

Abstract
Claudin-low breast cancer is a molecular subtype associated with poor prognosis and without targeted treatment options. The claudin-low subtype is defined by certain biological characteristics, some of which may be clinically actionable, such as high immunogenicity. In mice, the medroxyprogesterone acetate (MPA) and 7,12-dimethylbenzanthracene (DMBA) induced mammary tumor model yields a heterogeneous set of tumors, a subset of which display claudin-low features. Neither the genomic characteristics of MPA/DMBA-induced claudin-low tumors, nor those of human claudin-low breast tumors, have been thoroughly explored.
The transcriptomic characteristics and subtypes of MPA/DMBA-induced mouse mammary tumors were determined using gene expression microarrays. Somatic mutations and copy number aberrations in MPA/DMBA-induced tumors were identified from whole exome sequencing data. A publicly available dataset was queried to explore the genomic characteristics of human claudin-low breast cancer and to validate findings in the murine tumors.
Half of MPA/DMBA-induced tumors showed a claudin-low-like subtype. All tumors carried mutations in known driver genes. While the specific genes carrying mutations varied between tumors, there was a consistent mutational signature with an overweight of T>A transversions in TG dinucleotides. Most tumors carried copy number aberrations with a potential oncogenic driver effect. Overall, several genomic events were observed recurrently, however none accurately delineated claudin-low-like tumors. Human claudin-low breast cancers carried a distinct set of genomic characteristics, in particular a relatively low burden of mutations and copy number aberrations. The gene expression characteristics of claudin-low-like MPA/DMBA-induced tumors accurately reflected those of human claudin-low tumors, including epithelial-mesenchymal transition phenotype, high level of immune activation and low degree of differentiation. There was an elevated expression of the immunosuppressive genes PTGS2 (encoding COX-2) and CD274 (encoding PD-L1) in human and murine claudin-low tumors. Our findings show that the claudin-low breast cancer subtype is not demarcated by specific genomic aberrations, but carries potentially targetable characteristics warranting further research.
Author Summary Breast cancer is comprised of several distinct disease subtypes with different etiologies, prognoses and therapeutic targets. The claudin-low breast cancer subtype is relatively poorly understood, and no specific treatment exists targeting its unique characteristics. Animal models accurately representing human disease counterparts are vital for developing novel therapeutics, but for the claudin-low breast cancer subtype, no such uniform model exists. Here, we show that exposing mice to the carcinogen DMBA and the hormone MPA causes a diverse range of mammary tumors to grow, and half of these have a gene expression pattern similar to that seen in human claudin-low breast cancer. These tumors have numerous changes in their DNA, with clear differences between each tumor, however no specific DNA aberrations clearly demarcate the claudin-low subtype. We also analyzed human breast cancers and show that human claudin-low tumors have several clear patterns in their DNA aberrations, but no specific features accurately distinguish claudin-low from non-claudin-low breast cancer. Finally, we show that both human and murine claudin-low tumors express high levels of genes associated with suppression of immune response. In sum, we highlight claudin-low breast cancer as a clinically relevant subtype with a complex etiology, and with potential unexploited therapeutic targets.
Introduction
The claudin-low subtype of breast cancer (BC) is a distinct disease entity associated with a relatively poor prognosis, and with an inadequately understood clinical significance [1–3]. It is characterized by low expression of tight junction and cell-cell adhesion genes, low degree of differentiation, epithelial-mesenchymal transition (EMT) phenotype and high level of immune cell infiltration [2]. The claudin-low subtype represents 7-14% of all breast cancers, and despite its unique biological features, there are no therapies specifically targeting the subtype [2–5]. While claudin-low tumors are found in several large scale studies, there is a paucity of information regarding their specific genomic characteristics [6–9]. Thus, significant gaps remain in the understanding of the biology of claudin-low tumors, and there is a need for further research to explore how their unique features may be therapeutically targeted.
Accurate preclinical models are vital for research into novel treatment options. Mouse mammary tumors may be induced through exposure to medroxyprogesterone acetate (MPA) and 7,12-dimethylbenzanthracene (DMBA) [10]. The tumors generated by this protocol are diverse, and a subset of these show similarities to the human claudin-low subtype [11,12]. A homogeneous primary in vivo model of claudin-low breast cancer does not currently exist [11]. While the mechanisms of MPA [10,13] and DMBA [14–17] have been described, there is still contention regarding the suitability of a chemically induced model of cancer for a disease that is not primarily caused by carcinogens in humans [18]. Evaluating the claudin-low subset of MPA/DMBA-induced tumors as a model for human disease is therefore an important step toward advancing preclinical research of claudin-low breast cancer.
In this study, we identified and comprehensively characterized claudin-low-like mouse mammary tumors generated by MPA/DMBA-induced carcinogenesis. Through genomic and transcriptomic analyses, we evaluated these tumors as a model for human claudin-low breast cancer and showed these tumors to be phenotypically accurate representations of their human counterparts. In parallel, we analyzed the previously unexplored genomic features of human claudin-low breast cancer. Our findings highlighted several features of claudin-low breast cancer with potential therapeutic implications, including a low tumor mutational burden, high expression of the immune checkpoint gene CD274 (encoding PD-L1) and high expression of PTGS2 (encoding cyclooxygenase-2).
