

Abstract
Axons are the slender, cable-like, up to meter-long projections of neurons that electrically wire our brain and body. In spite of their challenging morphology, they usually need to be maintained for an organism’s lifetime. This makes them key lesion sites in pathological processes of ageing, injury and neurodegeneration. To better understand how axons are formed and maintained long-term, we focus here on the parallel bundles of microtubules (MTs) which form their indispensable structural backbones and highways for life-sustaining cargo transport and organelle dynamics. Many MT-binding and -regulating proteins in axons have prominent hereditary links to axon degeneration, but knowing their molecular roles is usually insufficient to explain their roles during axon morphogenesis, maintenance or pathology. Such understanding requires deciphering how these proteins interact in regulatory networks to implement observed cellular phenomena. Here we propose the model of local axon homeostasis as a conceptual framework that attempts to combine current knowledge into one coherent interactome. According to this model, each area of an axon is self-sustaining through local auto-regulatory networks; these networks maintain a balance between (1) the enormous mechanical challenges posed by the life-sustaining intra-axonal motor dynamics and (2) the maintenance activities required to sustain MT bundles as the highways needed for those dynamics. This model offers a new level of explanation, and we hope that it will help to raise the interest in axonal MTs and lead to the generation of more data that can help to decipher the important contributions of MTs to axon biology and pathology.
Introduction
Axons are the slender, cable-like extensions of nerve cells which form the nerves and nerve tracts that wire our brain and body, sending neuronal messages in highly regulated manners. With diameters of only 0.1-15µm (Hoffman, 1995), they extend over distances of up to a meter in humans. To adopt such a unique morphology and physiology, axons display many specialised features (Fig.1).

Specific properties of axons. Axons during the growth cone stage are shown in (A) and after synaptic maturation in (B), differing primarily in certain stage-specific specialisations including growth cones, synapses, electrical properties and glial interactions (here myelination; Pan and Chan, 2017). The core machinery in the axon shaft can be expected to be similar at both stages: parallel continuous bundles of extended but discontinuous MTs run all along axons serving as a structural backbone (see Fig.2), a transport highway for axonal trafficking (driven by motor proteins), and a source for ‘off-track’ MTs contributing to morphogenetic processes including branch formation, directed axon growth and synapse formation/plasticity (green, orange, blue curved arrows); MT bundles are interspersed with longitudinal actin trails (Leterrier et al., 2017), continuous networks of (smooth) ER (Gonzalez and Couve, 2014), and other membranous organelles including mitochondria (Saxton and Hollenbeck, 2012); axonal membranes display regularly spaced periodic rings of cortical actin (Qu et al., 2017; Xu et al., 2013), an unusually high number of ion-specific channel proteins and transporters to conduct nerve impulses (Kandel et al., 2012), as well as adhesions with external structures including parallel axons (not shown), glial processes (Pronker et al., 2016) and synaptic partner cells (Koper et al., 2012); a degree of independence from cell-body derived proteins is provided by local translation machinery (Cioni et al., 2018; Giuditta et al., 2002b) or supply from surrounding glia cells (not shown; Court et al., 2011; Frühbeis et al., 2013; Giuditta et al., 2002a; Rajendran et al., 2014). Note that the axon diameter in the region between glia cells in B (referred to as Node of Ranvier) usually has a much smaller diameter than the rest of the axon (Hoffman, 1995).
Axons are indispensable for nervous system function, as illustrated by paralysis in spinal cord injury caused by the interruption of ascending and descending axon tracts (Bichenback, 2013; Tedeschi and Bradke, 2016). Axons are key lesion sites in injury-induced trauma and coma (Gaetz, 2004; Medana and Esiri, 2003; Smith et al., 2000; Tang-Schomer et al., 2012), and axon decay is believed to be the trigger for neuronal loss in ageing and many neurodegenerative disorders (Adalbert and Coleman, 2012; Salvadores et al., 2017). Notably, most neurons cannot be replaced, and compensation of lost axons through collateral branching of intact neighbouring axons has obvious limitations (Adalbert and Coleman, 2012; Sturrock, 1987).
This means that most axons have to be maintained for an organism’s life time, i.e. up to a century in humans; unsurprisingly, mammals tend to lose almost half their axon mass towards high age (Calkins, 2013; Marner et al., 2003). This trend is severely enhanced in neurodegenerative disorders, as illustrated by gradually increasing paralysis in spastic paraplegia or motorneuron disease (Blackstone et al., 2011; Riancho et al., 2019).
Research into neurodegenerative disorders typically approaches the problem by describing observed phenotypes and unravelling the molecular mechanisms performed by proteins linked to the disease. However, it seems that this approach rarely leads to satisfactory explanations of the pathology (Aguzzi, 2019). We believe that more profound understanding will arise when widening the scope from molecular to cellular mechanisms, by studying how proteins work within regulatory networks to underpin observable processes of axon biology - thus performing investigations at the level of complexity at which pathology becomes manifest. Here we will illustrate this approach by focussing on the axonal cytoskeleton.
The importance of microtubule bundles for axon biology
As illustrated in Fig. 1, the cytoskeleton of the axon shaft consists of straight parallel bundles of MTs, which are interspersed with intermediate filaments (not shown) and longitudinal actin fibres called ‘actin trails’ - all running through evenly spaced periodic rings of cortical actin (Qu et al., 2017; Xu et al., 2013); significant deviations from this organisation, that will not be addressed in this review, exist at axon initial segments (not shown), growth cones and synapses (Dent et al., 2011; Leterrier, 2018; Leterrier et al., 2017; Prokop, 2013).
Of the three cytoskeleton classes, intermediate filaments were suggested by anatomical, developmental and genetic studies to regulate axon diameter, and their axonal aggregation is a hallmark of many neurodegenerative diseases (Friede and Samorajski, 1970; Hoffman, 1995; Perrot et al., 2008; Rao et al., 2003; Sakaguchi et al., 1993). However, intermediate filament accumulations are not necessarily the cause, but can be the consequence of axon decay (Eyer et al., 1998; Nguyen et al., 2000; Perrot et al., 2008). Notably, Neurofilament-H-lacZ mutant mice or Quiver mutant quail completely lacking axonal intermediate filaments, develop and breed fairly normally (Eyer and Peterson, 1994; Yamasaki et al., 1991), and various arthropods form axons of defined diameters in the absence of any axonal intermediate filaments (Allen et al., 2006; Hirokawa, 1986; Voelzmann et al., 2016a). These examples suggest that intermediate filaments play no key roles in axon growth and maintenance. In contrast, the actin and microtubule (MT) cytoskeleton are essential for all stages of neuronal development and maintenance (Sakakibara et al., 2013; Voelzmann et al., 2016a) and this review will focus on the role and regulation of MTs.
