

Abstract
The survival of any microbe relies upon its ability to respond to environmental change. Use of Extra Cytoplasmic Function (ECF) RNA polymerase sigma (σ) factors is a major strategy enabling such signal transduction. Streptomyces species harbour a large number of ECF σ factors; nearly all of which regulate genes required for morphological differentiation and/or response to environmental stress, except for σAntA, which regulates starter-unit biosynthesis in the production of antimycin, an anticancer compound. Unlike a canonical ECF σ factor, whose activity is regulated by a cognate anti-σ factor, σAntA is an orphan, raising intriguing questions about how its activity may be controlled. Here, we reconstitute in vitro ClpXP proteolysis of σAntA, but not a variant lacking a C-terminal di-alanine motif. Furthermore, we show that the abundance of σAntA in vivo is enhanced by removal of the ClpXP recognition sequence, and that levels of the protein rise when cellular ClpP-protease activity is abolished. These data establish direct proteolysis as an alternative and thus far unique control strategy for an ECF RNA polymerase σ factor and expands the paradigmatic understanding of microbial signal transduction regulation.
Importance Most antibiotics are derived from secondary metabolites produced by Streptomyces species. The recent rise in the number of bacterial infections resistant to antibiotics has led to renewed interest in discovery of new secondary metabolites produced by these microbes. An average species of Streptomyces harbours ~30 biosynthetic pathways, but the majority of them are not in the laboratory. A key approach is therefore activation of these “silent” pathways, but new insights into how their expression is regulated are required. Our findings reveal that the ECF σ factor (σAntA) that regulates antimycin biosynthesis lacks an anti-σ partner and instead is controlled by the Clp-protease system. These data establish direct proteolysis as a novel strategy for the control of ECF RNA polymerase σ factors and will aid the pursue of silent biosynthetic pathways.
Introduction
The survival of any organism relies upon its ability to respond to environmental change. This feature is especially true of bacteria, which often live in hostile and fluctuating environments. Streptomyces bacteria thrive in soils. The success of this genus of filamentous, sporulating bacteria is linked to their complex lifecycle and keen ability to sense and respond to its surroundings. Notably, a multitude of bioactive secondary or specialised metabolites are produced in response to environmental cues1. More than half of all small molecule therapeutics critical for human health and wellbeing are derived from or inspired by Streptomyces natural products2.
Streptomyces species typically harbor a large number of biosynthetic pathways, but only a few of them are expressed under common laboratory conditions. The biochemical diversity encoded by these silent pathways is a tremendous untapped resource for discovery of new antibacterial agents and other therapeutics. All data available indicates that the production of natural products is controlled predominantly at the level of transcription. Although there are complex regulatory cascades that tightly control expression of biosynthetic genes, they are ultimately activated, repressed or de-repressed by so-called cluster-situated regulators—regulatory protein(s) encoded within the biosynthetic gene cluster (BGC)3,4. Major roadblocks preventing the exploitation of silent biosynthetic pathways are a lack of insight into their regulation and limited technology for activating their expression.
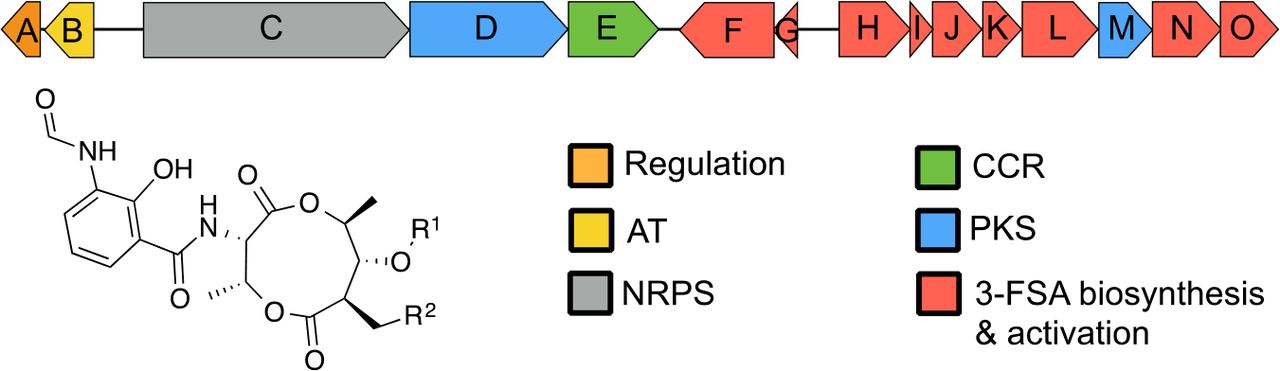