Results
Gene expression subtyping reveals two distinct tumor clusters
We determined the murine transcriptomic subtypes of 17 MPA/DMBA-induced mammary tumors from 13 mice (S1 File) by performing a hierarchical clustering of gene expression data using the mouse intrinsic gene list [11]. This revealed nine murine subtypes in the cohort (Fig 1, Table 1, S2 File). The tumors separated into two distinct clusters. One cluster consisted of claudin-lowEx and squamous-likeEx tumors, both of which have been shown to resemble the human claudin-low subtype [11]; this is therefore referred to as the claudin-low-like cluster. The other cluster contained tumors from seven different subtypes and is referred to as the mixed cluster. In four instances, two tumors from different mammary glands were harvested from the same mouse. These were classified as different subtypes in all cases and are presumed to be distinct primary tumors. All normal mammary gland samples were classified as normal-likeEx, and clustered separately from the tumors.

Hierarchical clustering of MPA/DMBA-induced mouse mammary tumor gene expression levels revealed two distinct clusters, one containing claudin-low-like tumors and the other containing a transcriptomically heterogeneous set of tumors. Normal mouse mammary gland samples formed a separate cluster.
Subtype distribution of MPA/DMBA-induced tumors and normal mouse mammary gland tissue
Histopathological analysis corroborated the intertumor heterogeneity that was demonstrated by subtyping (S1 File). Five of the eight claudin-low-like tumors, including both squamous-likeEx tumors, showed a squamous appearance, while no tumors in the mixed cluster displayed this histological phenotype (p = 0.009, Fisher’s exact test). There was also a higher frequency of claudin-low-like tumors showing marked neutrophil infiltration (p = 0.002, Fisher’s exact test) and displaying a marked or partial spindloid appearance (p = 0.050, Fisher’s exact test) compared to tumors in the mixed cluster.
Mutations in MPA/DMBA-induced mammary tumors are independent of gene expression subtype
To determine the genetic characteristics of the tumors, we performed exome sequencing to a mean depth of 58, with 84% of bases being sequenced to a coverage of 20x or higher. We identified a mean of 589 mutations per tumor (range: 288 to 1795), corresponding to a mean mutation rate of 11.9 mutations per megabase (range: 5.8 to 36.2) (Fig 2B). This is substantially higher than the average 1.3 mutations per megabase found in human breast cancer [19]. There was no significant difference in mutational burden between the tumors in the claudin-low-like and the mixed cluster, and the only subtype specific trend was a particularly high mutational burden in the two squamous-likeEx tumors (Fig 2B).
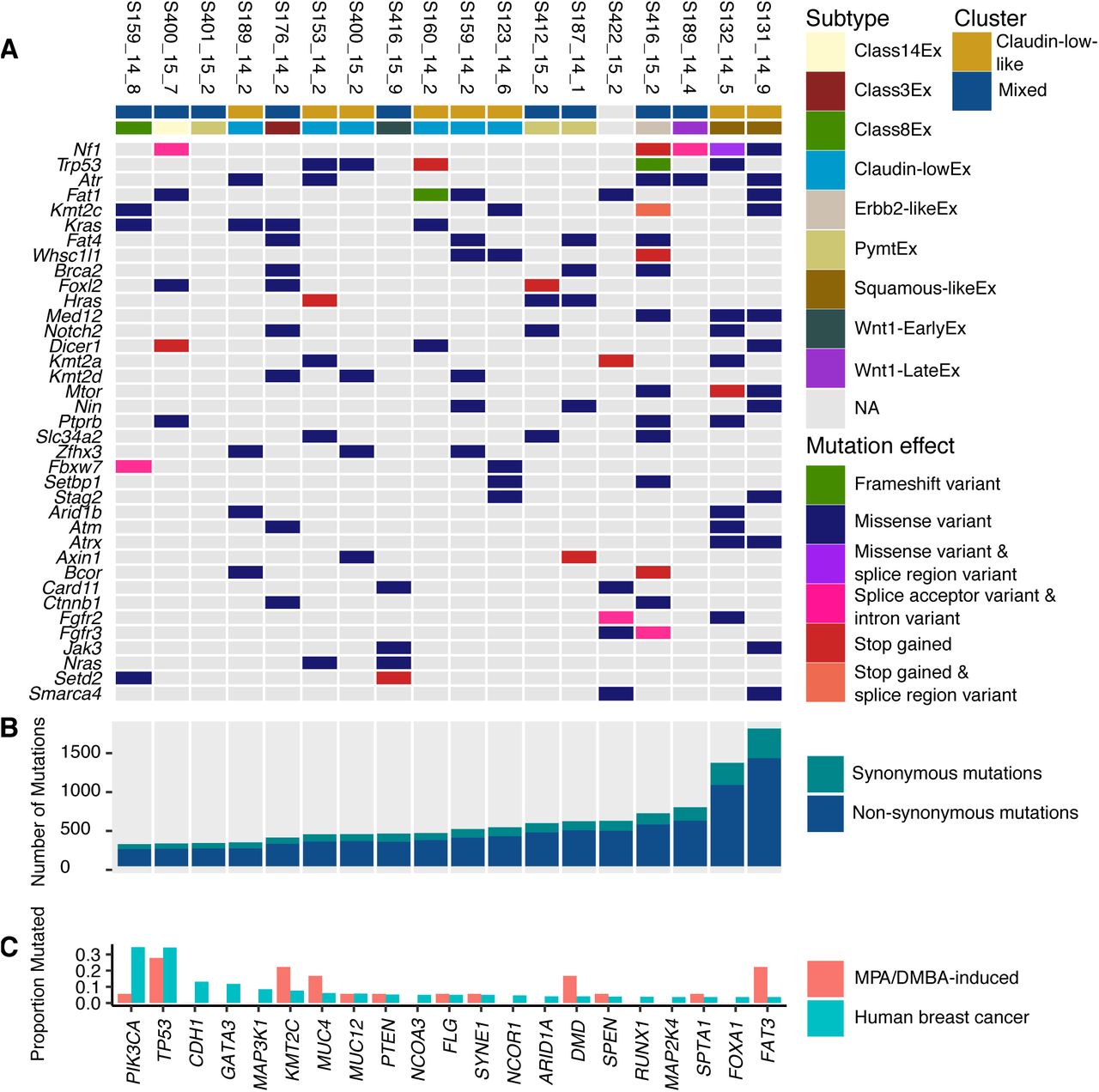
A Nf1, Trp53, Atr and Fat1 were the most frequently mutated driver genes in the MPA/DMBA-induced tumor cohort. No specific mutations accurately delineated the tumor clusters. B The MPA/DMBA-induced tumors carried between 288 and 1795 exonic mutations. No significant differences in mutational burden were found between the clusters, however a high mutational rate was observed in the two squamous-likeEx tumors. C MPA/DMBA-induced tumors generally showed divergent mutational rates compared to human breast cancer in the genes most frequently mutated in human breast cancer. TP53 mutations occurred at a similar rate in MPA/DMBA-induced tumors and human breast cancer.