MTs in axons are arranged into bundles which run all along axon shafts and are essential for their biology in at least three ways. Firstly, they serve as structural backbones, comparable to the vertebral column of a snake; since MTs in these bundles are discontinuous and expected to be interlinked via flexible connections (see section on cross-linkers), they are ideally suited to respond to longitudinal stretch and compression (similar to a half-extended telescope ladder), but also to torsion and flexure (Fig.2).

Axonal response to mechanical challenges. Continuous bundles of discontinuous MTs which are flexibly cross-linked (likely involving slip-bonds) are thought to provide a structural element that can respond to different forms of mechanical impact (as indicated in blue).
Secondly, MT bundles provide the highways for life-sustaining axonal transport between cell bodies and the axonal compartment. This transport is driven anterogradely by kinesins and retrogradely by the dynein/Dynactin complex; the cargoes include mRNAs, cytoplasmic proteins including signalling factors, vesicles delivering synaptic proteins, adhesion factors, neuropeptides and/or membrane lipids, as well as entire organelles including mitochondria (Fig. 3A-D; Goldstein et al., 2008; Gondre-Lewis et al., 2012; Gonzalez and Couve, 2014; Hirokawa et al., 2010; Pfenninger, 2009). Furthermore, local dynamics of organelles, such as fission or fusion of mitochondria, can be expected to require forces generated by MT-associated motor proteins (Fig. 3E; Saxton and Hollenbeck, 2012).

An interactome of MT-regulating and -associated mechanisms expected to contribute within the model of local axon homeostasis. Developing and mature neurons are shown at the bottom indicating that the close-up (magenta frame) might apply in both contexts. 1-16) Potential mechanisms that can ‘tame’ MTs into bundled conformation: MT polymerisation (blue stippled arrows) is driven by molecular machinery centred on Eb1 (blue balls), further influenced by the tubulin-supply machinery (not shown) and shaft-binding proteins (7); polymerisation generates new MTs required for bundle formation (8) and turn-over (14); to integrate into bundles, extending MTs require guidance via actin-Eb1 cross-linkage along the axonal surface (5; Shot) or along pre-existing MTs through MT-MT cross-linkers (9; brown L). The same or other cross-linkers provide the structural glue that holds MT bundles together (12; brown L); some of them can also bind to actin (2), they protect from (or recruit) MT severing activity (15), and influence motor protein dynamics (a). MTs which have escaped any cross-linkage are eliminated by cortical collapse factors when approaching the axonal surface (4; Efa6) or by MT severing factors at MT-MT cross-points (6). The bundled MTs are discontinuous; their free minus ends are stabilised by CAMSAP/Patronin (Ptrn) together with katanin (black scissors; 13), whereas non-polymerising MT plus ends are stabilised by other factors (not shown; e.g. CLASP or the Dynactin subunit p150/Glued; Hur et al., 2011; Lazarus et al., 2013). The dynein/Dynactin complex is believed to link cortical actin to MT bundles and drive them anterogradely (10), whereas Ptrn at minus ends may anchor MTs via spectraplakins to the axon cortex (1); spectraplakins may also link MTs directly to cortical actin (2) or to transmembrane receptors (3), and they are expected to perform further, still unexplored actin-independent bundle-promoting roles through their PRR domains (11). Tear-and-wear damages MTs (dashed green line), potentially affecting interaction with MT-binding proteins (16; red X); MT severing proteins might selectively eliminate such MTs (16; scissors) or MTs undergo repair (not shown). A-E) Mechanisms closely ‘associated’ with MT bundles: MT-associated motor proteins (‘motor’, solid orange arrows) drive axonal transport of (protein-loaded) vesicles (A), cytoplasmic factors including proteins, translational machinery (ribosomes) or RNAs (B), move other MTs (B, sliding), and position/rearrange organelles including mitochondria (C, mitos), ER, peroxisomes and endosome (D) - and this likely includes mitochondrial fission and fusion (E). a-e) The motor-associated functions all act downstream of MT bundles because they require them to walk on; but they also act upstream: for example, the forces they generate (stippled orange arrows) are the potential cause for MT disorganisation (buckling shown in d); transport delivers required regulators and building blocks for bundle-maintaining processes (b); the proper regulation of organelles/endocytic compartments provides systemic factors that can orchestrate taming mechanisms, including intracellular free calcium or reactive oxygen species (Ca2+, ROS; yellow cloud) as well as ATP required for many processes including actin dynamics, MT severing and MT motor activity (red stippled arrows; note that vesicular transport uses glycolysis to generate its own ATP; yellow star); vice versa, the MT severer spastin also regulates the ER through ATP-independent mechanisms (e), and MT-associated proteins (APC) regulate local translation events (c).
Third, axonal bundles provide a source for readily available MTs that can be used for other purposes (curved arrows in Fig.1); for example, splaying MTs can trigger axon extension processes in growth cones (Dent et al., 2011; Prokop et al., 2013), induce collateral branch formation along the axon shaft (Kalil and Dent, 2014), or support physiological changes at synapses (Bodaleo and Gonzalez-Billault, 2016).
Maintaining MT bundles is therefore crucial for axon longevity. Accordingly, there are prominent and numerous genetic links from MT regulators to hereditary neurodegenerative disorders (Suppl. Mat. in Prokop et al., 2013), and axon decay is a frequent side effect of MT-targeting chemotherapies (Prior et al., 2017; Wozniak et al., 2018; Wu et al., 2014). Of particular interest for this review are reports of pathological axon swellings where MT bundles have disintegrated into loops or waves (bottom of Fig.3), occurring in ageing, after injury and in certain axonopathies (Adalbert et al., 2009; Bernier and Kothary, 1998; Dalpe et al., 1998; Denton et al., 2014; Fassier et al., 2013; Havlicek et al., 2014; Sorbara et al., 2014; Tang-Schomer et al., 2012; Tarrade et al., 2006; Yamasaki et al., 1991; Yin et al., 2016). Notably, one study suggests that MT aberration upon ageing could cause swellings that trap and damage mitochondria, thus triggering axon degeneration (Fiala et al., 2007). However, in the existing literature too little emphasis is given to MTs and there are simply not enough data to deduce meaningful correlations between axon degeneration and MT bundle decay.
