

Abstract
High-throughput sequencing of fetal DNA is a promising and increasingly common method for the discovery of all (or all coding) genetic variants in the fetus, either as part of prenatal screening or diagnosis, or for genetic diagnosis of spontaneous abortions. In many cases, the fetal DNA (from chorionic villi, amniotic fluid, or abortive tissue) can be contaminated with maternal cells, resulting in the mixture of fetal and maternal DNA. This maternal cell contamination (MCC) undermines the assumption, made by traditional variant callers, that each allele in a heterozygous site is covered, on average, by 50% of the reads, and therefore can lead to erroneous genotype calls.
We present a panel of methods for reducing the genotyping error in the presence of MCC. All methods start with the output of GATK HaplotypeCaller on the sequencing data for the (contaminated) fetal sample and both of its parents, and additionally rely on information about the MCC fraction (which itself is readily estimated from the HTS data). The first of these methods uses an explicit formula based on simple probabilistic assumptions to “recalibrate” the fetal genotype calls produced by MCC-unaware HaplotypeCaller. The other two methods “learn” the recalibration model from examples. We use simulated contaminated fetal data to train and test the models. Using the test sets, we show that all three methods lead to substantially improved accuracy when compared with the original MCC-unaware HaplotypeCaller calls. We then apply the best-performing method to three chorionic villus samples from spon-taneously terminated pregnancies.
1. Introduction
High-throughput sequencing of fetal DNA is increasingly being used in academic and clinical settings. It is a powerful tool with the potential for use in prenatal diagnosis based on chorionic villus or amniotic fluid sampling [13], or in the analysis of chorionic villi or products of conception for genetic diagnosis of an unsuccessful pregnancy. In prenatal diagnosis, whole-genome or whole-exome sequencing can discover novel clinically significant variants that are not present in SNP arrays or gene panels, resulting in higher diagnostic yield [1]. Prenatal sequencing can inform prenatal and postnatal care and counseling, and may lead to prenatal therapeutic interventions [1].
In standard practice, the DNA of both parents is sequenced together with fetal DNA (”trio sequencing”) in order to establish patterns of inheritance and inform variant prioritization and interpretation. A technical difficulty that may arise in the analysis of fetal DNA is the contamination of the fetal sample with maternal cells. The prevalence of such maternal cell contamination (MCC) can be significant, depending on the experimental technique and quality of the sample; for example, one study reported 9.1% of amniotic fluid samples as having detectable MCC [12], while another found MCC fraction > 5% in as many as 26% of amniotic fluid samples under some practices [15]. High-level contamination (over 20%) was detected by one study [9] in a small, but non-zero number of samples (0.3% of cultured amniotic fluid samples and 1.3% of cultured chorionic villi samples; it must be noted that cultured amniotic fluid samples generally have less MCC than direct samples). In traditional prenatal analysis, such as that aimed at detecting chromosomal aberrations, maternal cell contamination is assayed by special tests, such as the Short Tandem Repeat analysis, and, if detected at a sufficient level, may nullify the analysis [10].
Meanwhile, standard variant calling software that is used to analyze next-generation sequencing data relies on the expectation that each allele is represented by half of the reads. MCC disrupts this assumption, leading to errors in variant calling. In this work, we propose and evaluate computational methods for reducing the MCC-caused error. All of these methods begin with variants called in the fetal specimen, the mother, and the father by a standard variant-calling pipeline and then “recalibrate” the results from the fetal specimen. The first method uses a simple probabilistic heuristic to decide on the “true” fetal genotype based on the called genotypes of the trio. The other methods eschew making assumptions about the best way to uncover the “true” fetal genotype from the maternally-contaminated observed specimen data and instead solve this problem using machine learning. We train these methods on synthetic “mother-father-fetus” trios generated from real family trios by adding specified numbers of maternal reads to the child sample. We use these synthetic trios, where the child’s genotype is known, to demonstrate that MCC correction significantly improves the accuracy of variant calling compared to contamination-naive calling, especially for higher fractions of MCC. As an intermediate technical step, we present a simple heuristic algorithm for estimating maternal cell contamination fraction in the fetal sample. We then apply the trained model to real sequencing data from miscarried fetuses and their parents with MCC > 3%, changing the fetal calls for a substantial number of SNPs.