All tumors carried mutations in driver genes defined by the COSMIC cancer gene census list [20], with a mean of 13.8 driver genes carrying mutations per tumor (range: 4 to 29) (Fig 2A). Several driver genes were recurrently mutated, including Trp53, Kras, and Kmt2c (S3 File), but no driver genes carried mutations at a significantly different rate between the two clusters. We did, however, identify two notable trends which did not reach statistical significance: an elevated rate of Trp53 mutations in the claudin-low-like cluster (50% vs. 11%, p = 0.13, two-tailed Fisher’s exact test) and an elevated rate of Zfhx3 mutations also in the claudin-low-like cluster (37.5% vs. 0%, p = 0.08, two-tailed Fisher’s exact test). No mutations were significantly associated with histological features.
MPA/DMBA-induced tumors and human breast cancers display disparate gene mutational profiles
To narrow down potential driver mutations in the MPA/DMBA-induced tumors, we compared amino acid changes caused by mutations in driver genes to known amino acid changes in human cancers [20] (Table 2, S4 File). There were hotspot amino acid changes in all Ras genes, including Kras G12C, G13R, Q61H, Hras Q61L and Nras Q61L. In total, 8 of 18 tumors carried hotspot amino acid changes in Ras genes. There was one Pik3ca mutation in the cohort causing an H1047R amino acid change. This mutation is frequently found in human breast cancer and has previously been reported in DMBA-induced mouse mammary tumors [21].
Selected hotspot mutations in MPA/DMBA-induced tumors
There were marked disparities between the gene mutational profiles of human breast cancer [22] and MPA/DMBA-induced tumors (Fig 2C, S5 File). The two most frequently mutated genes in breast cancer are PIK3CA and TP53. While TP53 showed comparable mutation rates between human breast cancer and MPA/DMBA-induced tumors (34% and 28%, respectively), PIK3CA mutation does not appear to be a common event in MPA/DMBA-induced tumors (35% in BC, 6% in MPA/DMBA). Several frequently mutated genes in breast cancer, such as CDH1, GATA3 and MAP3K1, were not mutated in any MPA/DMBA-induced tumors. Conversely, many genes frequently mutated in MPA/DMBA-induced tumors, such as ATR, FAT1 and KRAS, are rarely mutated in breast cancer.
DMBA induces a characteristic mutational spectrum with a high frequency of T>A transversions in TG dinucleotides
To characterize the mutagenic profile of DMBA, we analyzed the mutational spectra of the MPA/DMBA-induced tumors. Mutations showed a majority of T>A transversions, which accounted for 63% of all mutations (S1A Fig). In their trinucleotide context, thymine mutations (T>N) were overrepresented in positions with a 3’ guanine nucleotide (S1B & S1C Fig, S6 File). This was statistically significant when compared to the proportion of thymine nucleotides in an NTG context in the mouse reference genome (p < 0.001 in all cases, two-tailed Wilcoxon rank-sum test). There was a similar overrepresentation of cytosine mutations in positions with a 3’ adenine. This was statistically significant for C>A and C>G mutations (p < 0.001), but not for C>T mutations (p = 0.089), when compared to the proportion of cytosine nucleotides in an NCA context in the mouse reference genome.
Mutation signature analysis revealed evidence of signatures 4, 6, 22, 24 and 25 [23] in the MPA/DMBA-induced tumors (S1D Fig). All tumors were associated with signature 22, while signatures 4 and 25 were found in 17 and 11 of the 18 tumors, respectively. Signatures 24 and 6 were only found in four and one tumor(s), respectively. Notably, none of the signatures found in MPA/DMBA-induced tumors have been associated with human breast cancer [23].
MPA/DMBA-induced tumors have diverse copy number profiles
Breast cancer is largely driven by copy number aberrations (CNAs) [24], yet the copy number profiles of MPA/DMBA-induced mammary tumors have not previously been described. We found a mean of 1299 genes with CNA per tumor (range: 90 – 3057), of which a mean of 65% were amplifications. There was a tendency for claudin-low-like tumors to have a lower burden of CNAs, with a mean of 919 genes carrying CNA, compared to the mixed group of tumors, with a mean of 1637 genes carrying CNA (Fig 3A). This trend did however not reach statistical significance (p = 0.139, two-tailed Wilcoxon rank-sum test).