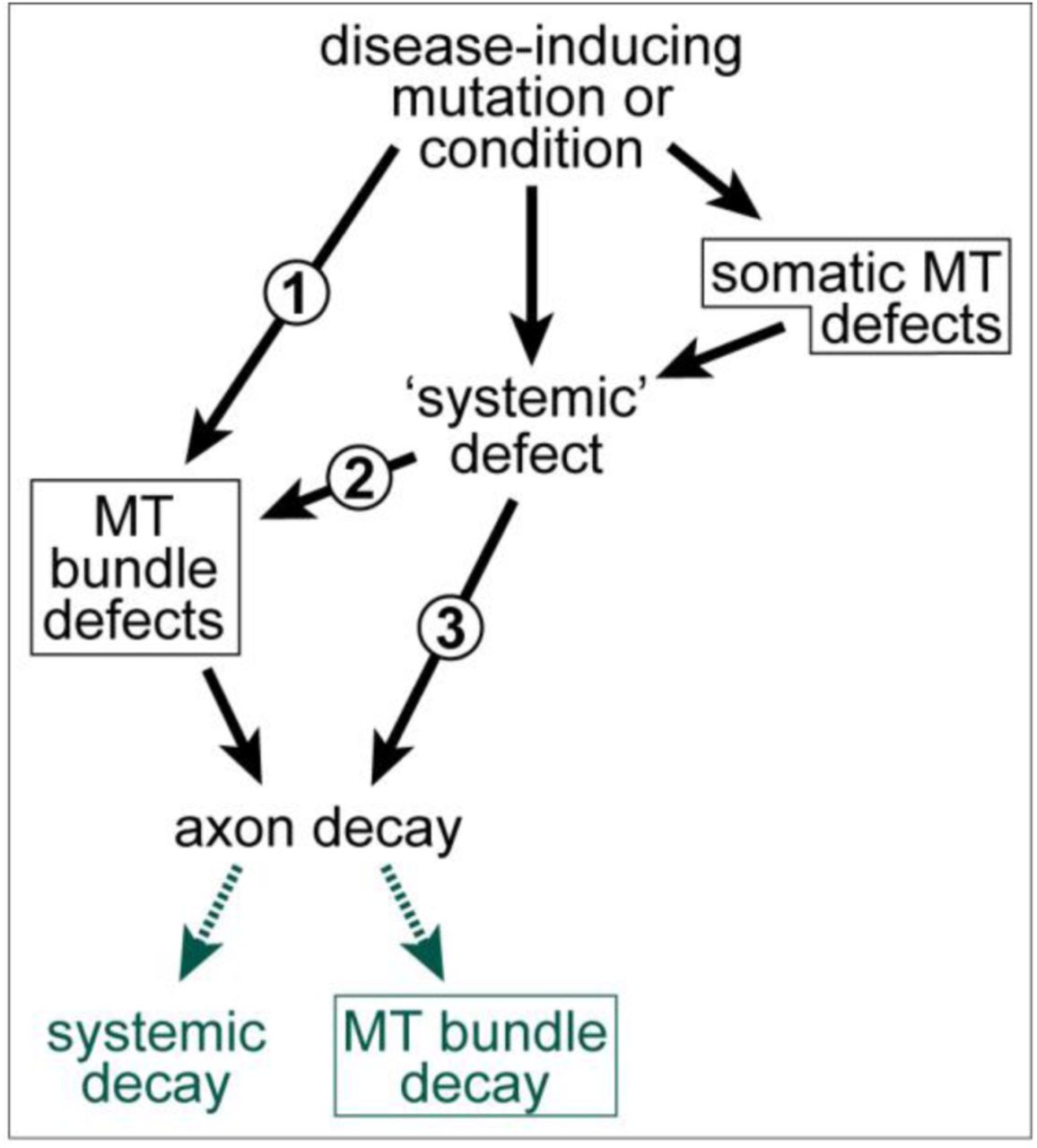
Even if there were a close correlation, this still does not exclude that, depending on the pathological condition, MT bundle deterioration may be a mere consequence rather than cause of axon decay (details in Fig.4). Ultimate clarification will only arise from developing a better understanding of MT bundle-forming and -maintaining machinery. Here we propose a conceptual framework that may facilitate such developments.

MT bundle defects as cause or consequence of axon decay. 1) Disease-inducing mutations/conditions can affect a MT-bundle regulator (e.g. dystonin; Voelzmann et al., 2017), thus causing MT bundle defects first which can, in turn, trigger axon decay. 2) Disease-inducing mutations/conditions can affect systemic factors which, in turn cause MT bundle defects as an intermediate causative step in the cascade leading to axon decay (e.g. axonal transport fails, leading to MT bundle defects which then contribute to axon decay (e.g. Alzheimer’s disease or ALS; Brandt and Bakota, 2017; Farah et al., 2003; Zempel and Mandelkow, 2015); this may occur even if MT regulators are affected, but these regulators mainly act in the cell body (e.g. dysregulation of the Golgi; Ferrier et al., 2013). 3) MT bundle deterioration may be a mere consequence of axon decay, although this case will be difficult to disentangle from option 2, since MT bundle disintegration and axonal disassembly may occur in parallel, as observed in developmental or injury-induced axon degeneration; Bradke et al., 2012; Wang et al., 2012; Yaron and Schuldiner, 2016). All MT-related phenotypes in this graph are emphasised with a frame.
The integrated model of local axon homeostasis
The foundations for this conceptual framework were laid when we took the decision to use the fruit fly Drosophila melanogaster as a means to study how cytoskeletal regulators collaborate in orchestrating the morphogenetic changes that drive axon growth (Sánchez-Soriano et al., 2007). Drosophila is not a miniature human, but it has many advantages and provides powerful means to uncover the regulatory concepts behind the roles and regulation of axonal MTs, which then often apply to higher organisms (Box 1; Aguzzi, 2019; Bellen et al., 2010; Elden et al., 2010; Prokop, 2018). Through using Drosophila neurons as a consistent standardised cell system, our group alone performed functional analyses of over 50 actin-and/or MT-binding or -associating regulators (Prokop et al., 2013); these studies form an unprecedented pool of data on the basis of which to develop novel concepts (Alves-Silva et al., 2012; Beaven et al., 2015; Gonçalves-Pimentel et al., 2011; Qu et al., 2018; Qu et al., 2017; Voelzmann et al., 2016b).
Box1 Why using Drosophila?