Antimycins have been known for 70 years and are the foundering member of a large class of natural products widely produced by Streptomyces species5,6. Recently, antimycins were shown to be potent and selective inhibitors of the mitochondrial Bcl-2/Bcl-XL-related antiapoptotic proteins that are overproduced by cancer cells and confer resistance to chemotherapeutic agents whose mode of action is activation of apoptosis7. The ~25 kb antimycin (ant) BGC harboured by S. albus is composed of 15 genes organised into four polycistronic operons antAB, antCDE, antFG and antHIJKLMNO (Fig. 1)8,9. The regulation of this ant BGC is unusual compared to other secondary metabolites. Its expression is regulated by FscRI, a cluster-situated LuxR-family regulator of candicidin biosynthesis; FscRI activates expression of antAB and antCDE10. Importantly, antA is a cluster-situated regulator that encodes an Extra Cytoplasmic Function (ECF) RNA polymerase σ factor (σAntA) that activates expression of the remaining operons: antGF and antHIJKLMNO (Fig. 1)9.

Schematic representation of the antimycin (ant) biosynthetic gene cluster. AT, acyltransferase; NRPS, non-ribosomal peptide synthetase; PKS, polyketide synthase; CCR, crotonyl-CoA carboxylase/reductase, 3-FSA, 3-formamidosalicylate. Antimycins: Antimycin A1, R1= COCH(CH3)CH2CH3, R2= (CH2)4CH3; Antimycin A2, R1=COCH(CH3)2, R2= (CH2)4CH3; Antimycin A3, R1= COCH2CH(CH3)2, R2= (CH2)2CH3; Antimycin A4, R1= COCH(CH3)2, R2= (CH2)2CH3.
σAntA, like all ECF σ factors, is similar to the housekeeping σ70 family, but only possesses two of the four highly characteristic sigma domains: domains σ2 and σ4; these regions of sigma bind the −10 and −35 promoter elements, respectively and are sufficient for recruitment of RNA polymerase11. Genes encoding ECF σ factors are almost always co-transcribed with their cognate anti-σ factor12. This class of anti-σ factors are transmembrane proteins that selectively bind to and inactivate a partner σ factor until its release is stimulated, usually by an exogenous signal12,13. After the σ factor is released it recruits RNA polymerase to express a defined regulon that usually includes the σ factor-anti-σ factor operon itself, which thus establishes a positive auto-feedback loop in the presence of the inducing stimulus. Streptomyces species encode a large number of ECF σ factors (>30 per strain) and nearly all of these regulate genes required for morphological differentiation and/or response to environmental stress and, in contrast to σAntA, are not dedicated regulators of one biosynthetic pathway9. In addition, unlike the canonical ECF σ factors, whose activities are controlled by cognate anti-σ factors, σAntA appears to be an “orphan”, lacking such a regulatory partner protein and thus has created curiosity about how its activity is controlled.
The Clp-protease system is essential for normal bacterial proteostasis and is best characterised in Escherichia coli14,15. The Clp protease is a multi-enzyme complex composed of a barrel-shaped peptidase, ClpP and a regulatory enzyme, either ClpA or ClpX (or ClpC in some organisms). ClpA and ClpX (and ClpC) are all AAA+-family protein unfoldases that recognise an N-and/or C-terminal recognition signal (degron) and utilise ATP to unfold and translocate proteins to the peptidase chamber where they are degraded into short peptides16. In Streptomyces species, the peptidase is specified by two genes instead of one and is redundantly encoded17. The primary peptidase is encoded by clpP1P2, whose corresponding proteins form a complex with ClpX or ClpA to facilitate normal proteostasis; the second peptidase is encoded by clpP3P4, but its expression only occurs when the primary system is compromised18,19. The best understood degron is the SsrA tag from E. coli (AANDENYALAA), which is added co-translationally to polypeptides stalled on ribosomes20,21. The E. coli SsrA tag has been comprehensively studied and the C-terminal Ala-Ala-COO− of this motif is essential for proteolysis by ClpXP22. Intriguingly, the C-terminus of σAntA harbours the sequence Ala-Ala-COO−, which previously led us to speculate that ClpXP may modulate its level/activity9.