A There was a trend toward a lower number of genes with copy number aberrations in the claudin-low-like cluster. B Copy number aberrations implicated in cancer were found in 14 of 18 MPA/DMBA-induced tumors. Two tumor sets (S422_15_2, S400_15_2 and S400_15_7, and S412_15_2, S176_14_2, S159_14_8 and S159_14_2) showed remarkably similar CNA profiles, but displayed different gene expression subtypes. CNA status of −2 is a homozygous deletion, CNA status of −1 is a heterozygous deletion, CNA status of 0 is copy number neutral, CNA status of 1 is a single copy amplification, and CNA status of 2 is a multi-copy amplification.
To determine CNAs in the MPA/DMBA-induced tumors with a potential oncogenic driver effect, we identified amplifications and deletions known to be associated with cancer [20] (Fig 3B). We found that 14 of the 18 tumors carried potential driver CNAs (range: 0 to 4, mean: 2.6). Three of the four tumors not carrying potential driver CNAs were claudin-low-like. There was however no statistically significant difference in the number of potential driver CNAs between the clusters. Several genes had recurrent CNAs, but none occurred at a statistically significant different rate in one cluster versus the other.
Only two of the CNA events identified in MPA/DMBA-induced tumors occur at a notable rate in human breast cancer; Mdm4 is amplified in 25% and Ppm1d is amplified in 10% of human BC [6,7].
We observed two sets of tumors carrying remarkably similar CNA profiles (Fig 3B). None of the tumors in these two sets displayed the same murine subtype as any other tumor within the same set.
The human claudin-low breast cancer genome is characterized by a low mutational burden, frequent TP53 mutations and a low rate of CNA
Little has been published specifically describing the genomic characteristics of human claudin-low breast cancer. We therefore analyzed the 218 claudin-low tumors found in the METABRIC dataset, for which DNA sequence data from 173 genes and whole genome copy number data is available [6,7].
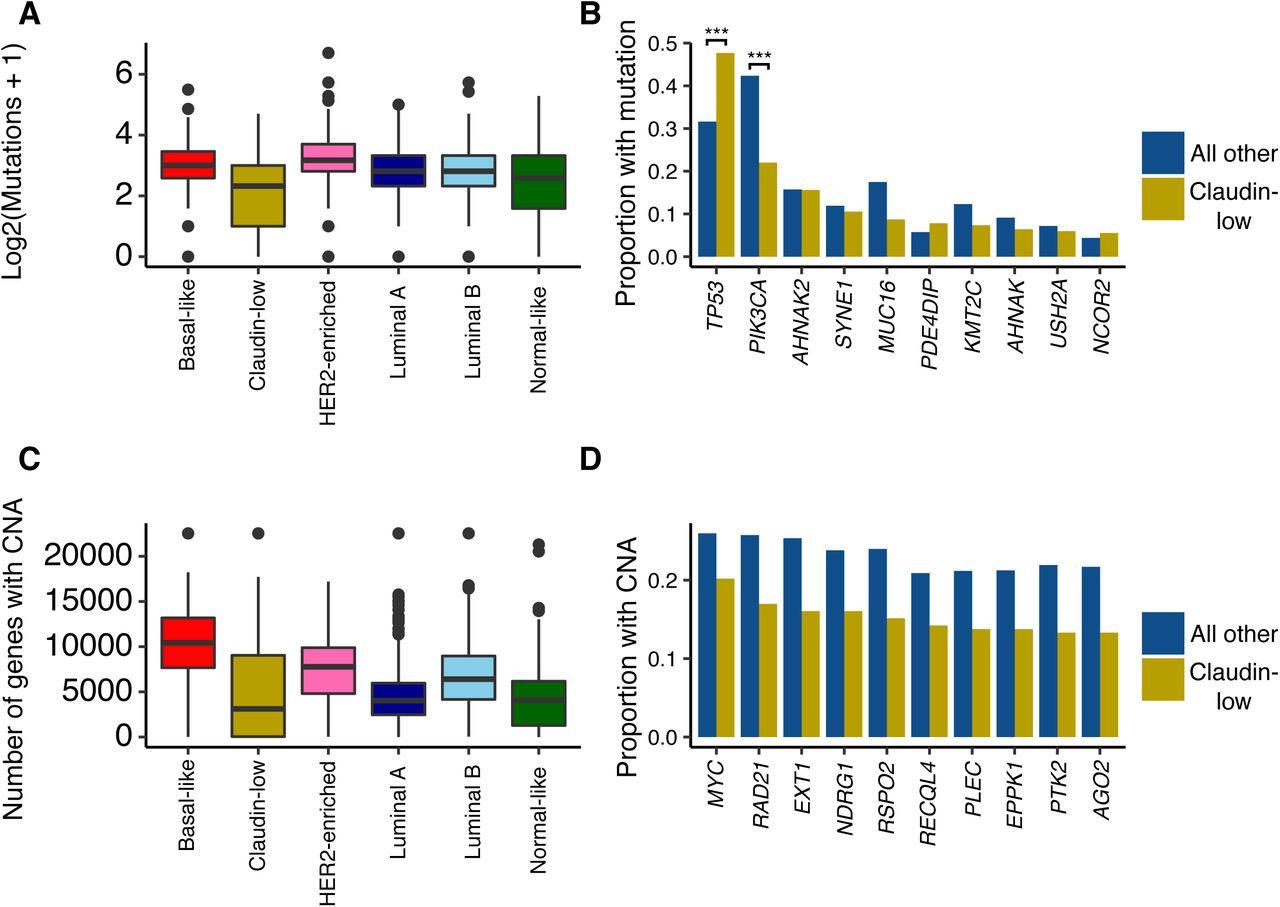
Claudin-low tumors, with a mean of 4.7 mutations per tumor, carried relatively few mutations compared to all other tumors, with a mean of 7.3 mutations per tumor (p < 0.001, two-tailed Wilcoxon rank-sum test) (Fig 4A). Claudin-low tumors share several characteristics with basal-like tumors and are often classified as such by the PAM50 assay [6,7,25]; however, basal-like tumors showed a significantly higher mutational burden than claudin-low tumors (mean: 8.08 mutations per tumor, p < 0.001, two-tailed Wilcoxon rank-sum test).