The use of Drosophila neurons to study the neuronal cytoskeleton has a number of advantages that were detailed elsewhere (Prokop et al., 2013). Key aspects are the high degree of conservation of cytoskeletal proteins, regulators and dynamics, the experimental amenability of neurons in primary cell culture and in vivo (Prokop et al., 2013; Prokop et al., 2012; Sánchez-Soriano et al., 2010), and the relative ease of genetic manipulation based on available resources and efficient combinatorial genetics (Hahn et al., 2016). The power of combinatorial genetics is rooted in the relative ease, speed and cost effectiveness with which genes can be manipulated and functionally analysed, facilitating also combined analyses of multiple factors in the same animals/cells (Prokop, 2018; Prokop et al., 2013; Roote and Prokop, 2013).
Combinatorial genetics has been extremely successful in overcoming problems of redundancy, and generating new conceptual understanding of co-operative networks of MT regulation (see main text). This can hardly be achieved through isolated work on individual factors.
Our loss-of-function analyses of 24 MT-binding or -associating (2nd order) proteins, revealed that more than half displayed significant MT disorganisation. Interestingly, the MT disorganisation found in these various conditions appears to display certain common characteristics: axons display areas in which their bundles are dissolved into chaotic, intertwined, crisscrossing arrangements of curled MTs (see examples in Fig.5). Notably, when using the same genetic conditions in vivo, comparable phenotypes were observed in the fly brain (Qu et al., 2018). Such in vivo phenotypes in the fly remind of the curled MT conformations in pathological axon swellings of mammalian models mentioned in the previous section. Potential evolutionary conservation of this phenomenon is also supported by the occurrence of MT curling and disorganisation in mouse and rat primary neurons (Ahmad et al., 2006; Sánchez-Soriano et al., 2009).

Disorganisation of axonal MTs upon loss of different MT regulators in Drosophila primary neurons. A) Normal neuron (wild-type, wt) with soma (asterisk), axon shaft (curved arrow) and growth cone (tip of most distal MT indicated by arrow head). B) Eb15 mutant neuron where the area of MT disorganisation is framed by a red stippled box and shown as close-up on the right. C-E) Similar close-ups shown for Efa6GX6[w-], Khc27 and shot3 mutant neurons. Note that the four mutated factors perform fundamentally different molecular functions, with Eb1 being a MT plus-end binder (‘8’ in Fig.3), Efa6 a cortical collapse factor (‘4’ in Fig.3), Khc a kinesin-1 motor protein (‘A-E’ in Fig.3) and Shot a multi-functional cross-linker (‘1-3, 5, 11’ in Fig.3). All neurons were derived from wild-type or homozygous mutant embryos, mechanically and chemically dissociated, kept for 7days in pre-culture in a centrifuge tube to deplete any maternal gene product, mechanically and chemically dissociated again, cultured on concanavalin A-coated glass coverslips for 1day at 21°C, fixed and stained with anti-α-tubulin (DM1A, Sigma; procedures detailed elsewhere: Prokop et al., 2012); images were taken using STED (stimulated emission depletion) microscopy. Scale bar in A represents 10 µm for the two neurons and 4 µm in close-ups.
As an attempt to explain this surprising phenotype across mutant conditions and animal groups, we developed the model of ‘local axon homeostasis’ (Prokop, 2016; Voelzmann et al., 2016a). The model states that MTs, which usually behave like rigid rods (Hawkins et al., 2010), are challenged to buckle (‘d’ in Fig.3) and/or curl up by the force-enriched axonal environment (‘A-E’ in Fig.3; detailed in the next two sections). Through this bias towards MT curling, the parallel axon bundles are at risk of becoming disorganised and turning into pathological swellings. The model therefore proposes that this destructive tendency of axonal MTs is contained through the action of different classes of MT-associating and -regulating proteins, which co-operate and complement each other to form robust machinery that ‘tames’ MTs into bundles (‘1-16’ in Fig.3).
In this model, each axon segment uses local mechanisms to maintain its bundled MT organisation (hence ‘local axon homeostasis’). Hereditary or acquired loss of single regulators would therefore be expected to weaken this machinery and increase the statistical risk of MT disorganisation. Such heightened probability might explain why many axonopathies affect primarily long axons (Prior et al., 2017), and why certain disorders linked to MT regulators display late onset axon decay (Voelzmann et al., 2017).
In the next two sections, we describe potential mechanisms underlying deteriorating destructive MT behaviours in axons and their relationship to molecular motor proteins. We will then summarise experimentally demonstrated MT maintaining and ‘taming’ mechanisms, and speculate about further potential maintenance mechanisms based on existing knowledge of known classes of axonal MT-regulating proteins.
Understanding the unusual curling behaviour of MTs in axons
MTs are polar polymers composed of α/ß-tubulin heterodimers which are arranged in a head-to-tail fashion into linear protofilaments; mostly 13 of these protofilaments are laterally aligned forming a straight tube of roughly 25 nm diameter (Fig.6A, C). But MTs can deviate from this norm: for example, axonal MTs were reported to contain 13 protofilaments in frog olfactory or goldfish brain axons, 11 or 15 in C. elegans, and 12 in Drosophila, crayfish and lobster (Benshalom and Reese, 1985; Burton et al., 1975; Savage et al., 1989). Deviation from the straight 13 protofilament conformation equips MTs with distinct, functionally relevant physical properties (Chaaban and Brouhard, 2017; Chalfie and Thomson, 1982). Importantly, protofilaments in these deviating MTs are skewed and cause a supertwist of the tubule (Fig.6D; Chrétien and Fuller, 2000; Chrétien et al., 1996; Chrétien and Wade, 1991); this supertwist forces motor proteins to rotate around MTs (Ray et al., 1993) and is the likely explanation for supercoil of entire axons observed upon MT bundle destabilisation (Krieg et al., 2017; Shaw and Bray, 1977).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A molecular perspective of microtubule properties. A) Cross-section of a MT with 14 protofilaments (PF) and lateral view of a 13 PF MT, both in B-lattice configuration, where α-tubulins make lateral bonds with α-tubulins and ß with ß, except at the seam (magenta line: seam; dashed red line: PF). B) Close-up of an α/ß-tubulin heterodimer showing the various post-translational modification sites as indicated; note that the GTP of ß-tubulin in lattices is usually hydrolysed (GDP). C) A 13 PF MT (top), cut open at the seam and rolled out (bottom); the yellow line shows the diameter, the blue line follows the helical rise of laterally bonded tubulins; in 13 PF MTs, tubulins are precisely aligned at the seam (yellow arrow head) but shifted by three positions (3-start helix). D) When deviating from the 13 PF prototype, tubulins are misaligned at the seam (orange arrow head); when forced into alignment, the PFs skew, causing a super-twist of the MT as described by the ‘lattice accommodation model’ (Chrétien and Fuller, 2000; Langford, 1980); for certain PF numbers, MTs can form two alternative alignments, of which usually the version with the lower helix start value (left) has a left-handed super-twist, the higher value is right-handed (Chrétien and Fuller, 2000). E) MTs behave like rigid rods with a persistence length of up to 10 mm, but can be bent down to diameters of ~1µm before they break; it has been reported that their cross-sectional profile may flatten above a certain threshold (black arrow head), thus softening the tube. F) Lattices of GDP-tubulin are 1-3% shorter than MTs that were polymerised with the non-hydrolysable GTP analogue GMPCPP, or stabilised with taxol (orange structure binding α-tubulin 1:1, according to Nogales et al., 1995); binding of kinesin-1 causes similar lengthening of tubulin (and additional compactions in the tubulin structure: yellow stars) which may cause cooperative binding of further kinesins and induce curvature if occurring only on one side of the MT; in extended taxol-bound MTs, bending forces were suggested to transfer tubulins on the concave side into their short conformation as an energetically favoured condition. For further references see main text.