Here, we reconstitute ClpXP proteolysis of σAntA in vitro and show that it is dependent upon the C-terminal Ala-Ala. We also show that the abundance of σAntA in vivo is higher when Ala-Ala is changed to Asp-Asp and that abundance σAntA is elevated in the absence of genes encoding the primary peptidase, ClpP and its unfoldases, ClpA and ClpX. These data establish direct proteolysis as an alternative, and thus far unique, control strategy of ECF RNA polymerase σ factors, expanding the paradigmatic understanding of microbial signal transduction regulation.
Results
σAntA orthologues are a new subfamily of ECF σ factor that regulate production of the antimycin biosynthetic starter unit
Since its initial discovery six years ago, more than 70 ant BGCs have been identified within actinomycete genera, including in Actinobacteria, Actinospica, Saccharopolyspora, Streptacidiphilus and Streptomyces5. Each of these BGCs harbours a single regulator, σAntA (53-100% shared amino acid identity across all orthologues), which lacks a cognate anti-σ factor partner5,9. Our previous work with S. albus S4 established that σAntA orthologues comprise a new subfamily of ECF σ factors9,23. We demonstrated σAntA is required for expression of antFG and antHIJKLMNO, which encode a standalone ketoreductase (AntM) and proteins required for the production/activation of the starter unit, 3-formamidosalicylate (3-FSA) (Fig. 1). We also mapped the transcriptional start sites and identified conserved promoter sequences for these operons in all known antimycin BGCs at the time9. The conservation of σAntA and target promoters within ant BGCs from taxonomically diverse species, suggests that σAntA-mediated regulation of these genes is direct. To verify this hypothesis, we performed ChIP-sequencing with a S. albus S4 ∆antA mutant complemented with an N-terminal 3xFLAG-tagged version of σAntA. The number of reads that mapped to the promoters of antGF and antHIJKLMNO was enriched for both biological replicates of ∆antA/3xFLAG-antA compared to that of the wild-type mock-immunoprecipitated control, indicating that σAntA directly activates the production of the 3-FSA starter unit during antimycin biosynthesis (Fig. 2).
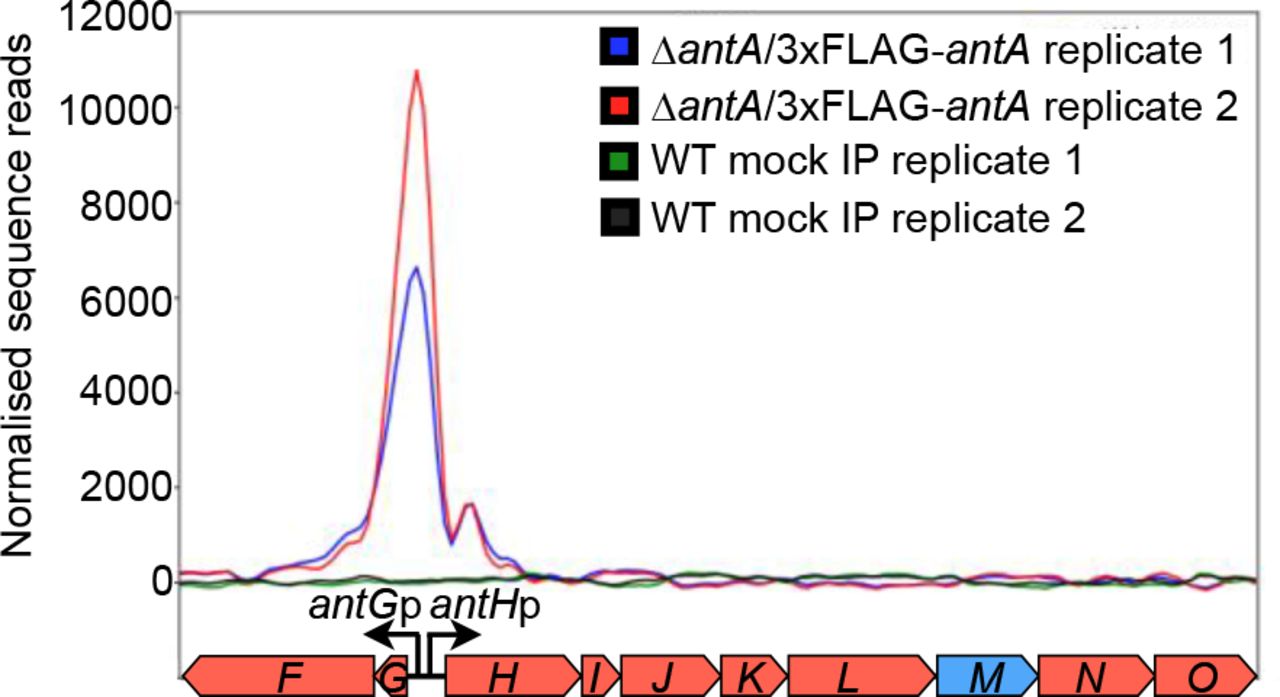
Shown is a graphical representation of normalised sequence reads mapped to the intergenic region of antG-antH (shown at bottom). The genomic coordinates depicted are nucleotides 43,148 to 51,448 of contig CADY01000091.1 of the S. albus S4 genome49. WT, wild-type; IP, immunoprecipitation.
