

Abstract
We study the number Nk of (spaced) word matches between pairs of evolutionarily related DNA sequences depending on the word length or pattern weight k, respectively. We show that, under the Jukes-Cantor model, the number of substitutions per site that occurred since two sequences evolved from their last common ancestor, can be esti-mated from the slope of a certain function of Nk. Based on these considerations, we implemented a software program for alignment-free sequence comparison called Slope-SpaM. Test runs on simulated sequence data show that Slope-SpaM can estimate phylogenetic dis-tances with high accuracy for up to around 0.5 substitutions per po-sitions. The statistical stability of our results is improved if spaced words are used instead of contiguous k-mers. Unlike previous methods that are based on the number of (spaced) word matches, our approach can deal with sequences that share only local homologies.
1 Introduction
Phylogeny reconstruction is a fundamental task in computational biology [12]. Here, a basic step is to estimate pairwise evolutionary distances between protein or nucleic-acid sequences. Under the Jukes-Cantor model of evolution [20], the distance between two evolutionarily related DNA sequences can be defined as the (estimated) number of nucleotide substitutions per site that have occurred since the two sequences have evolved from their last common ancestor. Traditionally, phylogenetic distances are inferred from pairwise or multiple sequence alignments. For the huge amounts of sequence data that are now available, however, sequence alignment has become far too slow. Therefore, considerable efforts have been made in recent years, to develop fast alignment-free approaches that can estimate phylogenetic distances without the need to calculate full alignments of the input sequences, see [15, 36, 42, 3] for recent review articles. Alignment-free approaches are not only used in phylogeny reconstruction, but are also important in metage-nomics [9, 31, 25] and in medical applications, for example to identify drugresistant bacteria [4] or to classify viruses [41, 2]. In all these applications, it is crucial to rapidly estimate the degree of similarity or dissimilarity between large sets of sequence data.
Some alignment-free approaches are based on word frequencies [32, 35] or on the length of common substrings [38, 21, 18]. Other methods use variants of the D2 distance that is defined based on the number of shared k-mers [33, 39, 37, 2]; a review focusing on these methods is given in [34]. kWIP [29] is a further development of this concept that uses information-theoretical weighting. Most of these approaches calculate heuristic measures of sequence (dis-)similarity that may not be easy to interpret. At the same time, alignment-free methods have been proposed that can accurately esti-mate phylogenetic distances between sequences based on stochastic models of DNA or protein evolution, using the length of common substrings [17, 27] or so-called micro alignments [40, 16, 23, 22].
Several authors have proposed to estimate phylogenetic distances from the number of k-mer matches between two sequences. The tools Cnidaria [1] and AAF [11] use the Jaccard index between sets of k-mers from two genomes to estimate the distance between them. In Mash [31], the MinHash [5] technique is used to reduce the input sequences to small ‘sketches’ which can be used to rapidly approximate the Jaccard index. In a previous article, we proposed another way to infer evolutionary distances between DNA sequences from the number of word matches between them, and we generalized this to so-called spaced-word matches [28]. A spaced-word match is a pair of words from two sequences that are identical at certain positions, specified by a pre-defined binary pattern of match and don’t-care positions. Theoretically, this previous approach is based on a simple model of molecular evolution with-out insertions or deletions. In particular, we assumed that the compared sequences are homologous to each other over their entire length. In practice, the derived distance values are still reasonably accurate if a limited number of insertions and deletions is allowed, and phylogenetic trees could be obtained from these distance values that are similar to trees obtained with more tra-ditional approaches. Like other methods that are based on the number of common k-mers, however, our previous approach cannot produce accurate results for sequences that share only local regions of homology.
Recently, Bromberg et al. published an interesting new approach to alignment-free protein sequence comparison that they called Slope Tree [6]. They defined a distance measure using the decay of the number of k-mer matches between two sequences, as a function of k. Trees reconstructed with this distance measure were in accordance to known phylogenetic trees for various sets of prokaryotes. Slope Tree can also correct for horizontal gene transfer and can deal with composition variation and low complexity sequences. From a theoretical point-of-view, however, it is not clear if the distance measure used in Slope Tree is an accurate estimator of evolutionary distances.
In the present paper, we study the number Nk of word and spaced-word matches between two DNA sequences, where k is the word length or the number of match positions of the underlying pattern for spaced words, respectively. Inspired by Bromberg’s Slope Tree approach, we study the decay of Nk as a function of k. More precisely, we define a function F (k) that depends on Nk and that can be approximated, under a simple probabilistic model of DNA evolution, by an affine-linear function of k. The number of substitutions per site can be estimated from the slope of F.
2 The number of k-mer matches as a function of k
We are using standard notation from stringology as used, for example, in [13]. For a string or sequence S over some alphabet, A, |S| denotes the length of S, and S(i) is the i-th character of S, 1 ≤ i ≤ |S|. S[i..j] is the (contiguous) substring of S from i to j. We consider a pair of DNA sequences S1 and S2 that have evolved under the Jukes-Cantor substitution model [20] from some unknown ancestral sequence. That is, we assume that substitution rates are equal for all nucleotides and sequence positions, and that substitution events at different positions are independent of each other. For simplicity, we first assume that there are no insertions and deletions (indels). We call a pair of positions or k-mers from S1 and S2, respectively, homologous if they go back to the same position or k-mer in the ancestral sequence. In our model, we have a nucleotide match probability p for homologous positions and a background match probability q for non-homologous nucleotides; the probability of two homologous k-mers to match exactly is pk.
In our indel-free model, S1 and S2 must have the same length L = |S1| = |S2|, and positions i1 and i2 in S1 and S2, respectively, are homologous if and only if i1 = i2. Note that, under this model, a pair of k-mers from S1 and S2 is either homologous or completely non-homologous, in the sense that none of the corresponding pairs of positions is homologous, and for a pair of non-homologous k-mers from S1 and S2, the probability of an exact match is qk. Let the random variable Xk be defined as the number of k-mer matches between S1 and S2. More precisely, Xk is defined as the number of pairs (i1, i2) for which
 holds. There are (L – k + 1) possible homologous and (L – k + 1) (L – k) possible background k-mer matches, so the expected total number of k-mer matches is
holds. There are (L – k + 1) possible homologous and (L – k + 1) (L – k) possible background k-mer matches, so the expected total number of k-mer matches is