MTs are structurally active, and their properties can change upon protein binding (see interactions with kinesins below) or when altering the ‘tubulin code’; the tubulin code is determined by the incorporation of different existing isotypes of α- and ß-tubulin into the MT lattice, and the addition of a range of distinct post-translational modifications (Fig.6B; Janke and Kneussel, 2010; Park and Roll-Mecak, 2018; Ti et al., 2018; Vemu et al., 2017). Some modifications influence the interaction with MT-binding proteins (e.g. poly-glutamylation attracting spastin; Valenstein and Roll-Mecak, 2016), others are believed to structurally protect MTs from damage or depolymerisation, such as poly-aminations on various residues (Song et al., 2013) or acetylation of luminal lysine 40 (Fig.6B; Baas et al., 2016; Howes et al., 2014; Soppina et al., 2012; Xu et al., 2017). Notably, mutation of lysine 40 in Drosophila α1-tubulin caused mild but physiologically relevant in vivo phenotypes (Jenkins et al., 2017; Yan et al., 2018). Furthermore, the MT lumen may contain MIPs (MT inner proteins) that likely also contribute to MT stability (Ichikawa and Bui, 2018).
Although curvature is a key driver of MT plus end dynamics during de-/polymerisation (Brouhard and Rice, 2018; van Haren and Wittmann, 2019), MT lattices in vitro usually behave as rigid rods with a persistence length of 1-10 mm (as compared to ∼12 µm measured for actin filaments; Fletcher and Mullins, 2010; Hawkins et al., 2010; Howard, 2001). However, in so-called gliding assays where MTs are moved around (minus-end-leading) on a carpet of active kinesins, they can undergo fishtailing or form micron-sized arcs or loops (Amos and Amos, 1991; Lam et al., 2016; Weiss et al., 1991). Interestingly, this phenomenon seems not to occur upon plus-end-leading movement on (axonemal) dynein carpets, although collisions are far more frequent in these assays (Sumino et al., 2012).
On kinesin carpets, single MT loops form through motor-driven buckling upon substrate pinning or collision, favoured by high MT and/or kinesin densities or exposure to non-polar conditions (n-heptane, air; Tab.1); loops can be astonishingly stable (frequently >5 mins, as reported in Liu et al., 2011). Their structural longevity is significantly enhanced when MTs are reversibly cross-linked with biotin-streptavidin so that MTs bundle up (analogous to Fig.2C), thus forming spools composed of up to dozens of MTs; spools appear to reproduce most behaviours observed for single MTs (Tab.1): (1) the smallest inner diameters for loops and spools usually lie in the range of 1-3 µm (with diameters of curvature below ~1 µm believed to break MTs; Odde et al., 1999; Waterman-Storer and Salmon, 1997); (2) the direction of loop rotation is a function of the left-versus right-handed supertwist of MTs (Fig.6D), and spools rotate according to the supertwist of their constituent MTs (Kawamura et al., 2008; Liu et al., 2008); (3) spool diameters increase with the degree of rigidity of its MTs (Wada et al., 2015). Interestingly, like off-track MTs in axons, MTs can escape the bundled conformation of spools which can sometimes trigger spool disassembly (Hess et al., 2005; Liu et al., 2008; VanDelinder et al., 2016b).
MT loop or spool formation in gliding assays under different conditions. a) primarily the lower range of mentioned diameters is listed; b) not clear from experimental section; c) measured from images. References [1] (Amos and Amos, 1991), [2] (Liu et al., 2011), [3] (Rashedul Kabir et al., 2012), [4] (Kawamura et al., 2008), [5] (Hess et al., 2005), [6] (Lam et al., 2014), [7] (Wada et al., 2015), [8] (Luria et al., 2011), [9] (VanDelinder et al., 2016b), [10] (Liu et al., 2008), [11] (Liu and Bachand, 2013)
Notably, key parameters promoting MT loops in gliding assays can also be found in the narrow axonal tubes: they are force-enriched due to the high density of MT-associated motor proteins, and growing/gliding MTs have a large probability to collide or get obstructed by the crowded organelle or protein content of axons (Fig.1). In line with this argument, the observed loop diameters in vitro roughly approximate observed diameters of disorganised MTs in axons (Tab.1, Fig.5), and even spool shapes can resemble MT conformations observed in growth cones of fly or mammalian neurons (Dent and Kalil, 2001; Hess et al., 2005; Sánchez-Soriano et al., 2010). Therefore curled MT conformations in axons might find explanations from in vitro work, and a number of mathematical models were put forward to describe loop or spool dynamics in gliding assays (Crenshaw et al., 2011; Gosselin et al., 2016; Luria et al., 2011; Pearce et al., 2018; Ziebert et al., 2015). Of these, two models provide mechanistic ideas for how bending through external forces can result in semi-stable loops.
The first model builds on known conformational changes of tubulin. Thus, tubulins in non-hydrolysed GMPCPP-MTs are 1-3% longer than hydrolysed GDP-tubulin, and taxol added after (but not during) polymerisation achieves a similar elongation (Fig.6F; Alushin et al., 2014; Amos and Löwe, 1999; Arnal and Wade, 1995; Castle et al., 2017; Hyman et al., 1995). Notably, this conformational change seems physiologically relevant, as its suppression by the T238A mutation in yeast ß-tubulin stabilises MTs in vivo and causes mitotic defects (Geyer et al., 2015; Machin et al., 1995). Since virtually all gliding assays use taxol-treated (hence extended) MTs (Tab.1), the first model proposes that kinesin-mediated MT bending switches tubulins located on the concave side of the tube into the shorter conformation. This conformation can be maintained as an energetically favoured state, which might be further assisted by MT-binding proteins (Fig.6F; Ziebert et al., 2015).
