

ABSTRACT
We screened a series of 4-anilinoquinolines and 4-anilinoquinazolines and identified novel inhibitors of Mycobacterium tuberculosis (Mtb). The focused 4-anilinoquinoline/quinazoline scaffold arrays yielded compounds with high potency and the identification of 6,7-dimethoxy-N-(4-((4-methylbenzyl)oxy)phenyl)quinolin-4-amine (34) with an MIC90 value of 0.63-1.25 μM. We also defined a series of key structural features, including the benzyloxy aniline and the 6,7-dimethoxy quinoline ring, that are important for Mtb inhibition. Importantly the compounds showed very limited toxicity and scope for further improvement by iterative medicinal chemistry.
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB) in humans,1 infects nearly a third of the earth’s population and caused 1.6 million worldwide deaths in 2017.2 With nearly ten million new cases of active disease each year, TB is now the leading cause of death from infectious disease globally.2 Current therapeutic strategies involve the use of a combination of anti-microbial agents including ethambutol, isoniazid, pyrazinamide and rifampicin (Fig. 1).3 However, more than 5% of Mtb infections now involve multidrug-resistant (MDR-TB) and extensively drug-resistant (XDR-TB) Mtb strains. MDR-TB is associated with a 50% mortality rate whereas XDR-TB is nearly always fatal.4 There is an urgent need for new therapeutic strategies.

Current therapeutic strategies for treatment of Mycobacterium tuberculosis infections.
Human protein kinases are pharmacologically tractable enzymes targeted by more than three dozen approved medicines.5 Hundreds of additional kinase inhibitors are under clinical and preclinical investigation. There is growing recognition that pathogen kinases may be targeted in the treatment of infectious diseases.6–7 Considering the conserved ATP-binding site across species, we looked to screen collections of ATP-competitive inhibitors of human kinases for their anti-tubercular activity.
To identify new chemical starting points against Mtb we looked to lapatinib, gefitinib, and erlotinib as starting points which have recently been revealed to have activity against Mtb (Fig. 2.).5,8 We tested the activity of lapatinib, gefitinib, and erlotinib against Mtb by measuring luminescence and growth on solid medium across a series of four two-fold dilutions starting at 20 μM (Tab 1).9–10 The reduction of visible growth on solid medium demonstrated the compounds to be bactericidal.

Structures of clinical quinazolines.
Results of clinical inhibitors.
Gefitinib treatment had no effect relative to the absence of compound. Erlotinib induced a modest effect that appeared to plateau at a signal of approximately 70 %. In contrast, lapatinib showed activity even at 5 μM and reduced the relative Mtb signal to below 10 % at 20 μM. This result suggested that the 4-benzyloxy aniline substituent might be important for anti-Mtb activity. These compounds demonstrate only limited toxicity in a human skin fibroblast cell line (WS-1) counter screen.11
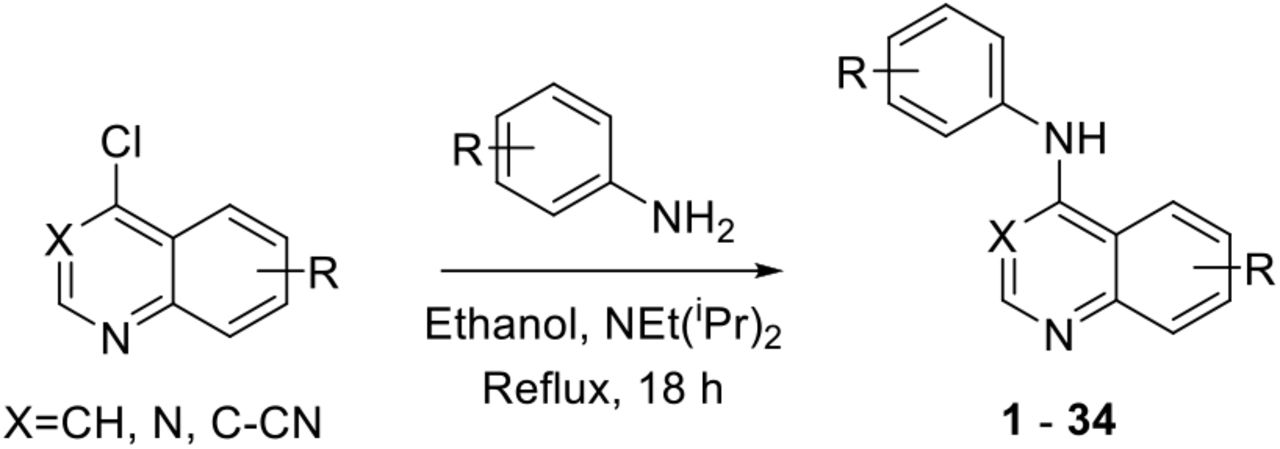
To further explore quinazoline Mtb activity, we profiled several focused arrays of compounds to probe the structure activity relationships of the quinoline/quinazoline. We hence synthesized a series of compounds (1-34) following up on the results listed in table 1, exploring the 4-anilinoquinoline and 4-anilinoquinazoline scaffolds through nucleophilic aromatic displacement of 4-chloroquin(az)olines. (Sch. 1) We were able to access products in good to excellent yields (55-91 %) consistent with previous reports and without protection of the alcohol substituted quin(az)oline starting material.11–13
