

Abstract
Fibroblast-to-myofibroblast transition (FMT) is central to fibrosis. A divergent member of the polo-like kinase family, PLK4 is known for its canonical role in centriole duplication. Whether this mitotic factor regulates cell type transitions was underexplored. Here we investigated PLK4’s activation and expression and regulations thereof in platelet-derived growth factor (PDGF)- induced FMT of rat aortic adventitial fibroblasts.
PLK4 inhibition (with centrinone-B or siRNA) diminished not only PDGF AA-induced proliferation/migration, but also smooth muscle α-actin and its transcription factor serum response factor’s activity. While PDGFR inhibition abrogated AA-stimulated PLK4 activation (phosphorylation) and mRNA/protein expression, inhibition of p38 downstream of PDGFR had a similar effect. Further, the transcription of PLK4 (and PDGFRα) was blocked by pan-inhibition of the bromo/extraterminal-domains chromatin-bookmark readers (BRD2, BRD3, BRD4), an effect herein determined via siRNAs as mainly mediated by BRD4. In vivo, periadventitial administration of centrinone-B reduced collagen content and thickness of the adventitia in a rat model of carotid artery injury.
Thus, we identified a non-canonical role for PLK4 in FMT and its regulation by a BRD4/PDGFRα-dominated pathway. This study implicates a potential PLK4-targeted anti-fibrotic intervention.
Introduction
Fibrosis is an adverse condition that can occur in essentially any organ including the vasculature. Ample evidence indicates important contribution of vascular fibrosis to cardiovascular diseases such as atherosclerosis and hypertension. Myofibroblasts are recognized as the major pathogenic cell population in fibrotic diseases1. Though the definition of myofibroblasts is still debated, these cells are generally characteristic of smooth muscle-like morphologies and proliferative/migratory behaviors2. Moreover, they often exhibit high levels of smooth muscle α-actin (αSMA), vimentin, platelet derived growth factor receptor α (PDGFRα), and extracellular matrix proteins (e.g. collagen). A variety of cell types can differentiate into myofibroblasts1. However, resident fibroblasts were confirmed as the main source in recent in vivo lineage tracing studies, at least in some vital organs such as heart2, 3. Thus, identifying novel molecular targets that regulate fibroblast-to-myofibroblast transformation (FMT) is critical for developing viable anti-fibrotic options.
FMT is a complex process involving extracellular and cell membrane signaling, cytosolic pathways, epigenetic/transcriptional reprogramming, and interactions of these networks2. The master regulators (e.g. epigenetic mechanisms) that govern FMT remain poorly defined1. The transforming growth factor (TGFβ1) signaling is the best known fibrogenic factor. By contrast, the PDGF pathways are less well-understood in fibrogenesis2. There are four PDGF ligands (PDGF-A, B, C, D) that activate two different receptor tyrosine kinases, PDGFRα and PDGFRβ. The PDGF-AA homodimer selectively activates PDGFRα, which is abundant in mesenchymal and fibroblastic cells yet inadequately explored for its role in FMT and vascular fibrosis4.
Polo-like kinases regulate cell cycle entry and exit. Among the five PLK family members, PLK1 is the most studied, which participates in multiple steps of mitosis. By contrast, PLK4, the divergent family member with little homology to the other four PLKs, is much less studied5. Of note, reports on PLK4 are rapidly increasing. Though identified as a master regulator of centriole duplication6, PLK4’s non-canonical functions remain little known7. Whether this mitotic factor regulates FMT was not previously reported. Traces of recent evidence implicate a possible PLK/FMT link. For example, while FoxM1 is a critical driver of lung fibroblast activation8, PLK1 is one of its prominent target genes9. Both PLK4 and PLK1 have recently attracted considerable effort to develop their selective inhibitors for cancer therapy10-12. These inhibitors rendered studies readily practical to discover novel PLK functions.
In this study, we primarily focused on the unique and under-studied family member PLK4 to investigate its role in FMT and the associated regulators. We also included PLK1, the representative of the PLK family13. We found that PLK4 inhibition constrained the rat aortic fibroblast proliferative/migratory behaviors, and somewhat to our surprise, also the nuclear activity of serum response factor, a transcription factor key to myofibroblastic transitions2. We further revealed that PDGFR and a downstream kinase (p38) positively regulated PLK4 activation. In pursuit of its transcriptional regulators, we identified BRD4 as an epigenetic determinant. Hence, we have identified a non-canonical function of PLK4 in promoting FMT. Moreover, we also observed an effect of PLK4 inhibition on attenuating vascular fibrosis in a rat artery injury model. Thus, targeting PLK4 may hopefully develop into an interventional option for ameliorating fibrotic conditions.
Results
PLK4 inhibition blocks PDGF-AA stimulated FMT of rat aortic adventitial fibroblasts
Recent progress in developing highly selective PLK inhibitors provides powerful tools for deciphering PLK4 functions. For example, centrinone-B (herein abbreviated as Cen-B) is a novel PLK4-selective inhibitor (Ki = 0.6 nM) with very low affinities for other PLK and non-PLK kinases5. To induce FMT, we used PDGF-AA (hereafter abbreviated as AA) which predominantly activates PDGFRα vs PDGFRβ4. Compared to the best known FMT inducer TGFβ11, AA stimulated αSMA expression to a similar degree in rat aortic adventitial fibroblasts, validating its ligand functionality (Figure S1). Interestingly, whereas treatment of cells with AA induced a drastic morphological change and ~3-fold increases of proliferation and migration, all hallmarks of FMT, pretreatment with Cen-B concentration-dependently abrogated these changes (Figure 1, A-C). Furthermore, AA-stimulated upregulation of αSMA and vimentin proteins (2.5-5 fold) was also abolished by pretreatment with Cen-B (Figure 1D).