In [28], we used this expression directly to estimate the match probability p for two observed sequences using a moment-based approach, by replacing the expected number E(Xk) by the empirical number Nk of word matches or spaced-word matches, respectively. Although in equation (1), an indel-free model is assumed, we could show that this approach gives still reasonable estimates of p for sequences with insertions and deletions, as long as the sequences are globally related, i.e. as long as the insertions and deletions are small compared to the length of the sequences. It is not possible, however, to estimate p from Nk in this way in the presence of large insertions and deletions, i.e. if sequences are only locally related.
Here, we propose a different approach to estimate evolutionary distances from the number Nk of k-mer or spaced-word matches with weight k, by considering the decay of Nk if k increases. For simplicity, we first consider an indel-free model as above, and we study exact k-mer matches with k ≤ kmax for some maximal word length kmax. Also, to simplify the following equations, we first restrict ourselves to word matches starting in both sequences at positions ≤ L - kmax + 1. From equation (1), we obtain
 which is an affine-linear function of k with slope ln p. Substituting the expected value E(Xk) in the right-hand side of (2) with the corresponding empirical number Nk of k-mer matches for two observed sequences, we define
which is an affine-linear function of k with slope ln p. Substituting the expected value E(Xk) in the right-hand side of (2) with the corresponding empirical number Nk of k-mer matches for two observed sequences, we define
 We can now simply estimate p as the exponential of the slope of F.
We can now simply estimate p as the exponential of the slope of F.
The above considerations can be generalized to a model of DNA evolution with insertions and deletions. Let us consider two DNA sequences S1 and S2 of different lengths L1 and L2, respectively, that have evolved from a common ancestor under the Jukes-Cantor model, this time with insertions and deletions. Note that, unlike with the indel-free model, it is now possible that a k-mer match involves homologous as well as background nucleotide matches. Instead of deriving exact equations like (1) and (2), we therefore make some simplifications and approximations.
We can decompose S1 and S2 into indel-free pairs of ‘homologous’ sub-strings that are separated by non-homologous segments of the sequences. Let LH be the total length of the homologous substring pairs in each sequence. As above, we define the random variable Xk as the number of k-mer matches between the two sequences, and p and q are, again, the homologous and background nucleotide match probabilities. We then use
 as a rough approximation, and we obtain
as a rough approximation, and we obtain