σAntA is degraded by the ClpXP protease in vitro
The activity of almost all characterised ECF σ factors are modulated by a cognate anti-σ factor, which is typically a small transmembrane protein co-encoded within the same operon, so the absence of an anti-σ factor partner to control σAntA is particularly intriguing, and makes σAntA be considered an orphan regulatory protein. An inspection of σAntA amino acid sequences revealed a C-terminal Ala-Ala in 67 out of the 71 orthologues (Supplementary Fig. 1). A C-terminal Ala-Ala is an important component of a common class of degrons for the ClpXP protease22. This observation led us to hypothesise that the activity of σAntA could be modulated by proteolysis instead of by an anti-σ factor. To test this hypothesis, we performed in vitro proteolysis. Previous work indicated that S. albus S4 σAntA was insoluble when overproduced by E. coli, so we pursued the overproduction and purification of the orthologue from Streptomyces ambofaciens ATCC 23877, which is an experimentally demonstrated producer of antimycins24. S. ambofaciens σAntA (75% shared amino acid identity with S. albus S4 σAntA) was purified as an N-terminal (His)6-SUMO-fusion protein. The (His)6-SUMO tag increases solubility and eases purification of putative substrates, without altering recognition of C-terminal degrons by ClpXP. ClpX orthologues from E. coli and S. ambofaciens possess 60% shared amino acid identity and therefore likely recognise similar substrates for degradation. Thus, ClpXP from E. coli was purified and its ability to degrade (His)6-SUMO-σAntA was assessed. Degradation of (His)6-SUMO-σAntA was apparent as early as 2.5 min after addition of ATP and all of the sample was degraded by 15 min (Fig. 3). Substrates of ClpXP become resistant to proteolysis by specific alterations of the C-terminal Ala-Ala22. Therefore, to investigate degradation specificity in the above experiment we constructed and tested a variant of S. ambofaciens σAntA in which the C-terminal Ala-Ala was mutated to Asp-Asp ((His)6-SUMO-σAntA-DD). Strikingly, the Asp-Asp variant was stable against ClpXP degradation over the lifetime of the assay (Fig. 3). Thus, the degradation of (His)6-SUMO-σAntA and the characteristic resistance afforded by the Ala-Ala-to-Asp-Asp mutation demonstrates that σAntA is a direct substrate of ClpXP in vitro.

(A) SDS-PAGE analysis of proteolysis reactions containing 37 pmols (His)6SUMO-σAntA or (His)6SUMO-σAntA-DD. (B) Densitometry analysis SDS-PAGE images for three independent proteolysis experiments. The mean is plotted and error bars illustrate the standard error of the mean (±1 SEM).