2. Materials and Methods
In the rest of the paper, we use the term specimen to denote the obtained fetal sample which may be contaminated with maternal cells. In practice, it can be a chorionic villus or amniotic fluid sample or an abortus sample.
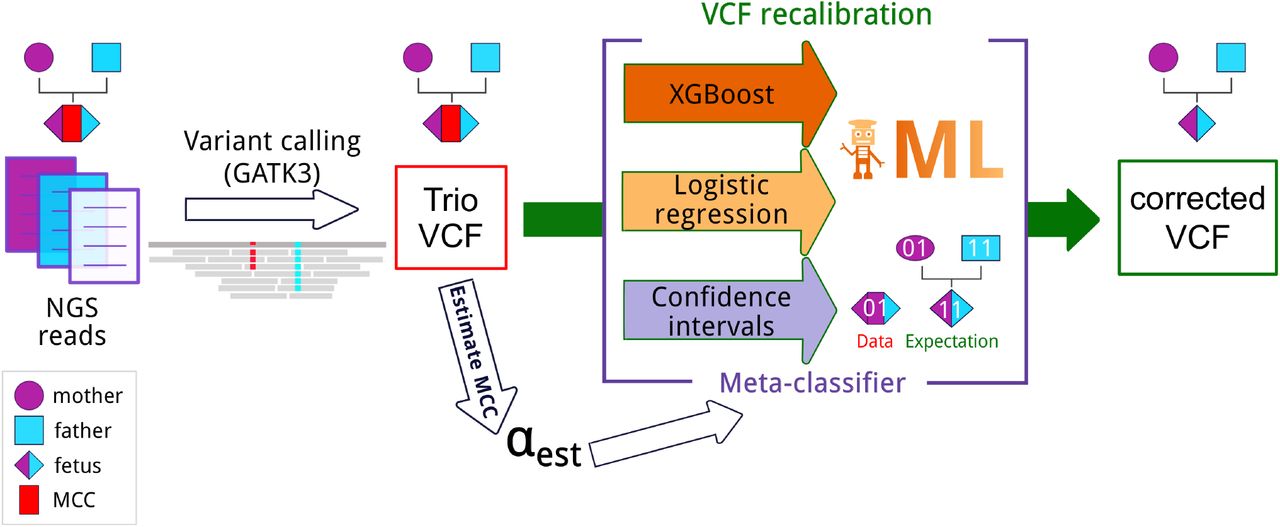
We assume that DNA samples are available for all three members of the trio. We expect the trio to be sequenced, and the reads mapped and variant-called according to a standard bioinformatics pipeline, e.g., one involving the Genome Analysis Toolkit (GATK, [5]), and that called genotypes are available along with accompanying quality information, such as allelic depths, genotype likelihoods, and so on. The VCF file produced by the pipeline is the starting point for all our analyses (see Figure 1).

Pipeline for accurate fetal variant calling in the presence of maternal cell contamination (MCC). NGS reads for each sample in the trio are mapped to the reference genome, and then variants are called with GATK v3. In the presence of MCC, the resulting VCF file contains incorrect calls for the fetus. To overcome this, we estimate MCC from this VCF and utilize the estimated value to recalibrate the VCF file and correct the calls for the fetus. Three different approaches for recalibration were utilized (see Materials and Methods section): confidence intervals and two machine learning-based approaches, namely, logistic regression and Gradient Boosted Decision Trees (XGBoost). The meta-classifier combines outputs of all previous methods. ML - machine learning.
2.1. Estimating the contamination fraction
We define the maternal cell contamination (MCC) fraction, which we denote by α ∈ [0, 1], to be the share of maternal DNA in the DNA of the fetal specimen. MCC accounts for the discrepancy between the results of variant calling on the contaminated specimen sample and the true fetal genotype. The MCC-aware variant calling and recalibration methods presented below all rely on knowing the value of α, and therefore it is vital to be able to estimate it accurately.