The second model is based on work showing that kinesin-1 has a preference for convex MT surfaces and can stabilise MT curvature by extending their lattice to similar degrees as taxol (translating into a diameter of curvature of 3.2 μm; Peet et al., 2018). The conformational changes imposed by kinesins include compaction of tubulin that goes beyond taxol-or GMPCPP-induced effects (Krebs et al., 2004; Morikawa et al., 2015), and kinesins were found to bind cooperatively to MTs potentially causing a snowball effect (Cross, 2019; Muto et al., 2005). The model proposes therefore that binding of kinesin-1 to the convex side can drive a bias towards curvature (Pearce et al., 2018). This model is particularly attractive for the non-extended GDP-MTs in axons, and it could apply to further axonal lattice-associating proteins such as tau and doublecortin, which also seem to bind differently to straight and curved MTs (Balabanian et al., 2017; Bechstedt et al., 2014; Ettinger et al., 2016; Samsonov et al., 2004).
Naturally, these models are in their infancy and will have to be refined by gradually incorporating further reported findings. For example, MTs behave as elastic cylinders (comparable to a garden hose) and can undergo softening through cross-sectional flattening when strongly bent (Fig.6E; Kononova et al., 2014; Memet et al., 2018). In this same vein, conformational changes of MTs upon kinesin-1 binding were reported to soften MTs locally rather than increase their rigidity (Kabir et al., 2014). If confirmed, this would have important implications for any existing models; together with the kinesin-induced tubulin compaction (yellow asterisks in Fig.6F), it might be a mechanism to absorb energy and reduce the shear force load on MTs.
In conclusion, formations of axonal areas of disorganised curled MTs could be seen as processes of ‘active self-organisation’, for which insights from gliding assays provide attractive explanations (Lam et al., 2016): extrapolation from in vitro work would suggest that MT curling could be caused through an interdependent relationship between the force-enriched environment and the responses of MTs as a function of their intrinsic properties. It is now important to challenge this view and perform thorough analyses across genetic conditions and animal models, by obtaining neutral parametric descriptions of MT disorganisations observed in the different conditions (Fig.5), and of the dynamics through which they are initiated and maintained. If such studies revealed comparable parameters across different conditions, this would support the idea of a common concept behind the unusual MT behaviours in axons - as proposed by the model of local axon homeostasis. In the next section we will summarise roles of axonal MT-associated motors during axon pathology and explore whether they might be the key drivers of MT disorganisations.
The intricate relationship between MTs and their associated motor proteins
MT-associated motors comprise the minus end-directed dynein/Dynactin complex and the mostly plus-end directed proteins of the kinesin family (Hirokawa et al., 2010). Several kinesins display direct roles in MT regulation (Sturgill and Ohi, 2013). These include active MT depolymerisation (kinesin-8, −13; Walczak et al., 2013) as well as MT polymerisation (kinesin-2, −5; Chen and Hancock, 2015; Gumy et al., 2013; Guzik-Lendrum et al., 2017), MT-cross-linkage (kinesin-5, −6, - 12; see section on bundling), and roles in promoting MT orientation as a feature of neuronal polarity (Tas et al., 2017; Zheng et al., 2008).
However, most attention is given to the active cargo and organelle transport and dynamics in axons (Fig.3A-E; see section on axonal cytoskeleton), which is driven retrogradely by dynein/Dynactin (Allan, 2011) and anterogradely by kinesins (primarily kinesin-1, −2, and −3; Hirokawa et al., 2010). The forces imposed by these dynamics and/or the size of cargoes moved, poses an obvious challenge to MT bundles (Appert-Rolland et al., 2015) and might be the main correlate of the bending forces generated by kinesin carpets in gliding assays (see previous section).
Clearly, there is an intricate mutual regulatory relationship and finely tuned balance between the amount of transport, and the structural properties of the transport highways (Appert-Rolland et al., 2015; Prokop, 2013). For example, MT density is higher in small calibre axons than in large axons (~15 versus ~150 MTs/μm2), and mathematical modelling suggests that this is required to achieve the same transport efficiency as in large axons (Wortman et al., 2014; and references within). The tubulin isotype composition of MTs, their posttranslational modifications, and the physical presence of other MT-binding proteins influence motor protein dynamics (‘a’ in Fig.3; Balabanian et al., 2017; Monroy et al., 2018; Sirajuddin et al., 2014; Subramaniyan Parimalam et al., 2016). Vice versa, it has been reported that motor proteins cause damage to the MTs they walk on (Dumont et al., 2015; Peet et al., 2018; Triclin et al., 2018; VanDelinder et al., 2016a), which likely triggers maintenance responses including MT repair (Akhmanova, 2018) or potentially even replacement (‘14’ in Fig.3).
Tipping the balance in this mutual relationship can easily be imagined to cause reciprocal deficiencies in transport rate and MT bundle organisation. For example, disorganisation or partial breakage of MTs has been reported to cause pathological transport deficits (Fiala et al., 2007; Tang-Schomer et al., 2012). Vice versa, immunological lesioning experiments initially caused transport defects, which were then followed by MT disorganisation (Sorbara et al., 2014). Analogously, we observe severe MT disorganisation in Drosophila primary neurons upon loss of kinesin-1 or −3 (kinesin-1 shown in Fig.5E).
How loss of these kinesins can cause MT disorganisation can currently only be hypothesised. For example, it has been reported for dendrites that kinesin-1 migrates on acetylated and kinesin-3 on tyrosinated MTs (Tas et al., 2017). Provided the same is true in axons, the loss of kinesin-1 would relieve acetylated MTs, but tyrosinated MTs would still bear their full transport load - and vice versa. Such imbalances in transport distribution across MTs could lead to shear forces that buckle MTs and seed MT disorganisation. In the same vein, MT disorganisation was reported to be triggered by directional changes in motor traffic upon deficiency of the dynein regulator NDEL1 at the axon initial segment (Kuijpers et al., 2016). Furthermore, the movement of large cargoes likely induces dynamic rearrangements of local MT-MT crosslinking networks (see section on cross-linkage); in this scenario, violating the balanced proportion between cross-linkers and transport load may become a path to bundle aberration.
Alternatively, transport defects might affect MTs through biochemical routes, simply caused by the fact that the bundle-maintaining machinery runs out of supply. There would be expected distribution gaps (a) of tubulin heterodimers as building blocks, (b) of the proteins required to execute MT bundle maintenance work (‘b’ in Fig.3), and (c) of organelles. Organelle deficiencies can trigger systemic changes that would likely affect MT maintenance, as detailed in Box 2.