Similar as in the indel-free case, we define
 where Nk is, again, the empirical number of k-mer matches. As above, we can estimate p as the exponential of the slope of F. Note that this estimation can still be applied if LH is small compared to L2 and L2, since LH appears only in an additive constant on the left-hand side of (4). Thus, while the absolute values F (k) do depend on the extent LH of the homology between S1 and S2, the slope of F only depends on p, q, L1 and L2, but not on LH.
where Nk is, again, the empirical number of k-mer matches. As above, we can estimate p as the exponential of the slope of F. Note that this estimation can still be applied if LH is small compared to L2 and L2, since LH appears only in an additive constant on the left-hand side of (4). Thus, while the absolute values F (k) do depend on the extent LH of the homology between S1 and S2, the slope of F only depends on p, q, L1 and L2, but not on LH.
3 The number of spaced-word matches
In many fields of biological sequence analysis, k-mers and k-mer matches have been replaced by spaced words or spaced-word matches, respectively. Let us consider a fixed word P over 0, 1 representing match positions (‘1’) and don’t-care positions (‘0’). We call such a word a pattern; the number of match positions in P is called its weight. In most applications, the first and the last symbol of a pattern P are assumed to be match positions. A spaced word with respect to P is defined as a string W over the alphabet {A, C, G, T,∗} of the same length as P with W (i) = if and only if P (i) = 0, i.e. if and only if i is a don’t-care position of P. Here, ‘*’ is interpreted as a wildcard symbol. We say that a spaced word W w.r.t P occurs in a sequence S at some position i if W (m) = S(i + m - 1) for all match positions m of P.
We say that there is a spaced-word match between sequences S1 and S2 at (i, j) if the same spaced word occurs at position i in S1 and at position j in S2, see Fig. 1 for an example. A spaced-word match can therefore be seen as a gap-free alignment of length |P| with matching characters at the match positions and possible mismatches a the don’t-care positions. Spaced-word matches or spaced seeds have been introduced in database searching as an alternative to exact k-mer matches [7, 26, 24]. The main advantage of spaced words compared to contiguous k-mers is the fact that results based on spaced words are statistically more stable than results based on k-mers [19, 10, 9, 8, 28, 30].

Spaced-word match between two DNA sequences S1 and S2 at (2,3) with respect to a pattern P = 1100101 representing match positions (‘1’) and don’t-care positions (‘0’). The same spaced word TA * * A * C occurs at position 2 in S1 and at position 3 in S2.
Quite obviously, approximations (3) and (4) remain valid if we define Xk to be the number of spaced-word matches for a given pattern P of weight k, and we can generalize the definition of F (k) accordingly: if we consider a maximum pattern weight kmax and a given set of patterns {Pk, 1 ≤ k ≤ kmax} where k is the weight of pattern Pk, then we can define Nk as the empirical number of spaced-word matches with respect to pattern Pk between two observed DNA sequences. F (k) can then be defined exactly as in (5), and we can estimate the nucleotide match probability p as the exponential of the slope of F.
4 Implementation
It is well known that the set of all k-mer matches between two sequences can be calculated efficiently by lexicographically sorting the list of all k-mers from both sequences, such that identical k-mers appear as contiguous blocks in this list. Note that, to find k-mer matches for all k ≤ kmax, it is sufficient to sort the list of kmax-mers since this sorted list also contains the sorted lists of k-mers for k < kmax.
This standard procedure can be directly generalized to spaced-word matches. In order to calculate the numbers Nk efficiently, we start by generating a suitable set of patterns {Pk, 1 ≤ k ≤ kmax}. We first specify a pattern  of weight kmax, for example by using the tool rasbhari [14]. As usual, we require the first and the last position of P to be match positions, i.e. we have P (1) = P (kmax) = 1. Let ℓk be the k-th match position in the pattern P. We then define the k-th pattern as
of weight kmax, for example by using the tool rasbhari [14]. As usual, we require the first and the last position of P to be match positions, i.e. we have P (1) = P (kmax) = 1. Let ℓk be the k-th match position in the pattern P. We then define the k-th pattern as