σAntA is degraded by ClpXP protease in vivo
To investigate if the in vitro degradation σAntA demonstrated above is relevant to its regulation in vivo we adopted a genetic strategy to assess the abundance of σAntA in mutant strains with defects in Clp-proteolysis. First, we deleted the clpX, clpP1, and clpP2 genes from S. albus S4. The resulting mutant underwent a normal developmental cycle, albeit sporulation was less robust on ISP2 and MS medium (Supplemental Fig. 3). Next genes encoding the 3xFLAG-σAntA or 3xFLAG-σAntA-DD fusion proteins were generated and introduced into the parental strain and the ∆clpXclpP1clpP2 mutant so the abundance of these proteins could be assessed over a developmental time course by Western blotting with anti-FLAG antisera. This experiment was initially performed with the σAntA fusions integrated on the chromosome under control of the native protein. However, a reliable signal could not be detected for 3xFLAG-σAntA and only a trace amount of the Asp-Asp variant was observed, presumably indicating that the cellular level of σAntA is normally low because the native promoter is relatively weak. The experiment was therefore repeated with 3xFLAG-σAntA and 3xFLAG-σAntA-DD expression driven by a stronger, constitutive promoter, ermE*25. Analysis of the resulting immunoblot revealed that 3xFLAG-σAntA-DD was more abundant than 3xFLAG-σAntA in extracts prepared from vegetative mycelia (14h and 17h) of the parent and ∆clpXclpP1clpP2 strains (Fig. 4). Strikingly, 3xFLAG-σAntA and 3xFLAG-σAntA-DD could only be detected in extracts from aerial mycelia (24h and 30h) of the ∆clpXclpP1clpP2 strain and not the parent; the Asp-Asp variant was also present in greater relative abundance (Fig. 4). These data support the hypothesis that σAntA levels, and thus its ability to activate gene expression is modulated by the ClpXP protease, however the conspicuous absence of 3xFLAG-σAntA and the presence 3xFLAG-σAntA-DD in protein extracts prepared from the latest time point suggests the involvement of degradative factor(s) in addition to ClpXP.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cells from the indicated strains were cultivated over a developmental time course on agar media. Protein extracts were generated from 100mg of either vegetative mycelia (14 and 17 hours) or aerial mycelia (24 and 30hours) was harvested and lysed. Thirty micrograms of total protein were analysed by Western blotting with anti-FLAG antisera. The images shown are derived from uncropped original images shown in Supplementary Fig. 4.
σAntA is degraded by ClpAP protease in vivo
Taken together, the data presented above establishes that ClpXP likely acts degrades σAntA in vivo, but also suggested the existence of other factor(s) that affect σAntA levels, especially later in the morphological development cycle. ClpA is an alternative targeting protein that forms a proteolytic complex with ClpP capable of degrading SsrA-tagged proteins21. Indeed, an overlap in proteins comprising the ClpAP and ClpXP degradomes has been observed for E. coli26. Thus, we hypothesised that ClpAP may also be able to degrade σAntA. We therefore generated a ∆clpXclpP1clpP2clpA mutant and re-assessed the abundance of the 3xFLAG-σAntA and 3xFLAG-σAntA-DD by immunoblotting as above. Analysis of the resulting immunoblot revealed that 3xFLAG-σAntA and 3xFLAG-σAntA-DD were present in equal relative abundance within ∆clpXclpP1clpP2 and ∆clpXclpP1clpP2clpA lysate prepared after 14, 17 and 24hrs of growth (Fig. 4). Strikingly, 3xFLAG-σAntA was observed in lysate prepared after 30hrs of incubation only for the ∆clpXclpP1clpP2clpA strain. Taken together, these in vivo data indicate that σAntA is degraded by both the ClpXP and ClpAP proteases.
Discussion
ECF σ factors are a major component of bacterial signal transduction, are typically involved in responding to external stimuli and their activity is canonically understood to be controlled via a cognate anti-σ factor protein; the anti-σ is usually membrane bound and almost always encoded at the same locus12. In this study, we characterised in vitro and in vivo, an ECF σ factor named σAntA that does not possess any identifiable anti-σ factor partner and as a consequence has evolved a different mechanism of regulation.
We established that σAntA is a cluster-situated regulator of antimycin biosynthesis and showed by ChIP-sequencing that it directly binds upstream of genes required for 3-FSA production. Although abundant within Streptomyces species, the activity of ECF σ factors that have been characterised are involved in responding to environmental stress and/or regulating morphological differentiation. To our knowledge, σAntA is the only ECF σ factor that is a cluster-situated regulator in the genus Streptomyces. Indeed, cluster-situated ECF σ factors have only thus far been observed within BGCs for lantibiotics produced by so-called rare actinomycetes and these are controlled by anti-σ factors. In Microbospora corallina, MibR and σMibX regulate microbisporicin biosynthesis and σMibX is controlled by the anti-σ factor, MibW27; in Planomonospora alba, PspR and σPspX regulate planosporicin production and σPspX is controlled by the anti-σ factor, PspW28.