General synthetic procedure
The first set of compounds probed a replacement of the 6-postion morpholine segment of gefitinib with a simple alcohol on the lapatinib scaffold (Tab. 2).20 Although neither the 6-OH (1) or 7-OH (2) quinazoline showed appreciable activity, the 6,7-dihydroxy compound (3) began to inhibit Mtb growth at higher concentrations. The analogous set of methoxy-substituted compounds (4-6) had very similar activity profiles. The 6-OH quinoline (7) showed improved activity relative to the matched quinazoline (1). The inclusion of fluorine substitution on the phenyl ring distal to the quinazoline (8-10) led to markedly increased activity for all three isomers. At 20 μM, 8-10 all reduced the relative Mtb signal to 25-37 %, and a modest but discernable reduction in signal was observed at 1.25 μM. Interestingly, the effect of fluorine substitution led to a different activity pattern from changing quinazoline ring substitution as the modification of 1 to the 7-OH (11) or 6,7-(OH)2 (12) resulted in a significant loss of activity, even at 20 μM. These results demonstrated that Mtb activity was sensitive to changes at multiple parts of the template and that these changes were not necessarily additive. We counter screened 1-12 in human skin fibroblast cells (WS-1) and observed very limited toxicity with 3 and 7 the only compounds in the single digit micromolar range (IC50 = 8.8 and 2.5 μM respectively).15
Results of alcohol replacement of lapatinib and gefitinib (1-12).
The next set of quin(az)olines profiled was prepared to explore the contributions from both the aniline and the core heterocycle (Tab. 3).11,16 The larger 3,4,5-trimethoxyphenyl aniline was employed on several diversely substituted quinolines and quinazolines (13-18) which led to no observable activity except when paired with the 6,7-(OCH2CH2OMe)2-quinazoline of erlotinib (19) which had only slight activity at 20 μM. On the other hand, incorporation of the aniline fragment from erlotinib (3-ethynylphenyl) did yield several active compounds, including the quinoline analog of erlotinib (20). The erlotinib aniline with a 6,7-(OMe)2-substituted quinoline core (21) or quinazoline (22) showed activity but not with the 3-cyanoquinoline (23). The analogous 3-bromophenyl aniline compounds (24-26) showed higher activity than the paired 3-ethynylphenyl compounds. Finally, the aniline substitution from lapatinib (3-Cl-4-(2-F-PhO)Ph) yielded a marginally active quinazoline (27) and a highly active quinoline (28) that had 10 % Mtb signal at 20 μM. We counter screened 13-28 in WS-1 cells and observed limited toxicity in most compounds. However, the bromine substitution appeared to increase toxicity (24-26) along with the lapatinib derivatives (27-28).
Matched pair comparison of structures similar to erlotinib and lapatinib (13-28).
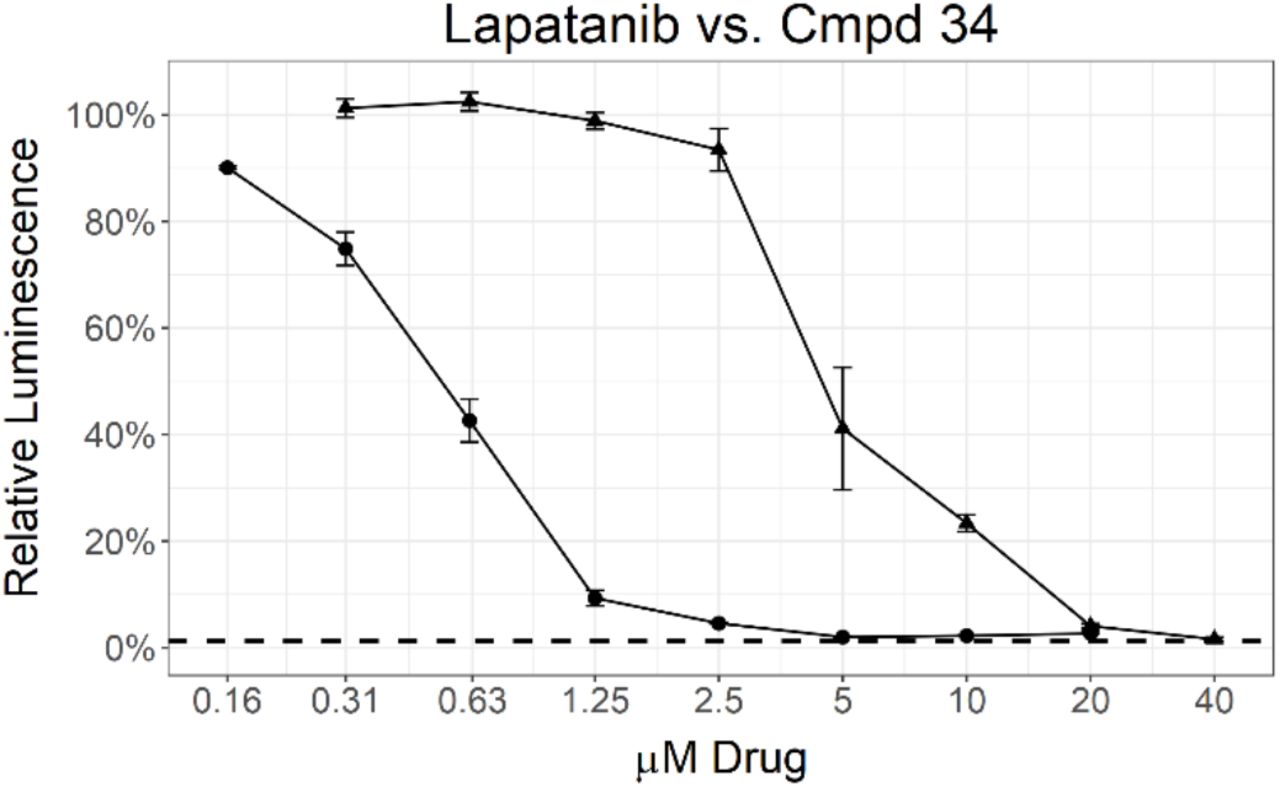
A subsequent set of compounds explored features of the 4-benzyloxyaniline portion of the quinoline template (Tab. 4).17–18 Variation of the ether linkage to an amide and addition of a hydroxy on the aniline portion revealed an activity pattern where 6,7-dimethoxy substitution with the benzylic arm was active (29-30). In contrast, truncation of the benzyl also removed the activity as in acetamide 31, which had no effect on Mtb. The 6-methoxy (32) and 7-methoxy (33) showed no reduction in Mtb signal at 20 μM. However, switching back to the 4-methyl benzyl ether linked compound 34 yielded the most potent activity observed with any compound in the present study, with a robust signal observed even at 1.25 μM. The Mtb MIC90 for 34 was in the 0.63-1.25 μM range. However, the kill curve plateaued at 5 μM, and no improved killing was observed at higher doses (98 % inhibition at 5 μM; 98 % inhibition at 20 μM) (Fig. 3).