A Claudin-low breast cancer was the subtype with the lowest mutational burden. Number of mutations displayed as log2(mutations + 1). B TP53 and PIK3CA were the most frequently mutated genes in human breast cancer. Claudin-low tumors carried TP53 and PIK3CA mutations at significantly higher and lower rates, respectively, compared to non-claudin-low breast tumors. *** = p < 0.001. C Claudin-low tumors carried relatively few CNAs compared to non-claudin-low tumors. D The ten genes which were most frequently affected by CNA in claudin-low tumors were all found to be copy number aberrant at a higher frequency in non-claudin-low tumors. MYC amplification is the most common CNA event in claudin-low breast cancer.
There was a high degree of overlap between the genes most frequently mutated in claudin-low breast cancers and the genes most frequently mutated in all other breast cancers (Fig 4B). Most of these genes carried mutations at similar rates between claudin-low and non-claudin-low tumors, albeit with a tendency towards a slightly lower rate in claudin-low tumors. There were however two notable differences in mutational frequency: a significantly higher rate of TP53 mutations and a significantly lower rate of PIK3CA mutations in claudin-low tumors compared to other tumors. Similarly, basal-like tumors also carried a high frequency of TP53 mutations and a low frequency of PIK3CA mutations [7,22].
Human claudin-low breast tumors carried significantly fewer genes with copy number aberration (mean: 4879) compared to all other tumors (mean: 6247; p < 0.001, two-tailed Wilcoxon rank-sum test) (Fig 4C). This difference was also marked when comparing claudin-low tumors with basal like tumors (mean: 10175 genes per tumor; p < 0.001, two-tailed Wilcoxon rank-sum test).
By gene, the most frequent copy number event in claudin-low breast cancer was MYC amplification, found in 20% of cases (Fig 4D). In comparison, this event was found in 26% of all other breast tumors. The ten most frequently amplified genes in claudin-low breast cancer were all located at chromosomal position 8q24, a region also frequently amplified in basal-like breast cancers [6,7].
Claudin-low-like MPA/DMBA-induced mammary tumors accurately reflect the gene expression characteristics of their human counterpart
We explored several established gene expression features of the claudin-low subtype and found that MPA/DMBA-induced claudin-low-like tumors accurately mirrored their human counterpart. Specifically, claudin-low-like tumors had low expression of genes involved in cell-cell adhesion, low expression of luminal genes, and high expression of genes related to EMT (Fig 5A, S2 Fig). Claudin-low-like tumors also showed a markedly lower degree of differentiation compared to tumors in the mixed cluster. In particular, the claudin-low-like cluster expressed significantly higher and lower levels of Cd44 and Cd24a, respectively, indicating a stem cell-like phenotype in these tumors [25,26] (S3 Fig). There was no significant difference in the expression of proliferation-related genes between the two clusters. Vascular content-related genes were expressed at a significantly higher level in claudin-low-like tumors compared to the tumors in the mixed cluster (S2 Fig), indicating a higher degree of neoangiogenesis in these tumors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A MPA/DMBA-induced claudin-low-like tumors recapitulated the gene expression characteristics of the claudin-low subtype as evidenced by the expression levels of relevant gene signatures. P-values are calculated for the claudin-low-like tumors versus tumors in the mixed cluster. B Cd274 and Ptgs2 are expressed at significantly higher levels in the claudin-low-like tumors than in the mixed cluster tumors. C Claudin-low is the breast cancer subtype with the highest expression of CD274 and PTGS2.
Immune cell admixture was significantly higher in the claudin-low-like tumors compared to tumors in the mixed cluster (p < 0.001, two-tailed Wilcoxon rank-sum test) and compared to normal mammary gland samples (p = 0.006). We also found higher expression of genes related to immunosuppression and interferons in the claudin-low-like cluster compared to both the mixed cluster and normal mammary gland samples. High immune cell infiltration combined with high expression of type 1 interferon-related and immunosuppressive genes are characteristics of tumors that may respond to immunotherapeutics [27,28].
We identified a significantly elevated expression of two potentially actionable genes related to immunosuppression in the claudin-low-like tumors: the immune checkpoint encoding gene Cd274 and the cyclooxygenase encoding gene Ptgs2 (Fig 5B). These features were also characteristic of human claudin-low tumors in the METABRIC cohort [6,7], which showed significantly higher expression levels of both PTGS2 and CD274 compared to non-claudin-low breast tumors (p < 0.001 for both, two-tailed Wilcoxon rank-sum test), and compared specifically to basal-like tumors (p = 0.004 and p < 0.001, respectively) (Fig 5C). These characteristics may indicate a susceptibility to immune checkpoint inhibitors and cyclooxygenase inhibitors in human claudin-low breast cancer [29,30].