Rat primary adventitial fibroblasts were cultured in the complete medium, starved in the basal medium overnight (see Methods), and pretreated for 2h with vehicle (equal amount of DMSO) or the PLK4-selective inhibitor centrinone-B (Cen-B) at indicated concentrations, followed by stimulation with 60 ng/ml PDGF-AA (abbreviated as AA throughout). Cells were harvested at 24h after stimulation (or otherwise specifically indicated) for various assays. A. Morphology. Cells were stimulated (or not) by AA for 24h without or with pre-treatment (1µM Cen-B). Green fluorescent calcein was used to illuminate cell morphologies. B. Proliferation. CellTiterGlo assay was performed after 72h AA stimulation of cells without or with pretreatment by Cen-B at increasing concentrations. C. Migration (scratch assay). Cells were stimulated (or not) by AA for 24h without or with pre-treatment (1 or 10 µM Cen-B). Calcein was used to illuminate the cells. D. Western blots of αSMA and vimentin. Protein band densitometry was normalized to loading control (β-actin), and then to the basal condition (DMSO, no AA), and finally quantified as fold change. Fold changes from at least 3 independent repeat experiments were averaged, and mean ± SEM was calculated. E. Effect of PLK4 silencing on αSMA. Cells were transfected with scrambled or PLK4-specific siRNA for 48 h in the complete medium, starved overnight, and then stimulated with AA for 10 min before harvest for Western blotting. F. Effect of Cen-B on MRTF-A. Cells were pretreated with 1µM Cen-B (or vehicle) and then stimulated (or not) with AA for 48 h. G. Luciferase reporter assay of SRF transcriptional activity. Cells (HEK293) were transfected with the empty vector control or the SRF-Luciferase vector, followed by luminescence reading. The condition with 5 µM tubastatin-A, an HDAC6 inhibitor and a novel SRF stimulator14, served as a positive control. Data are presented as mean ± SEM (n ≥ 3 experiments); *P <0.05.xx
Importantly, confirming a PLK4-specific function, silencing PLK4 with siRNA reduced αSMA protein levels (Figure 1E). While it was intuitive that PLK4 as a mitotic factor promoted cell proliferation5, a role for PLK4 in elevating αSMA expression was somewhat unexpected given its canonical association with the microtubule system (centrioles). We were thus prompted to further explore the mechanism underlying this new observation. MRTF-A is a powerful regulator that shuttles to the nucleus to activate SRF, the master transcription factor that governs αSMA gene transcription14. We therefore investigated the influence of PLK4 on MRTF-A protein and SRF transcriptional activity. Our data showed that while treatment with AA elevated MRTF-A protein levels, PLK4 inhibition with Cen-B averted this effect (Figure 1F). Further, the SRF transcriptional (luciferase) activity was diminished by Cen-B (Figure 1G). These results for the first time revealed that PLK4 promotes SRF activation and αSMA production at least in part by elevating MRTF-A protein levels in rat aortic adventitial fibroblasts.
Taken together, the results presented herein indicate that PLK4 inhibition attenuates FMT and somewhat unexpectedly, also SRF transcriptional activity. To the best of our knowledge, this non-canonical PLK4 function was not previously reported. It is also noteworthy that pretreatment with Cen-B largely preserved normal fibroblastic phenotypes and did not cause obvious cell death even at high (e.g.10 µM) concentrations (data not shown), suggesting a low cytotoxicity of this drug.
We also determined the effect of PLK1 inhibition on FMT (not previously known either) using the PLK1-selective inhibitor GSK461364 (Ki = 2.2 nM, hereafter abbreviated as G-4)11. The result (Figure 2) was similar to that of Plk4 inhibition. However, the PLK1 inhibitor at high concentrations (e.g. 0.5 µM) reduced cell viability to much below the non-stimulated basal level (Figure 2C), consistent with its cytotoxicity11.

Experiments were performed as described in Figure 1 except that the PLK1-selective inhibitor GSK461364 (abbreviated as G-4) was used for pretreatment prior to AA stimulation. A. Western blots of αSMA and vimentin. B. Morphologic comparison. Cells were stimulated (or not) by AA for 24h without or with pre-treatment (1µM G-4). C. Proliferation. CellTiterGlo assay was performed after 72h AA stimulation of cells without or with pretreatment by G-4 at increasing concentrations. D. Migration (scratch assay). Cells were stimulated (or not) by AA for 24h without or with pre-treatment (0.1 or 1 µM G-4). Data quantification followed the method in Figure 1; mean ± SEM (n ≥ 3 experiments); *P <0.05.
Blocking PDGFR kinase activity abrogates AA-stimulated PLK4 activation
Given the profound effects of blocking PLK4 kinase activity on FMT, it is important to understand how the activation and expression of PLK4 are regulated. Unfortunately, literature information in this regard is very limited. In our experimental setting, the FMT inducer PDGF-AA activates PDGFRα, a well-known “gateway” receptor on the cell surface that activates myriad of intracellular pathways2, 4. However, whether PDGFRα regulates PLK4 was not previously known. We addressed this question using a kinase inhibitor (Crenolatib; abbreviated as Crenol) selective to both PDGFRα and PDGFRβ (Ki <10 nM)15. PDGFRα-specific compounds are not yet commercially available.
As shown in Figure 3A, phosphorylation of PDGFRα (at Y754, commonly seen as an upper band)15 was quickly stimulated by AA within 5 min and declined to the basal level in 20 min. AA also quickly stimulated phosphorylation of MEK/ERK, JNK, and p38 with a time-course pattern similar to that of PDGFRα, consistent with literature evidence from other cell types16, 17. Thus, this result validated our experimental setting. Of note, phosphorylation of AKT and S6K responded to AA stimulation in a delayed manner (Figure 3A), probably as a secondary signaling event. Interestingly, treatment with AA activated PLK4 (phosphorylation at T170)18 and PLK1 (phosphorylation at T210)19 in the same time scale as did PDGFR〈(Figure 3A). Importantly, while the PDGFR-selective inhibitor Crenol blocked AA-induced activation of PDGFRα as well as MEK/ERK, JNK, AKT and S6K, validating the inhibitor functionality (Figure 3B), this inhibitor also abrogated AA-stimulated activation of PLK4 and PLK1 (Figure 3, C and D), placing them downstream of PDGFRα signaling. Of note, the phospho-PLK4 band most sensitive to AA and Crenol ran at a high position on the blot. Inasmuch as activated PLK4 dimerizes tightly to stabilize the protein20, it is tempting to speculate that a transient oligomer may form before its disassembly and degradation. Indeed, PLK4-positive bands higher than a theoretical motility position has been shown in other reports as well21, 22, and recent crystal structures showed a strand-swapped dimer of dimers of the PLK4 PB3 domain23, which is absent in other PLKs.