The C-terminal Ala-Ala present within σAntA orthologues served as a clue that instead of an anti-σ factor that ClpXP may regulate σAntA activity. We unambiguously demonstrated that ClpXP degraded σAntA in vitro, but not an altered σAntA variant in which Ala-Ala was changed to Asp-Asp. We also assessed the level of σAntA in vivo and showed that it was more abundant within vegetative mycelia than in aerial mycelia and was partially stabilised by the Asp-Asp mutation, which was consistent with our previous experiments that showed the ant BGC is downregulated at the level of transcription upon the onset of aerial growth9. We demonstrated that the abundance of σAntA and the Asp-Asp variant was higher in vivo in a ∆clpXclpP1clpP2 mutant strain and further so when clpA (orthologous to SCO7532 (clpC2)) was deleted. It was surprising that the Asp-Asp mutation did not fully protect σAntA from proteolysis in vivo, however enhanced abundance of σAntA-DD in ∆clpXclpP1clpP2 and ∆clpXclpP1clpP2clpA genetic backgrounds relative to the parent strain is consistent with previous studies indicating N-terminal and internal motifs can also be important for substrate recognition by Clp-proteases26,29. However, involvement of another protease, such as Lon, in the degradation σAntA cannot be excluded.
Direct ClpXP or ClpAP proteolysis of an ECF σ factor, as shown here, has not been reported previously. However, it has been linked to ECF σ factors in the past, where proteolysis of σS in E. coli and σT in S. coelicolor occurs via their association with an adapter protein or peptide, respectively30,31. In addition, ClpXP proteolysis of the anti-σ factors RseA and RsiW enables expression the σE and σW regulons in E. coli and Bacillus subtilis, respectively32–35. ClpXP has also been linked to the turnover of other transcription factor families. For instance, the λ repressor-like proteins (InterPro ID=IPR010982) PopR and its paralogue ClgR, which participate in a feedback loop regulating expression of the clp genes in S. lividans36,37, and the global oxygen-sensing regulator, FNR in E. coli 38.
Expression of the ant BGC is atypical compared to other BGCs in that it is expressed during vegetative growth, but downregulated upon the onset of aerial growth. Its expression is cross-activated by FscRI, a LuxR-family regulator, from the candicidin BGC, which activates expression of antBA and antCDE10. This regulation in turn enables direct activation of the 3-FSA biosynthetic operons (antGF and antHIJKLMNO) by σAntA. The cellular level of σAntA is antagonised by the Clp-protease system, for which it is a direct target and is ultimately responsible for clearing residual σAntA when FscRI is inactivated following the onset of morphological differentiation10. The above model (Supplementary Fig. 5) is intriguing and begs the question why is it important for σAntA to be actively cleared from cell? One possibility is that aberrant/excess production of 3-FSA is cytotoxic, however previous experiments in which the antA gene was artificially overexpressed did not adversely impact growth of the organism9. An alternative hypothesis for why σAntA must be rapidly removed from the cell is to prevent unnecessary consumption of L-Trp. Biosynthesis of L-Trp is biologically expensive and it is the most chemically complex and least abundant of the 20 common proteinogenic amino acids39. It is tempting to speculate that the evolutionary rationale underpinning this regulatory strategy is owed to the cell needing to dedicate more of this amino acid to production of proteins or metabolites involved in development. This is consistent with recent data showing that deletion of trpM, which controls precursor availability for L-Trp biosynthesis in S. coelicolor and presumably all streptomycetes, fails to undergo normal morphological development40.
In conclusion, here we establish direct proteolysis by the Clp-protease system as an alternative control strategy for ECF σ factors, which provides a new lens through which to examine microbial signal transduction and the regulation of natural product biosynthesis in Streptomyces species. Understanding the diversity of regulatory strategies controlling the expression of these pathways is critical for the development of new tools for exploiting the ‘silent majority’ of biosynthetic pathways harboured by these organisms.