To estimate α in a fetal sample, we look at the results of variant calling by GATK HaplotypeCaller [5] and consider positions in the genome where the mother and the father are homozygous for different alleles (i.e., one of the parents is homozygous for the reference allele, and the other for the alternative allele); we only consider biallelic sites. The fetus then should be heterozygous at these sites, with equal read coverage for the two parental alleles.
In the presence of MCC, however, the mother’s allele coverage fraction m should be higher, namely  . Then, we can use the value of m at the position obtained from the VCF file to compute α as 2m − 1. Since the actual coverage fluctuates, we average this ratio over all relevant sites to get the MCC fraction estimate. Let mi be the fraction of maternal reads at site i, Then we compute
. Then, we can use the value of m at the position obtained from the VCF file to compute α as 2m − 1. Since the actual coverage fluctuates, we average this ratio over all relevant sites to get the MCC fraction estimate. Let mi be the fraction of maternal reads at site i, Then we compute  to be the average of mi over all sites i where the mother and the father are homozygous for different alleles, and estimate
to be the average of mi over all sites i where the mother and the father are homozygous for different alleles, and estimate  .
.
Although this procedure should be symmetric with respect to which of the parents is homozygous for the reference allele at a site, we observe that estimating α when the mother is homozygous for the reference allele (“mo00”) systematically gives larger values than when the mother is homozygous for the alternative allele (“mo11”). We attribute this phenomenon to the reference bias of the variant calling pipeline [6]. We, therefore, estimate α separately over the two alternatives (“fa00_mo11” and “fa11_mo00”) and compute the average of the two estimates as the final result. We found this aggregated estimate to be more accurate than either alternative.
Using simulated data (see Section 2.4), we found that the MCC estimation error made by this method does not exceed 2% and in the vast majority of the cases is below 1% (Figure S1).
2.2. Confidence interval-based recalibration
As our first method, we consider a hypothesis-testing approach that carries out genotype call adjustment based on an explicit mathematical model. To construct the genotype prediction for the child, we first observe that the disagreement between the true variants of the child and the called variants of the fetal specimen is in the vast majority of cases due to the mislabeling of homozygous positions in the fetus as heterozygous in the contaminated fetal specimen. The maternal genotype must, of course, be heterozygous for that to happen. Following this observation, we set the recalibrated genotype prediction of the fetus to equal that of the contaminated specimen at all genome positions except for those where the contaminated specimen is called heterozygous, and mark these latter as candidates for readjustment to a homozygous call. At each site that is a candidate for readjustment, we compare the ratios of allelic depths in the mother and the fetal specimen to determine the homozygous readjustment target (00 or 11, with 0 denoting the reference allele and 1 denoting the alternative allele; only biallelic sites are considered). To check if the readjustment is indeed needed, we use the allelic depths of the fetal specimen, AD0 and AD1, to estimate the DNA contamination ratio under the hypothesis that the child is homozygous and so the extra allele comes from the heterozygous mother; e.g.,  when the candidate readjusted genotype is 00. Then we compare this value to the value of
when the candidate readjusted genotype is 00. Then we compare this value to the value of  , which we refer to here as
, which we refer to here as  (for “global”), estimated from the entire sample as described in Section 2.1 (for the present purpose, we view this latter value as exact). Using the binomial proportion confidence interval, we expect the site-specific contamination estimate
(for “global”), estimated from the entire sample as described in Section 2.1 (for the present purpose, we view this latter value as exact). Using the binomial proportion confidence interval, we expect the site-specific contamination estimate  to satisfy
to satisfy  , where z is a parameter corresponding to the confidence level. We have chosen z = 3 by tuning z experimentally to achieve a better prediction accuracy. If the contamination estimate at a site falls within the confidence interval, we attribute the heterozygosity observed at that site in the fetal specimen to maternal contamination and “correct” the call to be the appropriate homozygous genotype.
, where z is a parameter corresponding to the confidence level. We have chosen z = 3 by tuning z experimentally to achieve a better prediction accuracy. If the contamination estimate at a site falls within the confidence interval, we attribute the heterozygosity observed at that site in the fetal specimen to maternal contamination and “correct” the call to be the appropriate homozygous genotype.