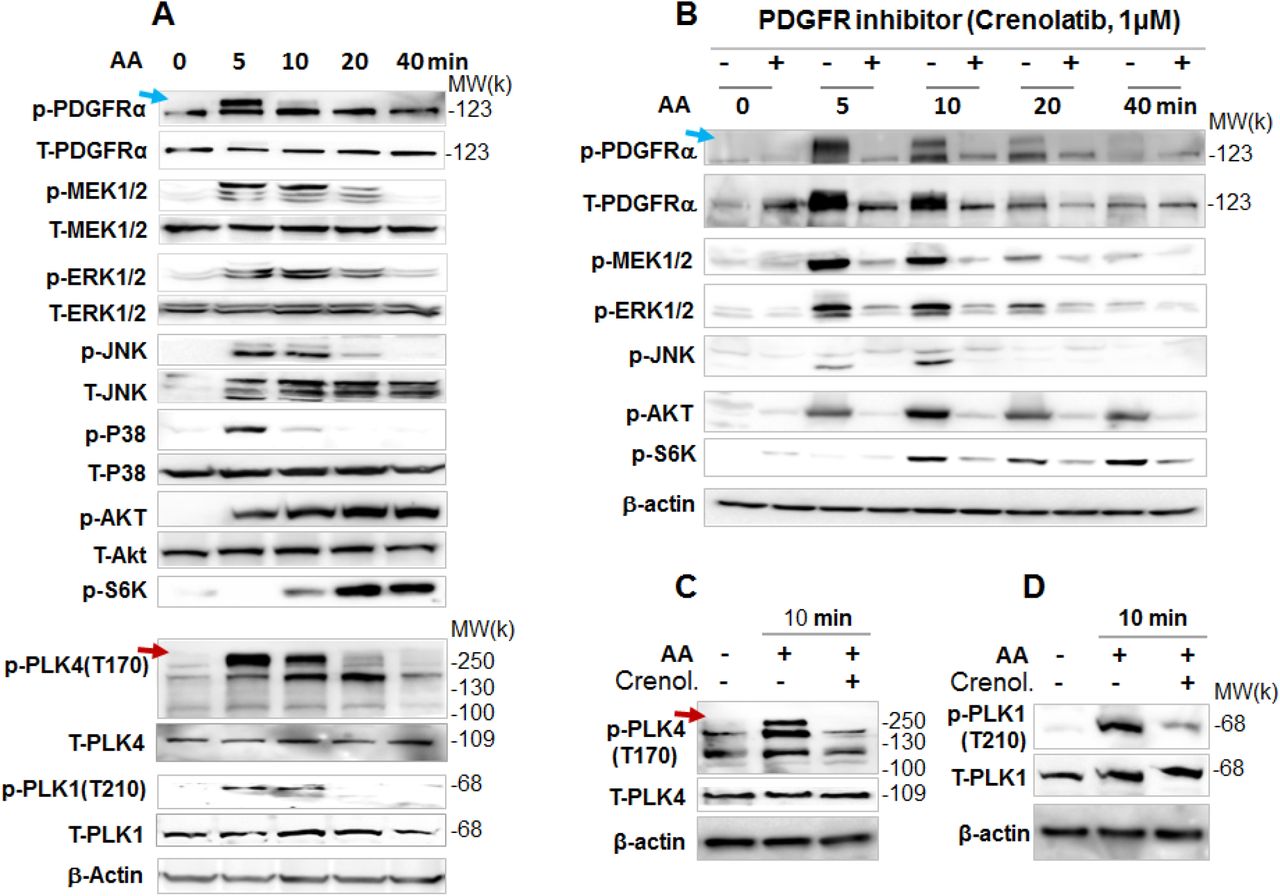
Rat primary adventitial fibroblasts were cultured, starved, and pretreated with vehicle (DMSO) or an inhibitor, as described in Figure 1. A. Time course of AA-induced activation (phosphorylation) of kinases. Western blots detect a phospho-protein (p-) and its respective total protein (T-). Arrows point to phospho-protein bands that were sensitive to AA stimulation. B. Blockade of AA-induced kinase activation by the PDGFR-selective inhibitor Crenolatib (abbreviated as Crenol). Starved cells were pretreated with vehicle or 1 µM Crenol for 30 min prior to AA stimulation. C and D. Blockade of AA-stimulated (10 min) activation of PLK4 (phosphorylation at T170) and PLK1 (phosphorylation at T210) by pretreatment with Crenol (1 µM, 30 min).
PLK4 activation is regulated by PDGFR downstream kinase activity but not vice versa
To delineate the position of PLK4 in the signaling cascade downstream of PDGFR, we first tested whether its blockade affects the activation of the MAPK and AKT/S6K pathways. Our data showed that pretreatment with either the PLK4 inhibitor (Cen-B) or the PLK1 inhibitor (G-4) did not appreciably alter AA-induced phosphorylation of the MAPK pathway kinases (MEK/ERK, JNK, p38) or AKT within 40 min (Figure 4, A and B). However, PLK1 (but not PLK4) inhibition abolished AA-initiated S6K phosphorylation (Figure 4B), suggesting differential functions of these two PLKs. We then dissected which of the PDGFR downstream kinase(s) regulated PLK4 activation, by using a panel of their respective inhibitors. Interestingly, the p38 inhibitor most prominently reduced AA-stimulated PLK4 phosphorylation whereas the other inhibitors did not produce a significant effect (Figure 4C). By contrast, PLK1 phosphorylation was effectively blocked essentially by every tested inhibitor (Figure 4D).

A and B. Lack of effect of PLK4 (or PLK1) inhibition on the activation of PDGFRα and downstream kinases. Time-course experiments were performed as described in Figure 3, except that 1 µM Cen-B (PLK4 inhibitor) or 1 µM G-4 (PLK1 inhibitor) were used for a 30-min pre-treatment prior to AA stimulation. C and D. Effects of various kinase inhibitors on the activation of PLK4 and PLK1. Cells were pretreated with an inhibitor of JNK (SP600125), PI3K (LY294002), mTOR (rapamycin), p38 (SB230580), or MEK1/2 (PD98059) for 30 min at 5 µM prior to a 10-min AA stimulation. Quantification: mean ± SEM, n = 4-5 independent experiments; *P <0.05.