Materials and methods
Growth media, strains, cosmids, plasmids, and other reagents
Escherichia coli strains were propagated on Lennox agar (LA) or broth (LB)41,42 and Streptomyces albus S4 strains were cultivated using LA, LB, and mannitol-soya flour (MS) agar or broth41. Development of clp mutants was assessed on MS and ISP2 medium41. Culture medium was supplemented with antibiotics as required at the following concentrations: apramycin, 50 μg/ml; carbenicillin, 100 μg/ml; chloramphenicol, 25 μg/ml; hygromycin, 50 μg/ml; kanamycin, 50 μg/ml; nalidixic acid, 25 μg/ml. Streptomyces strains were constructed by conjugal mating with E. coli ET12567 as previously described41. Enzymes were purchased from New England BioLabs unless otherwise stated, and oligonucleotides were purchased from Integrated DNA Technologies, Inc. All of the strains, cosmids, and plasmids used in this study are described in Supplementary Table 1, and all of the oligonucleotides used are provided in Supplementary Table 2.
Construction of plasmids
The insert for each plasmid generated in this study was prepared by PCR amplification with Q5 High-Fidelity DNA polymerase and oligonucleotides containing restriction sites. PCR-amplified inserts were restricted and cloned into the relevant plasmids cut with the same enzymes by standard molecular biology procedures. All clones were sequenced to verify the integrity of insert DNA. The restriction sites used for cloning are provided with the plasmid descriptions in Supplementary Table 1.
ChIP-sequencing and bioinformatics analyses
The antA coding sequence was amplified with RFS629 and RFS630, which contain KpnI and EcoRI restriction sites, respectively. The restricted PCR product was cloned into pSETNFLAG digested with the same enzymes. The resulting plasmid was then restricted with NotI and EcoRI to release ermE*p-3xFLAG-antA, which was subsequently cloned into pAU3-45 digested with the same enzymes. pAU3-45-3xFLAG-antA was mobilised to an apramycin-marked ∆antA strain9. Cultivation of the wild-type and ∆antA/pAUNFLAG-antA strains for ChIP-sequencing were performed exactly as described previously10. The pure DNA resulting from immunoprecipitates from two biological replicates of wild-type and ∆antA/pAUNFLAG-antA, as well non-immunoprecipitated chromosomal DNA, were sequenced with the Illumina HiSeq3000 platform with 150-nucleotide paired-end reads by the University of Leeds Next Generation Sequencing Facility at the St. James Teaching Hospital NHS Trust. The resulting reads were analysed exactly as described previously10. The graphic in Figure 2 was generated using DeepTools computeMatrix and plotProfile functions43.
Construction of S. albus S4 clp mutant strains
All deletions were performed by mutagenising cosmids using RecET recombineering in E. coli followed by their subsequent mobilisation to S. albus strains via conjugal transfer. The clpXclpP1clpP2-containing cosmid, cos117 and clpA-containing cosmid, cos251 were obtained by screening a previously constructed S. albus S4 Supercos1 cosmid library8 by PCR using oligonucleotides PBB001 and PBB002 (clpX) and PBB067 and PBB068 (clpA). Cos117 and cos251 were mutagenised as required using E. coli recombineering with strain GB05-red44 and a deletion cassette. Deletion cassettes were generated by PCR from paac-apr-oriT45 and consisted of the apramycin resistance gene, aac(3)IV and a conjugal origin of transfer (oriT), which was flanked by ΦC31-attL and -attR sites for excision of the cassette. Oligonucleotides used to generate deletion cassettes included 39 nt of homology upstream or downstream of the target open reading frame(s) and are listed in Supplementary Table 2. The resulting PCR products were digested with DpnI, gel purified and electroporated into arabinose-induced E. coli GB05-red harbouring cos117 or cos251. Transformants were screened for the presence of mutagenised cosmid by PCR using oligonucleotides listed in Supplementary Table 2 and the integrity of the locus was verified by DNA sequencing. Mutagenised cosmids were electroporated into E. coli ET12567/pUZ8002 and mobilised to a strain of S. albus S4 harbouring an entire antimycin BGC deletion (∆antall) by conjugation as described41. Transconjugants were screened for apramycin resistance and kanamycin sensitivity. The integrity of apramycin-marked mutants was verified by PCR using the oligonucleotides listed in Supplementary Table 2. The apramycin deletion cassette was subsequently excised from the chromosome by conjugal introduction of pUWLint31, which is a replicative plasmid with a temperature sensitive origin of replication that expresses the ΦC31 integrase required for removal of the cassette45. Transconjugants were screened for loss of apramycin resistance and excision of the cassette was verified by polymorphic shift PCR and DNA sequencing of the product.