2.3. Machine learning-based genotype recalibration
In this approach, the predictive model for genotype readjustment is not based on heuristics, but is trained using a machine learning algorithm. The input to the model consists of two components. The first is the estimated fraction of maternal DNA in the fetal specimen,  . The second is the vector of features characterizing the variants called in the fetal specimen and the parents at a particular position in the genome (practically, this is the line in the VCF file for that position; it includes the genotype likelihoods, genotype qualities, read depths, etc. for all three samples). The output of the model is the adjusted fetal genotype at this position in the genome.
. The second is the vector of features characterizing the variants called in the fetal specimen and the parents at a particular position in the genome (practically, this is the line in the VCF file for that position; it includes the genotype likelihoods, genotype qualities, read depths, etc. for all three samples). The output of the model is the adjusted fetal genotype at this position in the genome.
The approach assumes that we either consistently use a particular mapping and variant calling pipeline or train predictive models using a sufficiently rich training dataset combining data obtained using several pipelines, since a single mapping and calling pipeline may have bias dependent on the specifics of the read mapper and variant caller. In practice, it is necessary that the fields describing the variant (genotype likelihood, read depth, etc.) be the same in the training VCFs and the VCF to be recalibrated; therefore, the same, or closely related variant callers should be used to produce all VCFs. The “ground truth” to which the predictive model is fit is the genotype calls on the pure fetal sample made by the same pipeline as was used on the fetal specimen sample. To train and test the model, we simulate “virtual specimens” from a number of publicly available father-mother-child trios by randomly mixing mother and child reads at various MCC fractions, as described in Section 2.4, and then calling the variants in the simulated trios. Since none of the trios on which the “virtual specimens” were based have MCC, we can use them to obtain the true child genotypes. The predictive model is then trained and tested on the data set that maps, genomic position-wise, father-mother-virtual specimen variant data to the child variant data of the corresponding father-mother-child sample. The predictive models may overfit to particular variant calling pipelines or underlying trios, and care must be taken to ensure sufficient diversity of training samples.
Two machine learning algorithms were considered: logistic regression (as implemented in scikit-learn1) and Gradient Boosted Decision Trees (XGBoost implementation2, [2]). The inputs to the models are read from a VCF file and standardized, with categorical features being one-hot encoded (split into binary columns for each unique value the feature can take). L2 regularization is applied to the logistic models, while early stopping based on performance on a validation set is used to prevent XGBoost from overfitting. The hyperparameters of the models (regularization factor for logistic regression; maximum tree depth, etc. for XGBoost) were tuned using a grid search.
Additionally, an ensemble meta-classifier combining the results of all of the above methods (confidence intervals, logistic regression, XGBoost) was constructed. An ensemble makes predictions by combining ‘votes’ from multiple classifiers. Here we use soft-voting, i.e. voting based on the predicted probabilities of each class for the different classifiers, rather than a simple class prediction. This combined prediction helps mitigate the shortcomings of each individual classifier.
2.4. Contaminated trio generation
We obtained testing and training datasets by using publicly available exome reads from father-mother-child trios (“real-world trios”) to produce “virtual specimen trios” with predetermined maternal contamination fraction α. Namely, we mixed randomly selected reads from the “real-world child” and the “real-world mother” in such a proportion that the fraction of maternal reads would be α, and thus obtained “virtual specimen” reads. We used the remaining reads from the “real-world mother” to create the “virtual specimen mother”, while the “virtual specimen father’s” reads were identical to the original “real-world father’s” reads. For each “virtual specimen” trio, we also came up with a corresponding “pure child” trio, which was identical to the “virtual specimen” trio except that no mother’s reads were added to the child’s reads. Keeping everything but the presence of maternal contamination the same between the “virtual” and the “real” trios ensures that the genotypes called with and without contamination would be compared fairly.
We performed this procedure for four “real-world” trios with publicly available exome data: the Ashkenazim trio HG002_NA24385 from the Genome in the Bottle project [16] (“AJT”), the YRI NA19240 trio from the 1000 Genomes project [3] (“YRI”), the CHD trio [7] (“CHD”), and the Corpas family daughter trio [4] (“Corpas”). Since the child in the “real-world” trios was either a living individual (AJT, YRI, Corpas) or stillborn (CHD), there was no maternal cell contamination in the “real-world” reads. If the data were available only as an alignment, we first obtained raw reads using the bam2FastQ program of the bamUtil package [8]. The CHD trio had a large number of read duplicates, which distorted the contamination fraction, so we deduplicated the aligned reads using the biobambam package3, and used the reads from the deduplicated file to generate the “contaminated” trio.