Functional interdependencies of organelles with MTs may explain why different types of Charcot-Marie-Tooth disease or hereditary spastic paraplegias can be caused through motor proteins as well as regulators of membranous compartments (Blackstone, 2018; Bucci et al., 2012). In agreement with this line of argumentation, MT stabilising drugs have been beneficial in animal models of neurodegeneration, including SPG4 (Box 2) and Alzheimer’s disease (Brunden et al., 2014). Vice versa, axonal swellings induced by senile plaques in the Tg-swAPPPrp mouse model of Alzheimer’s disease were strongly enhanced when removing one copy of the KLC1 gene (a linker required for kinesin-1 mediated transport), and this effect was found to be conserved in Drosophila (Stokin et al., 2005).
Naturally, the argumentative framework presented here is highly speculative, given the enormous complexity of the relationships between MT bundle organisation, motor protein activity and systemic factors. But we hope that these reflections will motivate experimenters to have a closer look at MTs in future studies of axon biology and pathology. More data are urgently needed, which does often not require more than analysing neuronal morphology with antisera against MTs (rather than restricting to intermediate filaments), or increasing the magnification in ongoing ultrastructural studies to have a closer look at MTs. In the following sections we will explore the mechanisms that are potentially used to prevent motor-induced MT bundle aberrations.
Box 2. The intricate relationship between MTs and axonal organelles
Mitochondria are the main source for ATP (Sheng, 2017), required to fuel multiple processes relevant for MT regulation (red arrows in Fig.3); these include actin assembly and dynamics (Krendel and Mooseker, 2005; Skruber et al., 2018), protein phosphorylation (Bogoyevitch and Fairlie, 2007), GTP production required for MT polymerisation and signalling (Berg et al., 2002; Hall and Lalli, 2010; Voelzmann et al., 2016a), MT severing (McNally and Roll-Mecak, 2018), and MT-motor dynamics (Hirokawa et al., 2010; although vesicular transport uses local glycolysis to generate its own ATP; yellow star in Fig.3A; Hinckelmann et al., 2016; Zala et al., 2013). Secondly, the mitochondrial surface is an important signalling platform and could be used to orchestrate MT regulation locally (not shown in Fig.3; McBride et al., 2006). Thirdly, mitochondria cooperate with ER in the regulation of intracellular free calcium (yellow cloud in Fig.3; Rieusset, 2017; Wu et al., 2017) which has direct impact on MT regulators (e.g. spectraplakins, tau, kinesins; Kapur et al., 2012; McVicker et al., 2015) or even on MTs themselves (O’Brien et al., 1997). Fourthly, mitochondria collaborate with peroxisomes in the regulation of reactive oxygen species (‘ROS’ in Fig.3; Fransen et al., 2017; Pascual-Ahuir et al., 2017), which have known effects on MT regulation (Wilson and Gonzalez-Billault, 2015).
Furthermore, aberrations of axonal transport or MT-bundle organisation can cause mitochondrial damage or dysregulation of the mitochondria-peroxisome system, both leading to oxidative stress as a major path to axon pathology (Fiala et al., 2007; Liu et al., 2017; Pascual-Ahuir et al., 2017). Such causative relationships between MTs and oxidative stress can be experimentally demonstrated: for example the MT-stabilising drug epothilone B rescues oxidative stress caused by peroxisome transport deficiencies in a human iPSC model of SPG4 (spastin-linked spastic paraplegia 4; Wali et al., 2016).
Similar interdependencies would apply to other important organelles or membrane compartments that likewise depend on MT-binding motor proteins to undergo meaningful dynamics (Fig.3D); of particular importance are the ER with its multiple roles in calcium homeostasis, protein synthesis and lipidogenesis (Gonzalez and Couve, 2014), or the endolysosomal system required for proteostasis (Winckler et al., 2018). For example, drug-induced inhibition of the proteasome-ubiquitination system has been shown to induce alteration in MTs and axonal transport (Staff et al., 2013).
MT polymerisation as a fundamental requirement for bundle maintenance
As mentioned before, the numbers of axonal MTs have to be well adapted to the transport load (Wortman et al., 2014), and this requires a well-regulated machinery of MT polymerisation and disassembly (blue stippled arrows in Fig.3). On the one hand, MT volume has to be generated de novo during axon growth (‘8’ in Fig.3), and thereafter this volume has to be maintained at steady-state and prevented from MT senescence, which requires MT repair but likely also MT turn-over (‘14’ in Fig.1; Akhmanova, 2018; Triclin et al., 2018; Voelzmann et al., 2016a).
As we detailed in a previous review (Voelzmann et al., 2016a), the machinery of MT de-/polymerisation requires three sub-machineries: (1) dynamic protein complexes at the MT plus end (blue balls, ‘Eb1’ in Fig.3); (2) the supply of α/β-tubulin heterodimers as building blocks which occurs through a complex regulatory network in close co-regulation with MT dynamics (‘c’ in Fig.3; Gasic and Mitchison, 2018; Preitner et al., 2014); (3) proteins which bind or post-translationally modify the MT lattice, for example through stabilising MTs against depolymerisation (‘7’ in ‘Fig.3). Such complex machinery has to be orchestrated in tune with the wider systemic context of axons. This is illustrated by our recent work in Drosophila neurons, showing that loss of cortical actin rings in the axon shaft (Fig.1) causes a reduction in MT polymerisation speed, eventually affecting MT bundle integrity; simultaneous application of MT-destabilising drugs or removal of the MT-stabilising spectraplakin Short stop (Shot) exacerbated these effects, frequently even eliminating entire axons (Qu et al., 2017). Similar co-dependencies are suggested by other reports: (1) parallel loss of spectrin and tau causes axonal MT loss in C. elegans (Krieg et al., 2017); (2) axon-shortening induced by the MT-stabiliser taxol can be ameliorated through co-application of actin-destabilising drugs (in both chick and Drosophila neurons; Letourneau et al., 1987; Sánchez-Soriano et al., 2010, or vice versa Datar et al., 2019); (3) application of actin-destabilising drugs to PC12 cells changes the tubulin to microtubule ratio (Dennerll et al., 1988). Such co-dependencies of MT polymerisation on MT stabilisation and actin networks are currently best explained by biomechanical models as detailed in Box 3.
