance of neuronal architecture.
Our data suggest that the synaptic branches of the PLM neurons are particularly vulnerable to misregulation of MEC-17. Based on the position of the branches near the lateral surface of the cuticle, and the typical sinusoidal movements of C. elegans, it seems plausible that this susceptibility is a result of the mechanical stress associated with the continuous locomotory dorsal-ventral bending. Indeed, disruptions to microtubules and other components of the cytoskeleton within the C. elegans mechanosensory neurons have been reported to induce buckling of the axons and overt degeneration upon animal movement [36, 58–60]. Furthermore, the disruptions in protofilament number and MT structure caused by mec-17 loss of function [22, 23] may weaken the branch and make it more sensitive to mechanical stress. It is possible therefore, that excess acetylation may also sensitize the branch to damage by making it over-flexible such that it lacks the appropriate level of stability required for neuronal maintenance. Although the exact details about how MEC-17/αTAT1 enters the microtubule lumen remain to be resolved, it has been reported to enter through imperfections in the microtubule lattice [61–63]. Thus, MEC-17 may normally enter the microtubules within the PLM synaptic branches through mechanical stress-induced lattice holes, acetylating K40 and providing resistance against further stresses. Our findings illustrate that a correct balance of acetylated versus non-acetylated α-tubulin is required to generate an appropriate level of elasticity necessary to maintain neuronal structure in the face of mechanical stress.
Notably, we consistently observed PLML being more susceptible to damage than PLMR (compare orange and purple bars in Figures S2, S4 and S6). We are not aware of any structural differences between these neurons, and as C. elegans crawl on one of their lateral surfaces in laboratory conditions (meaning one PLM is underneath the animal adjacent the surface and the other is on top of the animal), these neurons should be exposed to the same mechanical stresses. One difference between these neurons however, is that their branches enter different sides of the ventral nerve cord, which is divided into a thick fascicle on the right and a thin fascicle on the left [64]. Whether this increased number of fibres on the right side is able to provide support to the synaptic branch of PLMR is not clear, but would offer an interesting area for future investigations.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
All panels display the proportions of animals with either both PLM synapses intact (gray), PLML disrupted (orange), PLMR disrupted (purple), or both disrupted (red). Data is replotted from that shown in Figure 4 and Supplementary Figure 5, with the combined mean proportions from three independent experiments shown (total n ≥ 86). (A) rpm-1 mutant lines: BXN249 [rpm-1(ju41); MEC-17 overexpression], BXN296 [rpm-1(ok364); MEC-17 overexpression], and single and double mutant lines between mec-17(ok2109) and rpm-1(ok364). (B) dlk-1 mutant lines: BXN287 [dlk-1(ju476); MEC-17 overexpression], BXN289 [dlk-1(km12); MEC-17 overexpression], and single and double mutant lines between mec-17(ok2109) and dlk-1(km12). (C) ptrn-1 mutant lines: BXN691 [ptrn-1(tm5597); MEC-17 overexpression], and single and double mutant lines between mec-17(ok2109) and ptrn-1(tm5597). (D) zyx-1 mutant lines: BXN450 [zyx-1(gk190); MEC-17 overexpression], BXN523 [zyx-1(ok834); MEC-17 overexpression], and single and double mutant lines between mec-17(ok2109) and either zyx-1(gk190) or zyx-1(ok834).
Interactions between microtubules and the actin network are increasingly being recognised as important facets controlling cytoskeletal dynamics. The coordinated activities of these major cytoskeletal components have been best described during neuronal development. Axon elongation from a growth cone is achieved by local depolymerization of the actin meshwork allowing polymerizing microtubules to protrude towards the leading edge and thereby extend the axon shaft [65]. Our discovery of an interaction between mec-17 and zyx-1 in the protection of the PLM synaptic branches suggests that microtubule-actin interactions are also important for the maintenance of neuronal structure. Various studies have demonstrated the importance of zyxin for actin remodelling, particularly under conditions of mechanical stress [51–54]. The function of zyx-1 in the same genetic pathway as mec-17 therefore implies that actin-microtubule interactions are essential for the preservation of synaptic branches.