For each “real-world trio”, we repeated this procedure with α = 0.01, 0.03, 0.05, 0.1, 0.15, 0.2, 0.25, 0.3, 0.35, 0.4, 0.45, 0.5.
We aligned the “virtual specimen” and the “pure child” trio reads to the GRCh38DH reference genome following the 1000 Genomes pipeline [11]. Variants were called using Genome Analysis Toolkit 3.8 HaplotypeCaller with the option -dontUseSoftClippedBases and restricting the sequence considered to Gencode v.27 protein-coding exons with 50-nt flanks.
2.5. Miscarriage samples
Internal Review Board approval was obtained from the Ethics Committee of the Institute of Information Transmission Problems (document 11616-2116/726, 27/11/2017). Families who suffered a miscarriage and requested chromosomal microarray analysis (CMA) of the embryo were offered the choice to participate additionally in the whole-exome sequencing study. Informed consent was obtained from the parents. Only cases where no copy-number anomalies were detected by the CMA were considered for the WES study. Chorionic villi were used for the abortus DNA sample, while blood was drawn from both parents for sequencing. DNA was extracted using the Gentra Puregene kit (Qiagen). The chromosomal microarray analysis was performed on the Thermo Fisher CytoScan Optima array. Copy-number analysis was performed using the ChAS 3.3 (Applied biosystems) software and an in-house database. Parenthood and the presence of fetal DNA in the chorionic villus sample were verified using short tandem repeat (STR) analysis with the COrDIS Plus system (GORDIZ, Russian Federation).
DNA was fragmented using Covaris S220 sonicator (Covaris, USA) using settings for 150-bp insert (peak power 175W, cycles per burst 200, duty cycle 10%, time 280 seconds). Libraries were prepared using the TruSeq DNA Library Prep for Enrichment kit (Illumina, USA) without size selection (this step was replaced by purification/concentration with 1.5x volume of Ampure XP beads). Exome capture was performed using the xGen Exome Research Panel v.1.0 (IDT, USA) using manufacturer instructions with pooling of 12 libraries per reaction. Library concentration was measured on Qubit fluorometer (Thermofisher, USA) and fragment length distribution was measured on Bioanalyzer2100 (Agilent, USA). 2.5 nM dilution of the resulting post-capture library was used for sequencing on the HiSeq4000 instrument (Illumina, USA) in paired-end mode with 150 cycles kit. The reads were aligned to the hg19 reference genome. The rest of the mapping and calling procedure was the same as for the “virtual specimen” trios (see Section 2.4).
3. Results
We compared the genotypes recalibrated by each of the MCC-aware methods on the “virtual specimen trios” (see Section 2.4) to the genotypes produced by the GATK Hap-lotypeCaller on the corresponding “pure child” trios, the latter being considered the ground truth. As a baseline, we also included the comparison to initial genotypes called by the GATK HaplotypeCaller on the “virtual specimen” trios (“no recalibration”). Since the “virtual specimen” generation procedure may be subject to hidden artifacts, e.g., arising from differences in the sequencing procedure of the original mother and the original child, we present the results separately for the four sets of “virtual specimen” trios generated from each “real-world” family.
The machine learning-based models were trained with a “leave-one-out” strategy, where every trio excluding the test trio was used for training. This ensures that we test on data derived from individuals unseen by the trained classifier.
Figure 2 summarizes the results separately for each publicly available family that served as the basis for the “virtual” contaminated trios, with the accuracy achieved by each method plotted against the contamination fraction. All three individual methods fare well, reducing the number of mis-called positions by 40-80% over the baseline of using the original MCC-unaware calls (the “no recalibration” approach). Both machine learning approaches outperform the confidence interval-based method and overcome some of the problems of that heuristic method. XGBoost is the best-performing individual method, while the meta-classifier out-performs all individual methods.