Discussion
In this study, we have performed a comprehensive analysis of mutations, copy number aberrations and gene expression characteristics of MPA/DMBA-induced tumors. We found marked inter-tumor heterogeneity, and showed that half of the tumors displayed a claudin-low-like phenotype, in line with a previous report [11]. Our findings demonstrate that these tumors provide a transcriptomically accurate representation of human claudin-low breast tumors, reflecting key features such as an EMT phenotype, high level of immune infiltration and a low degree of differentiation.
MPA/DMBA-induced tumors carried a mutational burden multiple times that of human breast cancer, a high frequency of activating Ras-mutations and a characteristic mutational spectrum. The specific genes carrying mutations varied widely between tumors, however all tumors had a consistent mutational signature. This indicates that the dominant mutational process in these tumors is DMBA-induced mutagenesis, and not aberrations occurring after tumor initiation, as a result of e.g. disrupted DNA-repair. Copy number aberrations in MPA/DMBA-induced tumors have not previously been explored, and we show here that most tumors carry potential driver CNAs. However, while we noted several genomic trends, such as a higher rate of Trp53 mutation and a lower burden of CNA in MPA/DMBA-induced claudin-low-like tumors, no individual genomic features accurately delineated the two gene expression-based tumor clusters. Further, several tumors carried similar sets of mutations and/or CNAs but displayed different subtypes. This suggests that no specific genomic event determines tumor subtype, and that other etiological models may be more appropriate, such as different cells-of-origin [31] or microenvironmental factors [32]. This finding concurs with recent reports showing that transgenic mouse mammary tumors display histological and transcriptomic phenotypes largely uncoupled from their underlying driver mutations [33,34]. One possible model for MPA/DMBA-induced tumorigenesis is therefore as follows: first, MPA induces a RANK-l mediated mammary gland proliferation [10,13]. DMBA then induces mutations in mammary cells in a pattern as elucidated by our mutation signature analysis, predominantly in TG and CA dinucleotides, stochastically distributed throughout the genome. The tumor is initiated when one or more driver mutations occur, for example Trp53 or Ras-mutation, with the tumor phenotype, however, determined by non-genomic factors. The biochemical mechanism of DMBA-induced mutagenesis has been described [14,15], whereas no causal mechanism for DMBA-induced copy number aberration is known; it is therefore likely that CNAs arise after tumor initiation.
Previous genomic analyses which included human claudin-low breast tumors have either not included specific analyses of the subtype [6,7], included few samples [3], or have been restricted to the triple-negative [35,36] or metaplastic [37] subsets of claudin-low tumors. We show here that human claudin-low tumors are characterized by a low number of mutations and a low burden of CNAs. This finding is surprising, given the apparent inverse correlation between CNA and mutational burden in cancer [24], and indicates that the claudin-low subtype is relatively genomically stable compared to other breast cancers. We also find similarities in genomic characteristics between claudin-low tumors and basal-like tumors, in particular a high frequency of TP53 mutations, a low frequency of PIK3CA mutations, and 8q24 amplifications as a common event. While the transcriptomic similarity between these two subtypes is established [25], these findings illustrate that there are also marked genomic similarities between claudin-low and basal breast cancer, albeit with a lower burden of genomic aberrations in claudin-low tumors.
Claudin-low tumors show high expression of immune-related genes and a high level of immune cell infiltration [3,25,38]. However, claudin-low tumors also express high levels of immunosuppressive genes. In MPA/DMBA-induced claudin-low-like tumors, we observed an elevated expression of two particularly notable genes involved in immunosuppression: Ptgs2 (encoding COX-2) and Cd274 (encoding PD-L1). This observation was consistent in human claudin-low breast cancer. COX-2 may be implicated in cancer development through several mechanisms: reducing apoptosis, increasing epithelial cell proliferation, promoting angiogenesis, increasing invasiveness of tumor cells and immunosuppression [39–41]. COX-2 may also be involved in vasculogenic mimicry, a process in which epithelial tumor cells form vascular channel-like structures without participation of endothelial cells, allowing nutrients to reach tumor cells without the need for neoangiogenesis [42]. Vasculogenic mimicry has previously been shown to occur in claudin-low tumors [43]. COX-2 and PD-L1 are clinically actionable through the use of COX-inhibitors [29] and checkpoint inhibitors [44], respectively. Further research into the potential use of checkpoint inhibitors and COX-inhibitors in claudin-low breast cancer is warranted, with promising future avenues including combinatorial Treg-depletion [38].
In summary, we have found that claudin-low-like MPA/DMBA-induced mouse mammary tumors are a transcriptomically accurate model for human claudin-low breast cancer. We did not find strong evidence that claudin-low-like MPA/DMBA-induced tumors are delineated by any specific genomic features, however the relatively small number of samples included in this study may have obscured possible associations. By analyzing publicly available data, we showed that human claudin-low breast cancer is a relatively genomically stable subtype. There is a high expression of genes related to immunosuppression in claudin-low breast cancers, a feature which is evident in claudin-low-like MPA/DMBA-induced tumors. Our observations suggest immunosuppression as a potential therapeutic target in claudin-low breast cancer and indicate MPA/DMBA-induced claudin-low-like tumors as an appropriate model for continued research.
Material and Methods
Ethics statement
The Norwegian Food Safety Authority approved all experiments in advance of their implementation (FOTS ID #4385). All mice were bred and maintained within a specific pathogen free (SPF) barrier facility according to local and national regulations, with food and water ad libitum. For invasive procedures, animals were anaesthetized with sevoflurane gas. Animals were euthanized by cervical dislocation under anesthesia with sevoflurane gas.