In other words, for k < kmax, the pattern Pk is obtained by cutting off P after the k-th match position, so each pattern Pk has a weight of k. To find all spaced-word matches with respect to all patterns Pk, 1 ≤ i ≤ kmax, it suffices to lexicographically sort the set of spaced words with respect to P in S1 and S2.
To estimate the slope of F, one has to take into account that the values F (k) are statistically stable only for a certain range of k. If k is too small, Nk and F (k) are dominated by the number of background (spaced) word matches. If k is too large, on the other hand, the number of homologous (spaced) word matches becomes small, and for Nk < L1 · L2 · qk, the value F (k) is not even defined. For two input sequences, we therefore need to identify a range k1,…, k2 in which F is approximately affine linear. To this end, we consider the differences
 and we choose a maximal range k1… k2 where the difference |δk - δk-1| is smaller than some threshold T for all k with k1 < k ≤ k2. In our implementation, we used a value of T = 0.2. We then estimate the slope of F as the average value of δk in this range, so we estimate the match probability as
and we choose a maximal range k1… k2 where the difference |δk - δk-1| is smaller than some threshold T for all k with k1 < k ≤ k2. In our implementation, we used a value of T = 0.2. We then estimate the slope of F as the average value of δk in this range, so we estimate the match probability as

Finally, we apply the usual Jukes-Cantor correction to estimate the Jukes-Cantor distance between the sequences, e.g. the number of substitutions per position that have occurred since they diverged from their last common ancestor. Examples are given in Fig. 2, 3 and 4.

F (k) plotted against k as in Fig. 2 and for simulated indel-free DNA sequences of length L = 10, 000 with a Jukes-Cantor distance of 0.5 for contiguous k-mers (top) and for spaced-word matches (bottom).

F (k) plotted against k as in Fig. 2 and 3, for simulated indel-free DNA sequences of length L = 30, 000. In a region of length LH = 10, 000, the two sequences are related to each other with a Jukes-Cantor distance of 0.2; this region contains around 20 indels at random positions. The ‘homologous’ region is flanked on both sides and in both sequences by unrelated random sequences of length 10,000 each. F (k) is plotted for contiguous k-mers (top) and for spaced-word matches (bottom).