Intriguingly, Ning et al. demonstrated that CAMSAP3/Patronin/PTRN-1 functions to anchor microtubule minus ends to actin filaments [49]. Given that there is also evidence of cargo switching between actin and microtubules [66], it is tempting to speculate that rescue of the mec-17 defect by mutation of ptrn-1 may point towards the need for transmission of damage signals between these networks. Thus, loss of ptrn-1 may disrupt the actin-microtubule interactions, and thereby prevent dissemination of the signal. The strong defect in animals with overexpression of mec-17, together with the significant worsening of the defects when these animals also carry mutations in mec-12, rpm-1, dlk-1, ptrn-1 or zyx-1, suggests that MEC-17 may function as a damage/remodelling signal. Further evidence for this comes from the findings that defects in mec-17 and zyx-1 [47] mutant animals are not suppressed by loss of the previously defined remodelling factor, DLK-1 [7]. Alternatively, it is possible that correct levels of MEC-17 are required to preserve the microtubule tracks necessary for retrograde signalling of DLK-1, and possibly other remodelling factors, to the soma following microtubule insult.
Our study contributes to the growing evidence linking microtubule post-translational modifications to neurodegeneration. Acetylation is linked to a number of neurodegenerative diseases, including Charcot-Marie-Tooth [67, 68], Huntington’s [27], amyotrophic lateral sclerosis, and Parkinson’s [29]. These studies have all demonstrated that disease symptoms could be suppressed with inhibitors of histone deacetylate 6 (HDAC6), an α-tubulin deacetylase [69, 70]. Our findings may encourage caution for these approaches, by demonstrating that misregulation of K40 acetylation levels can have dramatic consequences for neuronal structure.
In conclusion, this work establishes α-tubulin K40 acetylation as an essential mediator of synaptic development and maintenance.
Methods
C. elegans strains and genetics
Maintenance, crosses and other genetic manipulations were all performed via standard procedures [71]. Hermaphrodites were used for all experiments, and unless otherwise specified were grown at 20°C on nematode growth medium (NGM) plates seeded with OP50 E. coli. The atat-2(ok2415), dlk-1(ju476), dlk-1(km12), mec-12(e1607), mec-17(ok2109), ptrn-1(tm5597), rpm-1(ju41), rpm-1(ok364), zyx-1(gk190), zyx-1(ok834) mutant strains were used, together with the following transgenes: cjnEx036/cjnEx038(Pmec-4::mec-17 [10 ng/μL]; Pmyo-2::mCherry), cjnEx054/cjnEx055/ cjnEx056/cjnEx057/cjnEx066(Pmec-4::mec-17(D144N) [10 ng/μL], cjnEx068/cjnEx069/cjnEx070(Pmec-4::atat-2 [10 ng/μL]; Pmyo-2::mCherry), ekSi1(Pmec-12::mec-12::3′UTR), ekSi2(Pmec-12::mec-12[K40Q]::3′UTR), ekSi3(Pmec-12::mec-12[K40R]::3′UTR), jsIs37(Pmec-7::snb-1::GFP), uIs115(Pmec-17::tagRFP) [72], vdEx539(Pmec-4::mec-17 [10 ng/μL]; Plad-2::mCherry), zdIs5(Pmec-4::GFP). A full list of strains is provided in Supplementary Table 1.
Analysis of neuronal morphology
Animals were synchronized using a hatch-off method [44], whereby newly hatched larvae (0-30 min post-hatch) were collected from plates containing only eggs and transferred to new plates. L3-stage animals were scored 26-28 h post-hatch, while A1-stage animals were scored ~24 h after isolating them at L4 stage. For the analysis of PLM branch development in Figure 2A, animals were collected within 15 min post-hatch and grown at room temperature (~22°C).
Animals were immobilized in 0.05% tetramisole hydrochloride on 4% agar pads and imaged using a Zeiss Axio Imager M2 microscope with an Axiocam 506 mono camera and ZEN pro software. A synaptic branch was considered intact if it remained continuous from the main PLM axon to the ventral nerve cord. The synaptic branches of both PLM neurons were analysed in each animal and animals were scored as having intact synaptic branches only if both branches were intact. The proportions of animals with intact branches, disrupted PLML, disrupted PLMR, or both PLML and PLMR disrupted for each genotype analysed are shown in Supplementary Figures 2, 4 and 6. For analysis of branch development in Figure 2A, a branch was qualified as ‘present’ if it extended more than ~2 μm from the main axon shaft towards the ventral nerve cord, and as ‘complete’ if it extended to the ventral nerve cord.