Blocking PDGFR abrogates AA-stimulated PLK4 protein production
To investigate the long-term regulation of PLK4, we next determined whether PDGFR regulates PLK4 protein levels. As shown in Figure 5, while treatment with AA for 24h substantially elevated protein levels of PLK4 and also PLK1, pretreatment with the PDGFR blocker Crenol concentration-dependently inhibited PLK protein upregulation (Figure 5A). Serving as positive control, PDGFR inhibition markedly reduced the protein levels of MEK, ERK, JNK, and p38. In studies to dissect the PDGFR downstream pathways that possibly regulated PLK4 protein expression, we found that pretreatment with inhibitors of these kinases differentially altered PLK4 and PLK1 protein levels to varied extents (Figure 5, B-F). Whereas PLK1 was sensitive to JNK and mTOR inhibitors, PLK4 was more sensitive to the p38 inhibitor.
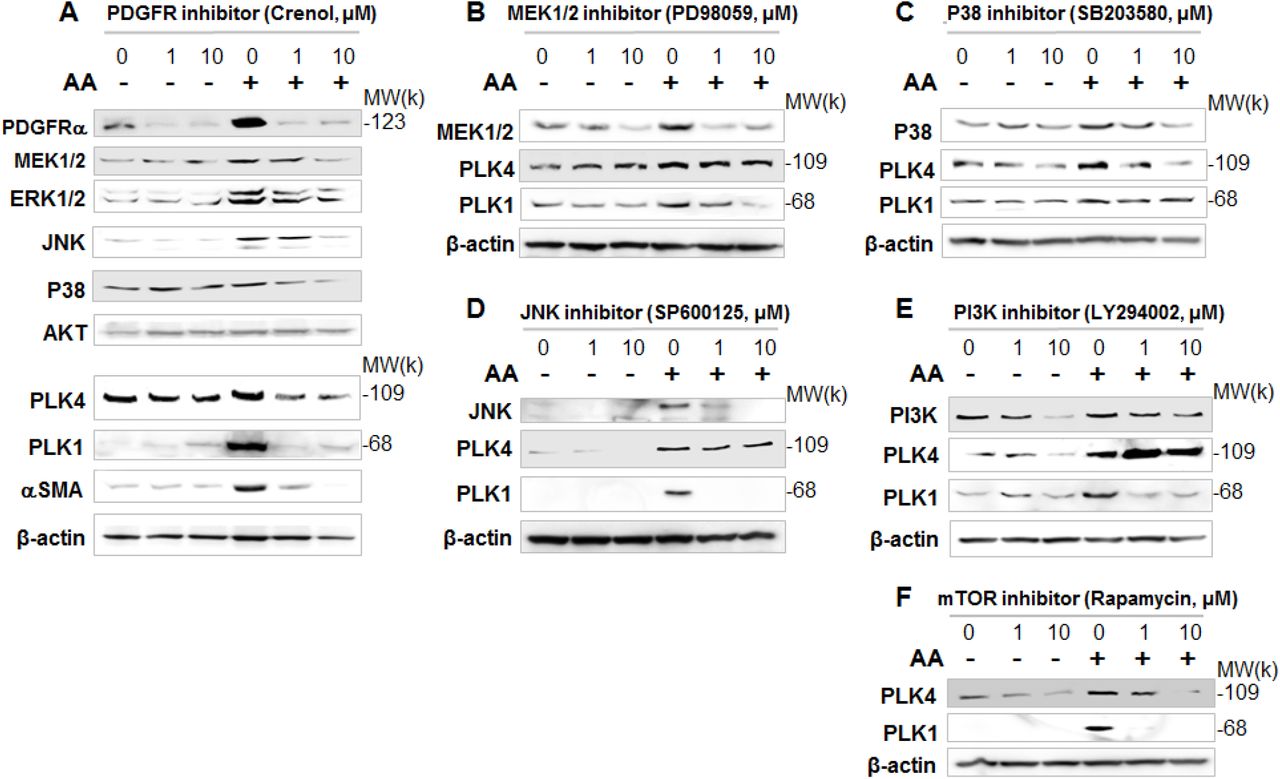
Rat adventitial fibroblasts were cultured and starved as described in Figure 1. Cells were pre-treated for 30 min with vehicle or an inhibitor for PDGFR (A), MEK1/2 (B), p38 (C), JNK (D), PI3K (E), or mTOR (F) at indicated concentrations, stimulated for 24h with AA, and then harvested for Western blot analysis. Shown are representative blots from two or three similar repeat experiments.
Up to this point, our data revealed a novel finding that PDGFRs (most likely PDGFRα) positively regulates PLK4 and PLK1 not only in a shorter term (PLK activation) but also in a longer term (protein production).
Silencing FoxM1 does not repress the expression of of PLK4
We next investigated the transcriptional regulation of PLK4. FoxM1 is a well-known transcriptional activator of mitotic regulatory genes, including cyclin B1, Topo2, Aurora B kinase, and also PLK19. It was therefore tempting to determine whether PLK4 is a FoxM1 target gene as well, a question not previously addressed. Our data showed efficient FoxM1 knockdown at mRNA and protein levels, which substantially inhibited PLK1 mRNA and protein expression (Figure 6), consistent with previous reports8, 9. However, FoxM1 knockdown did not reduce but rather increased PLK4 mRNA. Thus, these results implicated differential transcriptional regulations of PLK4 and PLK1.

Rat adventitial fibroblasts were transfected with scrambled or FoxM1-specific siRNA for 48h and then collected for qRT-PCR (A) and Western blot (B) assays. Quantification in A: Mean ± SD, n =3; one of two similar repeat experiments. Quantification in B: Densitometry was normalized to loading control (β-actin); data is presented as average of two repeat experiments.
Pan-BETs inhibition blocks AA-stimulated FMT of rat aortic adventitial fibroblasts
To further identify key regulatory mechanisms that control PLK4 transcription, we explored the role of BETs. While cell identities are governed by specific transcription programs, recent research discovered the BET family as a master epigenetic regulator that directs transcription programs in a cell type and environment dependent manner24-27. Moreover, recent evidence supports an important role of BETs in activation of fibroblasts28, 29 albeit with no data obtained from vascular fibroblasts. As shown in Figure 7 (A-D), pretreatment with JQ1 (the first-in-class inhibitor selective for BETs)30 abrogated AA-induced FMT phenotypes, including αSMA and collagen expression, and cell migration and proliferation. Apparently, pan-BETs inhibition with JQ1 “phenocopied” the effect of PLK4 inhibition with Cen-B (Figure 1). We were therefore motivated to determine the effect of BETs inhibition on PLK4 (and PLK1) expression.