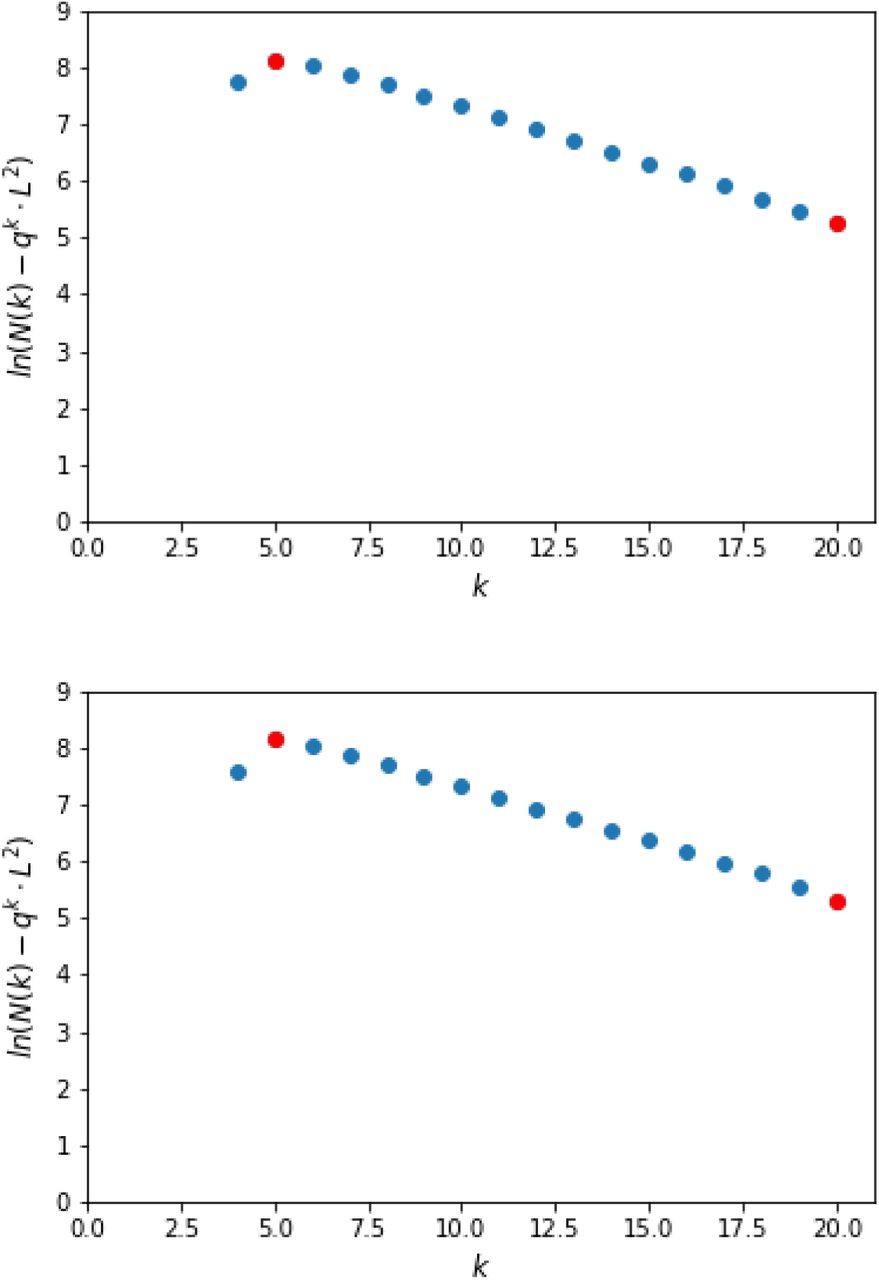
Test runs on simulated indel-free DNA sequences of length L = 10, 000, with a Jukes-Cantor distance of 0.2. Top: F (k) = ln (Nk - L2 · qk) plotted against k, where Nk is the number of k-mer matches. Bottom: for P20 = 11001110111001110111000101011011, patterns Pk were generated for k = 1,…, 19 as explained in section 4. F (k) is plotted against k, based on the number Nk of spaced-word matches with respect to Pk. In both cases, δk = F (k) -F (k - 1) is stable for k1 = 5 < k ≤ k2 = 20 (red dots).
5 Test results
We first simulated pairs of DNA sequences of length L = 10, 000 without indels, and with different phylogenetic distances. As a first example, we generated a pair of sequences with a Jukes-Cantor distance of 0.2, i.e with 0.2 substitutions per position. According to the Jukes-Cantor correction formula, this corresponds to a match probability of p = 0.82. In Fig. 2 (top), F (k) is plotted against k for these sequences based on exact k-mer matches; the difference F (k) - F (k - 1) is stable for k1 = 5 < k ≤ k2 = 20. With (6), we therefore estimate the slope of F from the numbers Nk of k-mer matches as

This way, we obtain an estimated match probability of  which is rather close to the correct value p = 0.82. A similar value was obtained when we used spaced-word matches instead of contiguous k-mer matches (Fig. 2, bottom). Results for indel-free sequences of the same length, with a Jukes-Cantor distance of 0.5 are shown in Fig. 3. Here, the slope of F was 0.449 when k-mer matches were used an 0.446 with spaced-word matches, leading to an estimated match probability of
which is rather close to the correct value p = 0.82. A similar value was obtained when we used spaced-word matches instead of contiguous k-mer matches (Fig. 2, bottom). Results for indel-free sequences of the same length, with a Jukes-Cantor distance of 0.5 are shown in Fig. 3. Here, the slope of F was 0.449 when k-mer matches were used an 0.446 with spaced-word matches, leading to an estimated match probability of  which corresponds to a Jukes-Cantor distance of 0.494, again very close to the true value.
which corresponds to a Jukes-Cantor distance of 0.494, again very close to the true value.
Fig. 4 shows the corresponding results for a pair of simulated sequences with insertions and deletions and with only local sequence homology. In a region of length LH = 10, 000, the sequences are homologous to each other, with a Jukes-Cantor distance of 0.2 substitutions per position, as in our first example (Fig. 2) – but this time, we included 20 insertions and deletions of length 50 each at random positions. In addition, the homology region was flanked on both sides and in both sequences by non-related random sequences of length 10,000 each. So both sequences had a length of around 30,000 where one third of the sequences were homologous to each other. Here, Slope-SpaM estimated a nucleotide match frequency of  corresponding to a Jukes-Cantor distance of 0.18, somewhat smaller than the true value of 0.2.
corresponding to a Jukes-Cantor distance of 0.18, somewhat smaller than the true value of 0.2.
For a more systematic evaluation, we simulated pairs of DNA sequences with Jukes-Cantor distances d between 0.05 and 1.0. For each value of d, we generated 20,000 sequence pairs of length 10,000 each and with distance d, and we estimated the distances with Slope-SpaM. In Fig. 5, the average estimated distances are plotted against the real distances. Our estimates are fairly accurate for distances up to around 0.5 substitutions per site, but become statistically less stable for larger distances.