The positions of the PLM synaptic branches shown in Figure 2B, were calculated using the NeuronJ plugin in ImageJ 1.52h. A line was traced from the edge of the soma to the branch point and this length was then divided by the total length of the axon (measured from the soma edge to the axon terminus).
Confocal microscopy
Animals were immobilized in 0.05% tetramisole hydrochloride on 3% agarose pads. To quantify the integrated fluorescence levels of SNB-1 at the synaptic region, jsIs37(Pmec-7::snb-1::GFP); uIs115(Pmec-17::tagRFP) worms were imaged using a Leica SP8 line scanning confocal microscope on an inverted platform equipped with Leica LasX software. All images were acquired with water immersion and a 40x/1.10 HC PL APO CS2 multi-immersive objective. The GFP and tagRFP-fluorophores were excited by a 448 nm (2% power) and 552 nm (1% power) optically pumped semi-conductor diode laser, respectively. Images were acquired sequentially in between frames with a Leica HyD hybrid detector, with gains set to 100% for 488 nm excitation and 15% for 552 nm excitation. Similarly, RAB-3 localization was imaged in vdEx262(Pmec-4::mCherry::rab-3; Punc-122::GFP); zdIs5 animals with GFP fluorescence visualized with a 488 nm laser (0.3% power; 10% gain) and mCherry excited by a 522 nm laser (1% power, 19% gain). An optimal z-step size of 0.211 nm was used for all z-stacks and a consistent pixel size of 56 nm was obtained across all images.
For the qualitative analysis of both synaptic markers, images were studied in ImageJ 1.52h. The size and the integrated fluorescence intensity levels of synapses were measured from maximal projections of the obtained z-stacks with CellProfiler-3.1.5. These measurements were performed solely with jsIs37(Pmec-7::snb-1::GFP), as this is an integrated transgene, whereas vdEx262(Pmec-4::mCherry::rab-3; Punc-122::GFP) is an extrachromosomal array, which does not provide consistent expression amongst animals. The size of the synapse was determined by firstly defining by hand the synaptic region labelled by the uIs115(Pmec-17::tagRFP) transgene and subsequently measuring the pixel count within this defined object. Integrated intensity values were determined within the object region. The relative integrated intensity levels were calculated by dividing the integrated intensity units obtained from jsIs37(Pmec-7::snb-1::GFP) by the integrated units obtained for uIs115(Pmec-17::tagRFP).
Drug treatment
Animals were grown on NGM plates containing either colchicine (Sigma-Aldrich) or paclitaxel (Sigma-Aldrich) dissolved in DMSO as previously described [36, 44]. Control animals were grown on NGM plates containing 1% DMSO, a concentration far exceeding the maximum amount used in the drug treatment plates (>0.05%). L4 stage animals were transferred to the drug plates and their F1 progeny scored at A1 stage.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7. Two-way comparisons were performed using unpaired t-tests, and ANOVA was used for comparing groups with more than two samples, followed by Tukey’s multiple comparisons post-hoc tests. Chi-square tests were used to compare the distribution of phenotypes between populations.
Author Contributions
B.N., J.S.T., W.W. and G.C. conducted experiments. B.N. and J.S.T. analysed the data. B.N. designed experiments. B.N. wrote the manuscript, with editing from all authors.
Strains used in this study.
Acknowledgements
We thank Michelle Yu-Ying Wong for intellectual input, generating strains and for comments on the manuscript; Hanadi Hoblos for assistance with generating strains; Massimo Hilliard, Martin Chalfie, Michael Nonet, and Jacek Gaertig for sharing strains; members of the Neumann and Pocock labs for valuable discussions and input. The authors acknowledge Oleks Chernyavskiy and Monash Micro Imaging, Monash University, for the provision of instrumentation, training and technical support. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by a MBio Postgraduate Discovery Scholarship to J.S.T., NHMRC Project Grant 1105374, NHMRC Senior Research Fellowship 1137645 and Victorian Endowment for Science, Knowledge and Innovation Fellowship VIF23 to R.P., and NHMRC Project Grants 1101974 and 1099690 awarded to B.N.
References