Rat adventitial fibroblasts were cultured and starved as described in Figure 1. Cells were pre-treated with vehicle control or JQ1 (1 µM or otherwise specified) overnight before AA stimulation and cell harvest for various assays. A and B. Western blots and qRT-PCR showing JQ1 blockade of AA-stimulated αSMA expression. Quantification: densitometry was normalized to GAPDH; mean ± SD of triplicates. C. Migration (scratch assay). Cells were stimulated (or not) by AA for 48h without or with pre-treatment (1 µM JQ1). Calcein was used to illuminate the cells. Mean ± SD of triplicates. D. Proliferation. CellTiterGlo assay was performed after 72h AA stimulation of cells without or with pretreatment by increasing concentrations of JQ1. Mean ± SD of triplicates. E. Western blots indicating that pretreatment with JQ1 (1 µM, 2h) abrogated AA-stimulated (24h) protein production of PDGFRα, PLK4, PLK1, and αSMA. Shown are representative blots of two similar repeat experiments. Densitometry was normalized to β-actin; mean ± SD of triplicates. F. qRT-PCR data indicating that pretreatment with JQ1 (1 µM, 2h) abrogated AA-stimulated (24h) mRNA expression of PDGFRα, PLK4, PLK1, and α SMA. Mean ± SD of triplicates.
BRD4 predominantly controls the transcription of PLK4 and PDGFRα
Interestingly, pretreatment with 1 µM JQ1 completely averted AA-induced upregulation of the mRNAs and proteins of PLK4 (and also PLK1), a result not previously reported (Figure 7, E and F). Moreover, AA-stimulated PDGFRα mRNA/protein expression was also effectively reduced by pretreatment with JQ1.
There are four BET family proteins, including BRD2, BRD3, BRD4, and BRD-T which is testis-specific and not relevant here. The inhibitor JQ1 binds to all of the BETs30. To dissect out which BET was responsible for the potent effect of the pan-BETs inhibitor JQ1, we performed genetic silencing experiments using siRNAs. Excellent knockdown efficiency of siRNAs respective for BRD2, BRD3, and BRD4 was shown in Figure S4. The data in Figure 8A showed that silencing BRD4 substantially reduced protein levels of PLK4, PLK1, PDGFRα, and αSMA. Silencing BRD2 slightly reduced the protein of PLK4 but not that of PLK1, PDGFRα, and αSMA. Silencing BRD3 did not reduce and appeared to have slightly increased these proteins. The robust inhibitory effect of silencing BRD4 on PLK4 and other three proteins is indicated by the quantitated data in Figure 8B. Silencing either BRD2 or BRD4 significantly reduced the mRNA levels of PLK4, PLK1, and PDGFRα (not significant with siBRD2), and αSMA but not that of FoxM1 (Figure 8, C and D). Given these results, BRD4 appeared to be the predominant BET governing the gene transcription of the two PLKs and PDGFRα.

Rat adventitial fibroblasts were transfected with a scrambled or BRD-specific siRNA for 48h and collected for Western blot (A) and qRT-PCR (B) assays. A. Comparison of the effects of silencing BRD2, BRD3, or BRD4 on PLK4 protein levels. Shown are representative blots of two similar repeat experiments. B. Silencing BRD4 reduces the proteins of PDGFRα, PLK4, PLK1, and αSMA. Quantification: densitometry normalized to β-actin; mean ± SEM; n=3-5 independent repeat experiments. C and D. Effects of BRD2 or BRD4 silencing on mRNA levels of PLK4, PLK1, PDGFRα, and αSMA (qRT-PCR data). Each bar value represents a mean ± SD of triplicates from one of two repeat experiments.
Periadventitial administration of PLK4 inhibitor ameliorates vascular fibrosis in the rat carotid artery injury model
After identifying the role for PLK4 in FMT and the regulators of its activation and expression, we finally determined whether PLK4 inhibition helps reduce fibrosis in vivo. We used a well-established rat arterial injury model where injury-induced fibrosis manifests as high collagen content the adventitia. Balloon angioplasty was performed in rat common carotid arteries, and the PLK4 inhibitor (Cen-B) or vehicle control was administered in a thermo-sensitive hydrogel distributed around the injured artery, following our previous report26. Cross-sections of injured arteries collected at day 7 were stained using the Masson’s trichrome method31 (Figure 9, A and B). Quantified data indicate that the collagen content (staining intensity normalized to artery perimeter) was significantly reduced in Cen-B treated arteries compared to vehicle (DMSO) control. Consistently, the adventitia thickness also significantly decreased after Cen-B treatment (Figure 9C). No significant change was observed in neointimal hyperplasia (measured as the neointima/media area ratio)26. These results indicate that in this model of rat carotid artery injury, periadventitial application of the PLK4 inhibitor Cen-B was able to ameliorate vascular fibrosis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Following balloon angioplasty of the rat common carotid artery, the PLK4 inhibitor (Cen-B, 100 µg per rat) dissolved in a hydrogel mix was applied around the adventitia of the injured artery. Arteries were harvested at day 7 post injury; cross sections were stained using the Masson’s Trichrome method. A. Representative sections from the arteries treated with vehicle (equal amount of DMSO) or Cen-B. Collagen is stained blue; the adventitia thickness is indicated by two arrows. The anatomy of the artery wall is labeled as A (adventitia), M (media), and N (neointima). B. Magnified portions of the images in A. C. Quantification. Collagen content (staining intensity) and thickness of the adventitia was normalized to the overall vessel size measured as the length of the external elastic lamina (the border between blue and red layers). Neointimal hyperplasia was measured as the intima/media area ratio (I/M). Student’s t-test: *P<0.05, n = 5 animals per group.