Mouse strains and tumor induction
Animals from an Lgr5-creERT2-EGFP;Rb/C transgenic mouse strain on a pure FVB/N background were purchased from Jackson Laboratory (www.jax.org), kindly gifted from Rune Toftgård (Karolinska Institutet, Sweden) and locally bred. The transgenes are considered biologically inert, and offspring from all potential genotypes were used (wildtype, single transgene and double transgene). Female mice were treated with medroxyprogesterone acetate (MPA) and 7,12-dimethylbenzanthracene (DMBA) in accordance with established protocol [10]. In brief: 90-day release MPA pellets (50 mg/pellet, Innovative Research of America cat.# NP-161) were implanted subcutaneously at 6 and 19 weeks after birth. 1 mg DMBA (Sigma Aldrich cat.# D3254) dissolved in corn oil (Sigma Aldrich cat.# C8267) was administered by oral gavage at 9, 10, 12 and 13 weeks after birth. Tumor growth was regularly monitored by manual palpation and measured by caliper. Tumor volume was estimated using the following formula: volume = (width2* length)/2. When the tumors reached the maximum allowed size of 1000mm3, or at the age of 32 weeks, tissue was collected at necropsy and fixed in 4% paraformaldehyde (PFA) or snap frozen and stored at −80 °C. 18 tumors from 14 mice, of which four mice carried two mammary tumors, were subject to genomic and transcriptomic analyses. Six normal mammary glands collected from mice not undergoing MPA/DMBA-treatment were included as controls. Mouse features and histopathological tumor features, can be found in S1 File.
Histopathology and immunohistochemistry
Mouse tissue was fixed overnight in 4% PFA, routinely processed and paraffin embedded. Formalin-fixed paraffin-embedded tissue was sectioned and stained with hematoxylin and eosin (HE). HE-stained tissue was classified by a certified veterinary pathologist.
Immunohistochemical staining was performed as previously described [45] with primary antibodies against K5 (Covance cat.# PRB-160P), K18 (Progen cat.# 61028), Ki67 (Novocastra cat.# NCL-Ki67p), ERα (Millipore cat.# 06-935), PR (Abcam cat.# ab131486) and Her2/Erbb2 (Millipore cat.# 06-562).
DNA and RNA isolation
DNA isolation for exome sequencing was carried out at Theragen Etex Bio Institute (Seoul, South Korea). DNA was isolated using QIAamp DNA Mini Kit (Qiagen cat.# 51306) per manufacturer’s protocol. DNA from two samples (S159_14_11 and S176_14_11) was isolated using CTAB Extraction Solution (Biosesang cat.# C2007) per manufacturer’s protocol. DNA integrity was assessed by electrophoresis and concentration was determined using the Nanodrop ND-1000 spectrophotometer (Thermo Scientific cat.# ND-1000) and Qubit fluorometer (Thermo Scientific cat.# Q33226). Total RNA and DNA isolation for gene expression microarrays was carried out using the QIAcube system (Qiagen cat.# 9001292) with the AllPrep DNA/RNA Universal Kit (Qiagen cat.# 80224) according to the protocol provided by the supplier, with 30mg tissue as input. The tissue was manually minced with a scalpel on ice followed by lysis and homogenization using TissueLyzer LT (Qiagen cat.# 85600) and Qiashredder (Qiagen cat.# 79654), respectively. Nucleic acid concentrations were measured by NanoDrop ND-1000 spectrophotometer and RNA integrity was analyzed using Agilent 2100 Bioanalyzer (Agilent Technologies cat.# G2939BA).
Gene expression microarrays
Gene expression profiling was performed using RNA isolated from eighteen snap frozen MPA/DMBA-induced tumors and six normal/untreated mouse mammary gland samples. Whole genome expression data was obtained using Agilent Sureprint G3 Mouse Gene Expression 8×60K microarrays (Agilent Technologies cat.# G4852B) with Low Input Quick Amp Labeling protocol (Agilent Technologies cat.# 5190-2331) and the Cy3 fluorophore. 40ng RNA was used for input. Microarrays were scanned using an Agilent SureScan Microarray Scanner (Agilent Technologies cat.# G4900DA) and data was extracted using Agilent Feature Extraction software. One tumor sample (S422_15_2) failed quality control and was excluded from further gene expression analyses.
Gene expression analyses
Gene expression data was analyzed using Qlucore Omics Explorer 3.2 (Qlucore AB) and R version 3.3.2 [46]. Gene expression values were quantile normalized and probes with a standard deviation of less than 2.8% of the largest observed standard deviation were filtered out. For genes represented by more than one probe, mean expression values were calculated to obtain one gene expression value per gene. Principal component analysis was performed to assess data quality and one normal mammary gland sample (S178_14_2) was identified as an outlier and removed from further analysis. Murine subtypes were determined by first calculating centroids for each subtype using the original data from the mouse intrinsic gene list [11], followed by calculating Spearman correlation for every sample to each of the subtype centroids. The subtype with the highest correlation coefficient was assigned as the sample’s subtype.
Unsupervised hierarchical clustering was performed using average linkage and Spearman correlation as the distance metric. Immune cell infiltration was inferred using ESTIMATE [47]. Scores for gene signatures relevant to the claudin-low subtype (adhesion, EMT, luminalness, proliferation, vascular content, immunosuppression and interferons [25,43,48–50]) were calculated using a standard (Z) score approach: for every gene in each signature, a standardized expression value was calculated by subtracting the mean across all samples, then dividing by the standard deviation. Calculation of the mean of the standardized expression values across all genes in the signature yielded the score. Gene lists included in the different signatures are found in S7 File. The degree of differentiation was calculated using a differentiation predictor [25]. Two-tailed Wilcoxon rank-sum tests were used for statistical testing of differences in scores between two groups.