Matched pair comparison of benzyloxyaniline.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MIC determination by two different assays for 34 (circle) and lapatanib (triangle). Data points represent the mean of 3 biological replicates with standard deviation. The dashed line labeled 1% inoculum represents an inoculation of 1 % of the number of cells used for compound testing. This control was used to determine 99 % inhibition of growth.
As with the other compound sets, we evaluated 29-34 in human skin fibroblast cells (WS-1) and observed moderate toxicity in the single digit micromolar range for most compounds.20 Importantly, the anti-Mtb effects of compounds appeared to be divergent from the toxic effects in WS-1 cells, suggesting that the Mtb effects were not driven by nonspecific cytotoxicity. The most potent anti-Mtb compound 34 had WS-1 IC50 = 5.4 μM, substantially higher than its Mtb MIC90 value and within threefold of the IC50 values for erlotinib and lapatinib. This result demonstrated that, in the human WS-1 cell line, 34 behaved comparably to two approved medicines.
These structure activity relationships between Mtb activity and the 4-anilinoquinoline/quinazoline scaffold have the potential to inform a medicinal chemistry strategy for enhanced Mtb activity. The most sensitive structural changes were found to be in the ring appended to the aniline rather than in the quin(az)oline core.
This body of work provides a number of exciting starting points for further optimization, with limited non-specific toxicity. However, the failure to achieve complete parasite kill led us to deprioritize the series due to the potential for resistance to develop. The mechanism of anti-Mtb activity of the quin(az)olines has yet to be defined. These compounds were originally prepared as inhibitors of human kinases targeting the ATP-binding site, a rational starting hypothesis for the mechanism of action is that the effects of these compounds are mediated by inhibition of Mtb kinases. However, it is possible that the observed phenotypes may originate from modulation of other, non-kinase ATP-binding proteins in the organism.
Gefitinib, erlotinib, and lapatinib have previously been reported to inhibit the intracellular growth of Mtb. Multiple lines of evidence were described suggesting inhibition of the host target epidermal growth factor receptor (EGFR) was responsible for this activity.15 However our results demonstrate that proteins within the pathogen itself may be targeted as well. The benzyl substituent present in the molecule showed a pivotal effect to potency, as is highlighted by the enhanced activity of 34 relative to 30. The present results help define a de-risked medicinal chemistry trajectory towards anti-tubercular compounds with targets in both the host and the parasite itself. Such dual acting compounds might offer advantages in efficacy and/or reduction in propensity for resistance.
Acknowledgments
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medi-cines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome [106169/ZZ14/Z]. C. G. is supported by R01 AI117023 from the NIH/NIAID. We are grateful Dr. Brandie Ehrmann for LC-MS/HRMS support provided by the Mass Spectrometry Core Laboratory at the University of North Carolina at Chapel Hill.
Footnotes
This version has been modified to include (a) compound evaluation in the WS-1 human skin cell line and (b) additional authors.