Accuracy of the recalibration methods at various MCC fractions. Machine learning methods were trained with a “leave one out” strategy. Each curve consists of twelve data points corresponding to accuracy of the method applied to “contaminated specimens” at various MCCs (the contamination values used are as given in Section 2.4). The accuracy is calculated on the intersection of the calls made on the virtual fetal specimen and the calls made on the real-world child.
We also compared the running times of the methods for both training and testing (Table 1). In terms of speed, the confidence interval-based method, which requires no training, is the clear winner. The machine learning methods take on the order of minutes to train. Once the models are trained, though, the running times for recalibrating a specimen are comparable.
Training and running times of our methods. Training includes training a model on the entire synthetic dataset excluding the test trio (Section 2.4). Running involves recalibrating a single VCF file with a pre-trained model.
3.1. Factors affecting the quality of machine learning-based genotype recalibration
We further explored the effect of various settings on the performance of the machine learning approaches.
As noted earlier, the training dataset needs to be sufficiently rich to succeed at genotype recalibration. To demonstrate this, the two machine learning algorithms were trained and tested on different pairs of families (a ‘one versus one’ approach), at all contamination fractions. The results are summarized in Figure S2. While all the methods lead to an improvement in accuracy, the gains are much more modest than when training with a ‘leave-one-out’ approach (Figure 2, contamination 0.3), due to overfitting to the features of a particular trio. To combat overfitting, we ultimately adopted the approach where we train on all available trios except for the test one.
The trained classifiers can tell us about the discriminative power of our input features. Table 2 shows the top 10 features ordered by discriminative power for both classifiers. The letter in the last parentheses represents which sample the feature belongs to (Specimen, Mother, Father).
Top 10 most important features for machine learning methods in descending order. The parameters learned by the trained models (the coefficients of the hyperplanes separating the different classes for logistic regression; the degree of decision tree branching on each feature for XGBoost) can give an estimate of relative feature importance.
Statistics on the miscarriage samples that were recalibrated. The numbers of raw calls and recalibrated raw calls, and filtered calls and recalibrated filtered calls are given.
It is interesting to note that the degree of contamination is not found among the top features for logistic regression. Additionally, the father’s features are prominently towards the bottom of the table for logistic regression, yet are included in the top 10 for XGBoost, suggesting some complex relationship that the linear model does not capture. Finally, the linear model gives importance to the one-hot encoded genotypes as well as to the likelihood scores, whereas XGBoost completely ignores the genotypes, implying that the information in the genotypes is redundant when the likelihood scores are known (which is indeed true).
3.2. Robustness of the recalibration method
We explored the robustness of the meta-classifier performance to variations in the setup.
First, since in practice, the MCC fraction is not known in advance, we investigated the robustness of the model to errors in MCC fraction estimate. We found that for the range of errors in the MCC estimate we obtained for the simulated specimens (Figure S1), the decrease in performance is close to negligible (Figure S4).
We then investigated the dependence of the recalibration method performance on read coverage and found that for positions covered by more than 80 reads, which is typical coverage for exome sequencing, the recalibration accuracy is close to maximal (Figure S3).
Finally, we investigated the effect of the choice of the reference genome on the performance of the meta-classifier. We tested the performance of the classifier with GRCh38 as the reference genome, and in settings where the classifier was trained on hg19-mapped data and tested on GRCh38-mapped data, and vice versa. As seen in Figure S5, the machine learning approach is robust not only to the choice of the reference genome build, but also to changing the reference genome build between training and evaluation. Therefore, a model trained on one reference genome can be used to recalibrate variants called with a different reference.
3.3. Application to clinical miscarriage data
We applied the meta-classifier recalibration to three spontaneously miscarried abortus samples where the MCC exceeded 3% (estimated as in Section 2.1). The abortus samples (labeled 3, 6, and 15) were sequenced together with the peripheral blood from both their parents. After mapping and variant-calling with the GATK HaplotypeCaller, we obtained 107004, 91682, and 102751 calls in the three trios, respectively. These include variants where the abortus has a 0/0, 0/1, or 1/1 phenotype, each of which is potentially subject to recalibration. The actual numbers of recalibrated variants were 964, 1190, and 775, respectively. Since the Haplotype-Caller results are meant to be filtered, we repeated the analysis after applying GATK-suggested hard filtering to the call set ([14] with the additional requirement that each trio member’s genotype have GQ ≥ 30). This procedure resulted in 55200, 56165, and 62638 MCC-naive calls in the three trios, of which 296, 515, and 131, respectively, were recalibrated. As we expected, the vast majority of readjustments affected the 0/1 abortus genotype. Also not surprisingly, recalibration predominantly affected heterozygous variants with skewed allelic depths and favored the “elimination” of the allele with the lower allelic depth (Figure S6).