Immunoblot analysis
Spores of parental strain, S. albus ∆antall, ΔclpXclpP1clpP2 and ΔclpXclpP1clpP2clpA mutants carrying pPDA or pPDD were grown on SFM agar (buffered with 50mM TES, pH 7.2) covered with cellophane discs. Protein extracts were prepared from mycelia collected at regular intervals during growth (14h, 17h, 24h and 30h) as follows: 100 mg of cells were resuspended in 200 μl lysis buffer (50 mM sodium phosphate buffer, pH 7.0, 150 mM sodium chloride, 10 mg/ml lysozyme, cOmplete, Mini, EDTA-free protease inhibitors (Roche) and 100 mg of 0.1 mm glass beads (PowerLyzer®)) and lysed by vortexing for 30 min at 2000 pm, 37°C, with a subsequent incubation for another 30 min at 37°C. The obtained suspension was centrifuged for 20 min at 20,000g at 18°C. Thirty micrograms of the clarified protein extract were subjected to SDS-PAGE and then transferred to nitrocellulose membrane (pore size 0.2 μm) for Western blot analysis. The membrane was probed with mouse monoclonal ANTI-FLAG® M2-Peroxidase (HRP) antibody (Sigma), 1:10 000, and the signals were detected using PierceTM 1-Step Ultra TMB Blotting Solution (Thermo Scientific).
Protein purification and in vitro ClpXP proteolysis assays
The wild-type antA gene was PCR amplified and cloned into the AgeI and HindIII sites of the pET23b-SUMO vector, which harbours an N-terminal (His)6-SUMO tag46. The plasmid for production of (His)6-SUMO-σAntA-DD was generated by site-directed mutagenesis (Agilent QuikChange) using primers listed in Supplementary Table 2. (His)6-SUMO-σAntA and (His)6-SUMO-σAntA-DD were produced by E. coli Rosetta(DE3) (Novagen) grown in LB at 37 °C until OD600 0.5, followed by induction with 0.4 mM IPTG and growth at 18 °C for 16 hours. Cells were resuspended in 50 mM sodium phosphate, pH 8, 1M NaCl, 20 mM imidazole, 10% glycerol, and 1 mM DTT and lysed by french press at 28 kpsi, followed by treatment with protease inhibitor cocktail set III, EDTA-free (Calbiochem) and benzonase (Millipore Sigma). (His)6-SUMO-σAntA and (His)6-SUMO-σAntA-DD proteins were purified by Ni-NTA affinity chromatography and Superdex-75 gel filtration and stored in 50 mM potassium phosphate, pH 6.8, 850 mM KCl, 10% glycerol, and 1 mM DTT. E. coli ClpX and ClpP proteins were purified as described previously46,47.
In vitro ClpXP proteolysis assays were performed at 30 °C by preincubating 0.3 μM ClpX6 and 0.8 μM ClpP14 with ATP regeneration system (4 mM ATP, 50 μg/mL creatine kinase, 5 mM creatine phosphate) in 25 mM HEPES-KOH, pH 7.5, 20 mM KCl, 5 mM MgCl2, 10% glycerol, 0.032% NP40, and 0.2 mM DTT and adding substrate to initiate the reactions. Samples of each reaction were taken at specific time points and stopped by addition of SDS-PAGE loading dye and boiling at 100 °C before loading on Tris-Glycine-SDS gels. Bands were visualized by staining with colloidal Coomassie G-250 and quantified by ImageQuant (GE Healthcare) after scanning by Typhoon FLA 9500 (GE Healthcare). The fraction (His)6-SUMO-σAntA remaining was calculated by dividing the (His)6-SUMO-σAntA density at a given time point by the density at time zero and normalized by ClpX density.
Data availability
The next-generation sequencing data obtained in this study are available under ArrayExpress accessions E-MTAB-7700 and E-MTAB-5122.
Funding information
B.B. was supported by a grant from the Biotechnology and Biological Sciences Research Council (BB/N007980/1) awarded to R.F.S. S.K. was supported by a National Science Foundation Graduate Research Fellowship and the Howard Hughes Medical Foundation; T.A.B is an employee of the Howard Hughes Medical Foundation.
Acknowledgements
We are grateful for helpful discussions with Matt Hutchings, Paul Hoskisson, Kenneth McDowall and Alex O’Neill.
References