Discussion
All major diseases involve fibrosis where FMT is a key fibrogenic event1. To date there remains a lack of effective clinical methods for treating fibrosis2. Identification of new FMT mechanisms is therefore imperative for the pursuit of viable anti-fibrotic therapy. Here we have found that PLK4 provides a novel target for molecular intervention of FMT. Specifically, our major findings are summarized as follows. (1) PLK4 inhibition blocked PDGF(AA)-induced FMT. (2) Though previously known as centriole-specific, PLK4 positively regulated SRF nuclear activity and αSMA transcription. (3) Upon AA stimulation, PLK4 was activated by PDGFR and downstream kinase p38. (4) The transcription of PLK4 was predominantly governed by the epigenetic reader BRD4. In aggregate, our results for the first time revealed a non-canonical role for PLK4 in promoting FMT and SRF nuclear activity. Further, we uncovered a BRD4/ PDGFR-dominated mechanism underlying PLK4 expression/activation. Importantly, in an arterial injury model PLK4 inhibitor attenuated vascular fibrosis, suggesting a potential anti-fibrotic approach.
A non-canonical role for PLK4 in FMT is intriguing. The PLK4’s canonical role in centriole duplication6 is being elaborated32. PLK4 phosphorylates and complexes with STIL which in turn recruits SAS6 forming the core module of centriole genesis. However, non-canonical PLK4 function is little known7, 33. Thus far, few PLK4’s substrates beyond centriolar proteins have been identified33. Thus, PLK4 is much less understood in contrast to PLK1, the best studied PLK member that mediates multiple mitotic processes5, 32. Functional difference between PLK4 and PLK1 is rationalized by the fact that PLK4 is a peculiar member within the PLK family because of its unique domain structure and divergent coding sequence5, 10. Likely for this reason, knowledge gained on other PLKs (mostly on PLK1) cannot be simply extrapolated to PLK4. In light of PLK4 being a mitotic factor, it is intuitive that PLK4 is pro-proliferative5 in vascular fibroblasts as observed herein, and in cancer progression as previously reported12, 34. PLK4 was recently also linked to cancer cell migration and invasion33, consistent with our result that PLK4 promoted vascular fibroblast migration. On the other hand, FMT is a process beyond cell proliferation and migration. It is remodeling in signaling and transcription programs, e.g. expression of non-fibroblastic proteins typical of the contractile protein αSMA2. As such, FMT represents cellular reprogramming that alters cell type with multiple phenotypic changes. In this regard, it is an unanticipated finding that PLK4, known as a centriole-specific actor, enables a profound cell type transformation (FMT) in a non-canonical manner.
Elevated expression of αSMA endows cells with a contractile function, which is effectuated by the master transcription factor SRF2. Consonantly, our data showed that while PLK4 inhibitor profoundly reduced SRF’s activity, PLK4 silencing substantially repressed αSMA levels. This result is unexpected and counter-intuitive, given that PLK4 is known as centriole-associated and hence cytosol-localized whereas SRF is a chromatin-associated nuclear protein. Interestingly, this paradox may be at least partially explained by our novel observation that PLK4 promotes protein levels (or stability) of MRTF-A. This co-factor of SRF is a cytosol-nucleus shuttling protein and may thereby convey PLK4’s cytosolic function to the nucleus where SRF activation and αSMA transcription occur.
To the best of our knowledge, this non-canonical PLK4 function has not been previously reported. The finding is provocative, particularly in view of that the SRF/αSMA transcriptional remodeling is both phenotypically and mechanistically distinct from cell proliferation and migration. Though indirect, supporting evidence can be traced to the literature. The closest is a recent report in cancer research where PLK4 was found to play a role in cytoskeleton re-organization and lamellipodia formation in HeLa cells33. PLK4 interacted with the Arp2/3 complex altering actin cytoskeleton dynamics, and the authors used the result to explain the PLK4 function in cancer cell migration. Similarly, an earlier study showed that PLK4 expression enhanced the polarity, spreading, and invasion of colon cancer cells35. However, whether PLK4 played a role in αSMA transcription was not examined in either of these studies. Another line of evidence is from a very recent report that PLK4 upregulation promoted epithelial cell state transition in neuroblastoma36. Although this report showed that PLK4 enhanced vimentin expression, there was no data on αSMA transcription nor on SRF activity. In addition, a new report demonstrated that PLK1 positively regulated angiotensin-II activation of RhoA and actomyosin dynamics in murine vascular smooth muscle cells13. However, caution must be taken into the analogy of PLK4 with PLK1. Literature evidence and our own results indicate that though in the same family, PLK4 and PLK1 functionally vary, especially in different cell types or states. Nevertheless, the positive regulation of MRTF-A/SRF by PLK4 in the FMT context represents a distinct mechanism that may or may not cross-talk with those previously reported. In this light, a PLK4 non-canonical role in regulating MRTF-A and SRF nuclear activities warrants future studies for further elaboration.
Given PLK4’s critical role in centrosome biogenesis and its potential non-canonical functions, this kinase is expected to be intricately regulated37. Hitherto relatively little is known as to how PLK4 is functionally and transcriptionally regulated in mammalian systems18, 38. We found that PLK4 activation (phosphorylation at T17018, 37) by AA treatment was blocked by PDGFR inhibition. Because AA specifically activates the PDGFR αα homodimer4, we inferred that it was primarily the activation of PDGFRα that led to PLK4 activation. We reasoned that PDGFRα should have not activated PLK4 directly because it is a receptor tyrosine kinase4 which is not expected to phosphorylate threonine-170 of PLK4. Downstream of PDGFR, p38 appeared to be an activator of PLK4, as evidenced by experiments using either a p38 inhibitor or siRNA. On the other hand, PLK4 inhibition did not affect phosphorylation of PDGFRα and p38, placing PLK4 downstream of the PDGFR/p38 signaling. Our data do not mandate on whether PLK4 is a direct substrate of the p38 kinase, as definitively addressing this question requires an assay using both purified proteins. Although a p38-mediated PLK4 activation was not previously reported, consistent evidence is from a recent in vivo study where p38 proved to be a critical activator of FMT and SRF-directed αSMA expression39. Consistent evidence is also provided by our result that inhibiting either PDGFR or p38 averted AA-induced PLK4 protein upregulation. In contrast, pretreatment with inhibitors of other PDGFR downstream pathways (e.g. MEK/ERK and JNK) did not produce an obvious effect. Taken together, our results have profiled a PDGFR->p38->PLK4->MRTF-A->SRF signal transduction pathway.