To illustrate how the ML model recalibrates the abortus trio data, we present a case in trio 6, where the abortus is initially called as heterozygous in the frameshift deletion chr12:14976417 CTA/C in the gene C12orf60, as are both parents (Figure 3). The classifier recalibrates this variant to a homozygous deletion call, with the reference allele predicted to have come from contamination with maternal DNA. In a manner consistent with the observation above, the allelic depth distribution in this locus is noticeably skewed in favor of the variant allele, which is predicted by the model to be the only allele observed in the abortus sample.

Integrative Genomics Viewer visualizaton of a deletion in Trio 6 that is called heterozygous by the MCC-naive GATK HaplotypeCaller but gets recalibrated to a homozygote by the ML method.
4. Discussion and Conclusions
As its costs are dropping, high-throughput sequencing is becoming increasingly routine in clinical and academic practice, and is gaining ground in prenatal diagnosis and in the diagnostic study of miscarried fetuses. While the bioinformatics toolkit for analyzing the sequencing data of individuals is well-developed and mature, it may encounter difficulties in the analysis of sequencing data for products of conception, which may be mixed with maternal tissue. We have shown that the accuracy of standard variant-calling pipelines does indeed degenerate as the maternal cell contamination increases, but that much of this decline can be alleviated by MCC-aware variant genotype correction. We show that the MCC fraction can be readily estimated from trio sequencing data and then used to inform the genotype correction. We show, using synthetic contaminated samples obtained from real trio data, that even a simple intuitive method based on confidence intervals does much to correct for the MCC-related loss of calling accuracy, and that the machine-learning methods trained on simulated data show even greater improvement, with the more sophisticated XGBoost having the better performance of the individual methods. The best performance, meanwhile, was obtained with the ensemble method that incorporates the results of all three methods that we tried.
We note that all methods discussed in this paper only recalibrate the variants that have been discovered by a general-purpose variant caller (we used GATK HaplotypeCaller which is commonly employed for human data analysis, but any variant caller which outputs sufficiently rich information about variants into a VCF file should work). Therefore, MCC-aware recalibration is a simple add-on to a standard bioinformatics pipeline, needing only the VCF file produced by it. The user can then compare the original VCF to the recalibrated VCF to see which genotypes had been changed. On the other hand, the reliance on standard variant calling implies that if MCC had caused the true fetal variant to be missed in all members of the trio, then that variant will not be called by the recalibration pipeline, either. We expect such cases to be rare.
We have applied our method to in-house sequencing data for three speciments from spontaneous miscarriages. While these particular samples had rather low MCC, this may not always be the case, as high levels of MCC do occur among aminocentesis and chorionic villi samples.
Currently, next-generation sequencing is applied to fetal DNA obtained by invasive methods which extract amniotic fluid or fetal or chorionic villi cells. Our work is aimed at the analysis of this sort of sequencing data, and we do not consider MCC fractions higher than 50%. As its costs drop, sequencing may become a practical option in non-invasive prenatal testing (NIPT) as well. NIPT analyzes fetal DNA circulating in maternal blood which constitutes a small fraction of the DNA sample obtained. Analyzing that sort of data would make MCC-aware variant calling a necessity, and may in fact give rise to more sophisticated and/or specialized algorithms, such as those that work directly with mapped reads.
Supplementary material
Supplementary material is available at bioRxiv online. We also present a repository with scripts and notebooks for reproducing our experiments as well as applying our methods: https://github.com/bazykinlab/ML-maternal-cell-contamination
Funding
The work was funded by the Skoltech Biomedical Initiative grant to GAB and DY.





{kind=link}
{kind=link}
{kind=link}