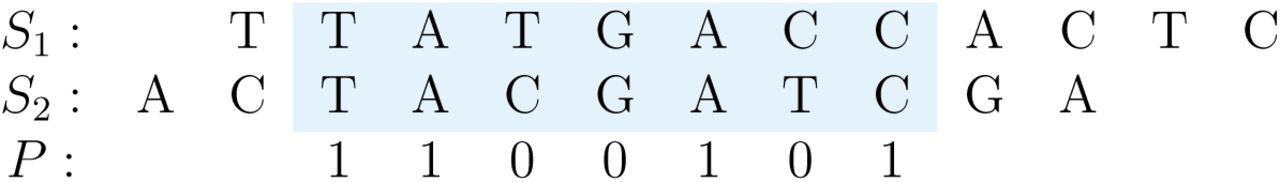{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
For Jukes-Cantor distances d between 0.05 and 1.0, we generated pairs of simulated DNA sequences of length L = 10, 000; for each value of d, 20,000 sequences pairs with a distance of d were generated. For each pair, we estimated the distance d with Slope-SpaM, using exact k-mers (top) and spaced-word matches (bottom). Average estimated distances are plotted against the real distances; standard deviations are shown as error bars.
6 Discussion
A number of alignment-free methods have been proposed in the literature to estimate the nucleotide match probability p for a pair of DNA sequences from the number Nk or D2 of k-mer matches for a fixed value of k. This can be done by setting Nk in relation to the total number of k-mers in the compared sequences [31] or to the length of the sequences [27]. A certain draw-back of these approaches is that, in order to accurately estimate the match probability p, not only Nk and the sequence lengths (or total number of k-mers, respectively), but also the extent of the homology between the compared sequences must be known.
Indeed, if two sequences share only short, local homologies with each other, an observed number Nk of k-er matches could indicate a high match probability p within the homologous regions – while the same value of Nk would correspond to a lower p if the homology would extend over the entire length of the sequences. Since it is, in general, not known which proportion of the input sequences is homologous to each other, the above methods assume, for simplicity, that the compared sequences are homologous over their entire length. Obviously, this assumption affects the accuracy of these approaches since genome sequences often share only local homologies.
In this paper, we introduced Slope-SpaM, another approach to estimate the match probability p between two DNA sequences – and thereby their Jukes-Cantor distance – from the number Nk of word matches; we generalized this approach to spaced-word matches, based on patterns with k match positions. The main difference between Slope-SpaM and previous methods is that, instead of using only one single word length or pattern weight k, our program calculates Nk, together with an expression F (k) that depends on Nk, for a whole range of values of k. The nucleotide match probability p in an alignment of the input sequences is then estimated from the slope of F. From the definition of F (k), it is easy to see that the slope of F and our estimate of p depend on the values of Nk, but not on the extent of the homology between the compared sequences.
Test results on simulated sequences show that our distance estimates are accurate for distances up to around 0.5 substitutions per sequence position. Our experimental results also confirm that Slope-SpaM still produces accurate distance estimates if sequences contain only local regions of homology. In Fig. 2 and Fig. 4, sequence pairs with a Jukes-Cantor distance of 0.2 were used. While the sequences in Fig. 2 are similar to each other over their entire length, in Fig. 4 a local region of sequence ‘homology’ is embedded in non-related random sequences twice the length of the homology region. Nevertheless, the slope of the function F is almost the same in both figure, leading to similar estimates of the match probability p. In fact, with the values shown in Fig. 4 (top), we would have obtained the same correct estimate for p that we obtained with the data from Fig. 2, if our program had chosen a minimum value k1 = 11 for the word length k, instead of k1 = 10. Alternatively, a sufficiently large maximal value k2 would have led to a more accurate estimate of p. A better way of selecting these parameters may further improve the accuracy of Slope-SpaM.
Given the good performance of our approach on simulated sequences, we expect that Slope-SpaM will also be useful to calculate phylogenetic trees for real-world sequences.
References