In pursuit of the molecular mechanism underlying transcriptional regulation of PLK4, we distinguished that transcription factor FoxM1 directs the mRNA (and protein) expression of PLK1 but not PLK4. Instead, we made another novel finding that BRD4 in the BET family played a predominant role in governing PLK4 gene transcription. This is consistent with previous reports indicating a positive role of BETs in FMT. These studies showed that BET family inhibitors mitigated FMT and fibrosis in vital organs such as lung, liver, pancreas, and heart27-29, 40. Our study distinguishes from these reports by addressing the unanswered questions: 1) Whether BETs regulate PDGFRs were not known; 2) a BET regulation of PLK4 (or PLK1) expression remained unexplored. In light of our data showing that silencing BRD4 effectively reduces mRNA and protein levels of PDGFRα, PLK4, and αSMA, BRD4 appears to play a master role in governing this entire PLK4 pathway. Indeed, in the recent literature BRD4 emerges as a master epigenetic regulator in a variety of cell state/type transitions25-28. While a cell type/state transition (e.g. FMT) often results from environmental perturbations, environmental cues (e.g. PDGF) and the resultant transcription reprogramming represent two ends of this event while an epigenetic regulator such as BRD4 functions at their interface. Upon stimulation, BRD4 acts as a key organizer of trans- and cis-elements (e.g. super-enhancers) and the core transcription machinery, and (re)localizes them to specific sets of genes to activate their transcription. The two tandem bromodomains of BETs (blocked by JQ1) “usher” this transcription assembly to target genes by binding to bookmarked (acetylated) chromatin loci. This BRD4-directed mechanism has been recently recognized as critical in effectuating cell state transitions associated with various pathobiologic contexts24-28. As indicated by our data, BRD2 also participated in regulating the PLK4 pathway but played a lesser role (vs BRD4). Due to a paucity of information regarding BRD2 molecular operations, the mechanism behind the functional difference observed here between BRD2 and BRD4 awaits future elucidation.
Another interesting finding derived from our data is the differential mechanisms involving PLK4 and PLK1. While both are under the control of BRD4 and both participate in FMT, there are differential regulations of these two PLKs. For example, in terms of kinase activation, whereas PLK1 is generally sensitive to the tested inhibitors of the PDGFR downstream kinase, PLK4 appears to be more specifically regulated by p38. At the transcription level, PLK1 but not PLK4 is regulated by FoxM1, a powerful transcription factor in mitotic regulation8, 9. These differential results seem to be reasonable considering that whereas PLK1 participates in multiple steps of mitosis, the canonical role of PLK4 has thus far been identified as specific for centriole duplication. While interesting, differential PLK4 and PLK1-associated molecular and cellular regulations are off the scope of the current study given our focus on PLK4.
Conclusions
We have made an unexpected finding that PLK4, a kinase traditionally known as a centriole duplication factor, regulates cell type transitions of vascular fibroblasts under PDGF-AA stimulation. The significance of our study is three-fold. First, a positive role of PLK4 in MRTF-A/SRF activation represents a novel non-canonical PLK4 function. In an extrapolated sense, PLK4 possibly moonlight other yet to be discovered functions as well. Second, while PLK4 is a central player in the PDGFRα->p38->PLK4->SRF pathway that prompts PDGF-induced αSMA expression, BRD4 as an epigenetic determinant governs the entire pathway by coupling the PDGF environmental stimulation to transcriptional remodeling. In this regard, PLK4 appears to be a novel signaling effector in sensing and transmitting environmental cues. Third, consistent with the in vitro role of PLK4 in FMT, PLK4 inhibition mitigates vascular fibrosis in vivo. Now that clinical tests are ongoing for PLK4 as an anti-cancer therapeutic target10, 12, 32, rectifying PLK4 activity may provide a new option for developing anti-fibrotic interventions.
Methods
Animals and ethics statement
Male Sprague–Dawley rats were purchased from Charles River Laboratories (Wilmington, MA), housed and fed under standard conditions, and used for in vivo experiments at body weights of 300–330 g. All animal studies conformed to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health) and protocols were approved by the Institutional Animal Care and Use Committee at The Ohio State University. Isoflurane general anesthesia was applied during surgery (through inhaling, at a flow rate of 2 L/minute), and buprenorphine was subcutaneously injected (0.03 mg/kg, ~0.01 mg/rat) after the surgery. Animals were euthanized in a chamber gradually filled with CO2.
Rat aortic adventitial fibroblast cell culture, induction of FMT, and pretreatment with inhibitors
Primary aortic adventitial fibroblasts were isolated from 6-8 weeks old male Sprague-Dawley rats. For cell expansion, the culture was maintained at 37°C/5% CO2 in Complete Fibroblast Medium (Cat. M2267, Cell Biologics Inc.) containing growth factor supplement and 10% fetal bovine serum (FBS, Cat. 6912, Cell Biologics Inc.); 0.25% Trypsin-EDTA solution (Cat. #25200114, Life technologies, Carlsbad, CA.) was used for cell detachment. The fibroblasts at passage 5 were used for experiments. For induction of FMT, cells were first starved overnight in Fibroblast Basal Medium (Cat. 2267b, Cell Biologics Inc.) that contains no FBS, and then stimulated with 60 ng/ml PDGF-AA (rat recombinant, R&D Systems Inc., MN) for specifically indicated (in figures) length of time. In the experiments using various inhibitors (Table S1), prior to adding PDGF-AA cells were pretreated with an inhibitor for 2h (at an indicated concentration) or vehicle control (equal volume of DMSO, Sigma-Aldrich, St. Louis, MO).
Cell proliferation and migration assays
Cell viability assay was performed using the CellTiterGlo kit (Cat. G7571, Promega, Madison WI), as we previously reported26. Briefly, 72h after PDGF-AA treatment, plates were decanted and re-filled with 50 μl of CellTiter-Glo reagent and 50 μl PBS per well. Plates were incubated at room temperature for 20 min and then read in FlexStation 3 Benchtop Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA) by using a 250-ms integration.
Cell migration was measured using the scratch assay following our previous report26. Briefly, cells were cultured to 90% confluence in 6-well plates and then starved for 24 h in Fibroblast Basal Medium. A sterile pipette tip was used to generate an ~ 1 mm cell-free gap. Dislodged cells were washed away with PBS. Plates were then refilled with Fibroblast Basal Medium containing 20 ng/ml PDGF-AA and no FBS and incubated for 24 h. For pre-treatment prior to PDGF-AA stimulation, an inhibitor or vehicle control (equal amount of DMSO) was incubated with the cells for 2h or otherwise specified. For illumination of the cells, Calcein-AM was added (final 2 μM) and incubated for 30 min at the end of the PDGF-AA treatment, and images were taken after 3 times of wash with PBS. Cell migration was quantified with ImageJ (NIH) based on the width of the cell-free gap.