Whole exome sequencing
Whole exome sequencing was carried out at Theragen Etex Bio Institute. Library preparation and target enrichment was carried out using the SureSelect XT Mouse All Exon Kit (Agilent cat.# 5190-4641) per manufacturer’s instructions. Sequencing was performed on an Illumina HiSeq 2500 (Illumina cat.# SY–401–2501). DNA was sequenced to an average depth of 58. Quality control was performed with FastQC [51].
Sequence alignment and processing
Adapter sequences were removed using CutAdapt, version 1.10 [52]. Low quality reads were trimmed using Sickle version 1.33 [53], in paired end mode with quality threshold set to 20 and length threshold set to 50 base pairs. Reads were aligned to the mm10 reference genome using the Burrows-Wheeler MEM aligner (BWA-MEM), version 0.7.12 [54]. Following alignment, duplicate reads were marked using Picard (https://broadinstitute.github.io/picard/) version 2.0.1. Base quality scores were then recalibrated using GATK version 3.6.0 [55–57]. Lists of known single nucleotide polymorphisms and indels for the FVB/N mouse strain, were downloaded from the Mouse Genomes Project, dbSNP release 142 and used for base quality score recalibration and mutation filtering (available at ftp://ftp-mouse.sanger.ac.uk/) [58].
Mutation calling and analysis
Somatic mutations were called using the MuTect2 algorithm in GATK [55–57] with a minimum allowed base quality score of 20. Mutations were filtered against variants found in matched normal liver tissue and known single nucleotide polymorphisms for the FVB/N mouse strain. Candidate somatic mutations which did not pass the standard MuTect2 filters were removed from further analysis. Mutations not meeting the following requirements were also removed from further analysis: minimum allele depth of 10, minimum allele frequency of 0.05, and presence of the mutation in both forward and reverse strands. Mutations were annotated using SnpEff [59] and filtered for downstream analysis using SnpSift [60]. Candidate driver mutations were defined as moderate or high impact mutations, as defined by SnpEff, in driver genes as identified by the COSMIC cancer gene census [20]. To identify hotspot mutations, mouse amino acid positions were aligned to the orthologous human amino acid position using Clustal Omega [61] through UniProtKB [62] and used to query mutations found in the COSMIC database [20]. Mutational spectrum and signature analysis was performed using the deconstructSigs framework [63] modified to allow the use of the mm10 mouse reference genome. The COSMIC mutational signatures were used for reference [23].
Copy number aberration analyses
Copy number aberrations were identified from exome sequence data using EXCAVATOR2 [64] using the mm10 reference genome. CNA calling was performed using standard settings, and a window size of 20000 base pairs. To identify potential driver CNAs, we filtered for CNAs associated with cancer in the COSMIC gene list [20].
Supporting information
S1 Fig. The mutational spectra and mutational signatures of MPA/DMBA-induced mammary tumors A T>A transversions were the most frequent mutation type in MPA/DMBA-induced tumors, followed by C>A transversions. B Heatmap of mutational frequencies by trinucleotide context. There was an overrepresentation of T>N mutations in positions with a 3’ guanine and C>N mutations in positions with a 3’ adenine. C Histogram of C>A and T>A transversions by trinucleotide context in a representative tumor (S159_14_8). D Mutation signature 22 was the predominant mutational signature in the MPA/DMBA-induced tumors and was evident in all tumors in the cohort.
S2 Fig. Gene expression scores by cluster in MPA/DMBA-induced tumors Expression scores by cluster for genes related to differentiation, adhesion, luminal features, proliferation, vascular content, EMT, immune features, interferon signaling and immunosuppression. Two-tailed Wilcoxon rank-sum test. ns = not significant, p > 0.05. * = p < 0.05. ** = p < 0.01. *** = p < 0.001.
S3 Fig. Expression of Cd24a and Cd44 by cluster in MPA/DMBA-induced tumors Claudin-low-like tumors had a lower expression of Cd24a and a higher expression of Cd44 compared to the mixed cluster of tumors (p = 0.003 and p = 0.005, respectively, two-tailed, Wilcoxon rank-sum test), indicating a stem cell-like phenotype in the claudin-low-like tumors.
S1 File. Mouse characteristics and histopathological data
S2 File. Subtype correlations for MPA/DMBA-induced tumors
S3 File. Mutations observed in MPA/DMBA-induced tumors
S4 File. MPA/DMBA-induced tumor driver gene mutations in the COSMIC database
S5 File. Comparative mutation rates in MPA/DMBA-induced tumors and human breast tumors in the TCGA cohort
S6 File. Mutational signatures for all MPA/DMBA-induced tumors
S7 File. Gene lists used for gene expression scores
Acknowledgements
We thank Phuong Vu, Eldri Undlien Due and Tina Brinks for helping with laboratory work, Prof. Rune Toftgård for providing the transgenic mouse lines, and the support staff at the Department of Comparative Medicine, Oslo University Hospital Radiumhospitalet for help with animal work. We are grateful to the members of the Department of Cancer Genetics, Institute for Cancer Research, Oslo University Hospital for insightful discussions, and in particular thank Tonje G. Lien for statistical input.
References