Western blotting to assess protein levels
Western blot analysis was done as we previously described26. Briefly, cells were harvested and lysed on ice in the RIPA buffer (Cat.89900, Thermo Fisher) that includes a protease inhibitor cocktail (Cat.87785, Thermo Fisher). Cell lysates were quantified for protein concentrations using the Bio-Rad DC™ Protein Assay kit (Cat.5000112, BioRad) and loaded to a 10% SDS-PAGE gel. The full primary antibody list is shown in Table S2. Beta-actin or GAPDH was used for loading control. The signals from primary antibodies were amplified by horseradish peroxidase (HRP)-conjugated immunoglobulin G (IgG) (Bio-Rad) and illuminated with Pierce ECL Western Blotting Substrates (Thermo Fisher). Blot images were immediately recorded with Azure C600 Imager (Azure Biosystems). Protein band densitometry was quantified using NIH ImageJ and normalized to loading control for statistical analysis.
Quantitative real-time PCR (qRT-PCR) to determine mRNA levels
We followed the method described in our previous report26. Briefly, total RNA was isolated from cultured cells using the Trizol reagent (Invitrogen, Carlsbad, CA). Potential contaminating genomic DNA was removed by using gDNA Eliminator columns provided in the kit. RNA was quantified with a Nanodrop NP-1000 spectrometer, and 1 µg was used for the first-strand cDNA synthesis. Quantitative RT-PCR was then performed using Quant Studio 3 (Applied Biosystems, Carlsbad, CA). The house keeping gene GAPDH was used for normalization. Each cDNA template was amplified in triplicate PerfeCTa SYBR® Green SuperMix (Quantabio) with gene specific primers listed in Table S3.
Gene silencing with siRNAs
siRNAs were ordered from Thermo Fisher (sequences listed in Table S4). Cells were grown to ~70% confluence in 6-well plates in Complete Fibroblast Medium. A gene-specific siRNA was added to transfect fibroblast cells for overnight using the RNAi Max reagent (Cat.13778150, Thermo Fisher). Cells then recovered in the complete medium for 24h. For FMT induction, the culture was changed to Fibroblast Basal Medium and incubated for overnight prior to AA stimulation.
Luciferase reporter assay for SRF transcriptional activity
We followed the manufacturer’s instruction and our recently reported protocol14. Briefly, the pGL4.34 vector plasmid containing the CArG box (SRF response element) was purchased from Promega (Cat.E1350). An empty vector was generated by removing the SRF response element. Cells were transfected with the empty vector (control) or pGL4.34 using Effectene Transfection Reagent (Cat.301425, Qiagen, Germantown, MD). Positively transfected cells were selected with hygromycin B (Cat.10687010, Thermo Fisher), seeded in 24-well plates at a density of 20,000 cells/well, and grown for 6 h. Cells were then starved in the basal medium overnight and treated with vehicle (DMSO) or 1 uM centrinone-B for 2 h, cells were then lysed in Bright-Glo (Cat.2610, Promega;) and luminescence was read in xx (instrument).
Model of rat carotid artery injury and peri-adventitial administration of PLK4 inhibitor
To induce adventitial fibrosis, balloon angioplasty injury was performed in rat common carotid arteries as we previously described26. Briefly, rats were anesthetized with isoflurane (5% for inducing and 2.5% for maintaining anesthesia). A longitudinal incision was made in the neck to expose carotid arteries. A 2-F balloon catheter (Edwards Lifesciences, Irvine, CA) was inserted through an arteriotomy on the left external carotid artery and advanced into the common carotid artery. To produce arterial injury, the balloon was inflated at a pressure of 2 atm and withdrawn to the carotid bifurcation and this action was repeated three times. The external carotid artery was then permanently ligated, and blood flow was resumed. Immediately following balloon injury, a PLK4 inhibitor (centrinone-B, 100 µg/rat) or DMSO control dissolved in a mix of two thermosensitive hydrogels was administered around the adventitia of injured arteries. The hydrogel mix (total 300 µl) contained equal volume of 25% AK12 (PolySciTech, AKINA Inc.) and 25% Pluronic gel (Sigma-Aldrich). One week after balloon injury, common carotid arteries were collected from anesthetized animals following perfusion fixation at a physiological pressure of 100 mm Hg. Throughout the surgery, the animal was kept anesthetized via isoflurane inhaling at a flow rate of 2 L/minute. Buprenorphine was subcutaneously injected (0.03 mg/kg, ~0.01 mg/rat) after the surgery. Animals were euthanized in a chamber gradually filled with CO2.
Morphometric analysis
Cross sections (5 µm thick) were excised from rat common carotid arteries embedded in paraffin blocks. Sections were stained for collagen and morphometric analyses using a Masson’s trichrome approach31 (reagents from xxx). Collagen was stained blue and smooth muscle actin (media layer) was stained red. Fibrosis was assessed by two parameters in parallel: the thickness and collagen content of the adventitia layer which was distinguishable from other tissue layers by distinct colors. Collagen content was measured as blue stain intensity normalized to the artery overall size (length of external elastic lamina). Planimetric parameters for assessing intimal hyperplasia (intima/media area ratio) were measured following our previous report26: area inside external elastic lamina (EEL area), area inside internal elastic lamina (IEL area), lumen area, intima area (= IEL area-lumen area), and media area (= EEL area – IEL area). All measurements were performed with ImageJ by a student blinded to treatment groups. The data from all 3-5 sections were pooled to generate the mean for each animal. The means from all the animals in each treatment group were then averaged, and the standard error of the mean (SEM) was calculated.
Statistical analysis
Differences in measured variables between experimental and control groups were assessed by Student’s t test (two-tailed) or one-way ANOVA with Bonferroni test; p < 0.05 was considered significant. Data were presented as mean ± SEM (from at least 3 independent experiments) or mean ± SD. Statistics and graphical data plots were generated using GraphPad Prism v.5.0 for Windows (GraphPad).
Footnotes
The authors declare no conflict of interest
References