Box 3. Biomechanical models of axon growth
The regulation of axonal growth dynamics has been explained with the concept of tensegrity (tensional integrity), an architectural principle based on structural nets that are under continuous tension whilst containing isolated components under compression (Buckminster Fuller, 1961; Ingber and Folkman, 1989). In axons, “actin is under tension supported in part by microtubules under compression” (Heidemann and Buxbaum, 1990). Tension is provided by pulling acto-myosin networks in growth cones (Fass and Odde, 2003; Heidemann et al., 1990), the rigid but contractile properties of cortical actin in the axon shaft (Fan et al., 2017; Heidemann and Buxbaum, 1990; Krieg et al., 2017; Xu et al., 2013; Fig.1), and the stiff nature of cross-linked MT bundles is well suited to oppose compressive forces up to a certain threshold (Fig.2).
In such a balanced system, destabilisation of MTs increases tension at the expense of compression, and a decrease in acto-myosin networks or contractility reduces tension in favour of compression; force-generating MT polymerisation is one component responding to and regulating this balance with ultimate impact on axon length (Buxbaum and Heidemann, 1988; Dennerll et al., 1988; Heidemann et al., 1990; Letourneau et al., 1987).
In further agreement with this hypothesis, pulling the tips of axons enhances their growth rate (Bray, 1984; Lamoureux et al., 2010; Zheng et al., 1991); even single MTs polymerise faster when being pulled on in vitro (Brouhard and Rice, 2018). The necessary translation of the mechanical stimulus into changes of MT polymerisation rates might therefore occur at the level of MT polymerases (Brouhard and Rice, 2018) and/or be mediated by mechano-sensitive calcium channels in the axonal membrane (Franze et al., 2009; He et al., 2019; Heidemann and Buxbaum, 1990).
Cortical guidance and elimination of polymerising MTs
Whilst well-equilibrated MT polymerisation is a requirement for axonal maintenance, it also poses a risk, in that extending MTs can accidentally project out of the bundle and seed MT disorganisation (‘4’ in Fig.3), potentially similar to loop formation upon MT obstruction in gliding assays (see section on curling). A key factor preventing this from happening is Eb1 (Alves-Silva et al., 2012; Figs.3 and 5B). Eb1 directly binds at extending MT plus ends where it promotes polymerisation (Zanic et al., 2013) and serves as a scaffold for many other proteins (Gupta et al., 2014).
One mechanism through which Eb1 maintains extending MTs in bundled configuration, is MT guidance mediated by Short stop (Shot). Shot is a well-conserved spectraplakin, able to cross-link cortical actin, MTs and Eb1 (‘5’ in Fig.3), thus guiding polymerising MTs in parallel to the axonal surface and laying them out into parallel bundles (Alves-Silva et al., 2012). Similar to Eb1 deficiency, also loss of Shot causes severe MT disorganisation in axons - and the same is true for its two mammalian homologues ACF7 and dystonin (Bernier and Kothary, 1998; Dalpe et al., 1998; Sánchez-Soriano et al., 2009; Voelzmann et al., 2017) - of which the latter links to the axonopathy HSAN6 (type 6 hereditary sensory and autonomic neuropathy; Edvardson et al., 2012).
Such cortical guidance is complemented by at least one control mechanism: if MTs (accidentally) leave their bundled arrangements and extend towards the cortex, they get eliminated by Efa6 (‘4’ in Fig.3), a cortical collapse factor that associates with the axonal membrane via its C-terminal plekstrin homology domain; consistent with the model of local axon homeostasis, loss of Efa6 causes three neuronal phenotypes, in culture as well as in fly brains: significant MT disorganisation (Fig.5D), longer axons, and more axonal branches (Qu et al., 2018); all three phenotypes are based on morphogenetic processes to which ‘off-track’ MTs can contribute (curved arrows in Fig.1). Our model would predict that mutant phenotypes caused by loss of Shot and Efa6 should enhance each other because they are caused through complementary mechanisms of MT bundle regulation. Accordingly, we found a clear increase in MT disorganisation when removing both Shot and Efa6 from the same neurons (Qu et al., 2018). We propose therefore that, Shot and Eb1 keep MTs away from the membrane, whereas Efa6 acts as a finely tuned quality control factor eliminating accidental off-track MTs, whilst still permitting enough MTs to get through to perform intended functions in axon growth and branching.
Interestingly, the cortical collapse function of Efa6 is not conserved in vertebrates (Qu et al., 2018). But the concepts derived from Efa6 studies still appears relevant, because loss of the unrelated neuronal cortical collapse factor KIF21A causes analogous phenotypes in mammalian neurons: KIF21A mutations linked to the neurodevelopmental disorder CFEOM1 (type 1 congenital fibrosis of the extraocular muscles) affect axon growth and axonal branching just like Efa6 (Qu et al., 2018; van der Vaart et al., 2013), and we would also predict potential increases in MT disorganisation (no data available).
Guidance along cortical actin seems not the only mechanism through which Eb1 and Shot keep MTs on track. This is illustrated by the simple fact that MT disorganisation observed upon loss of Shot or Eb1 (Fig.5B, E) does not occur when removing actin from axon shafts (Alves-Silva et al., 2012; Qu et al., 2017; Sánchez-Soriano et al., 2010). This suggests that both factors perform additional, actin-independent functions or interactions to promote MT bundles.
For example, the unusual Shot-PH isoform, which is highly enriched in the nervous system and harbours a plakin repeat region (PRR; conserved in mammalian spectraplakins), is a likely candidate for such roles, but still has to be investigated (‘11’ in Fig.3; Hahn et al., 2016; Voelzmann et al., 2017). Eb1 has a long list of protein interactors besides Shot (Gupta et al., 2014), and some of them might associate with MTs and guide extending plus ends along pre-existing bundles (‘9’ in Fig.3); for example, they could be proteins, such as APC-like or GAS2-LIKE family members (Pickled eggs/Pigs in Drosophila), known to bind both MTs and Eb1 in Drosophila and mammals (Beaven et al., 2015; Pines et al., 2010; Stroud et al., 2014).
Role of severing proteins and MT-destabilising kinesins
As indicated in the previous section, MT disorganisation, axon growth and collateral axon branch formation are driven by MTs leaving the bundled conformation (curved arrows in Fig.1; Kalil and Dent, 2014; Tint et al., 2009; Tymanskyj et al., 2017). Consequently, MT-disassembling factors should have the principal potential to negatively regulate either process. This is true for cortical MT collapse factors (Drosophila Efa6, mammalian Kif21A; see previous section) which can down-regulate axonal growth, branching and MT disorganisation. Also the MT-depolymerising kinesin-13 family member Kif2A (Homma et al., 2003) and MT severing proteins (spastin, katanin and fidgetin) were reported to inhibit neurite growth and/or branching (Leo et al., 2015; Mao et al., 2014; Tao et al., 2016). Even more, MT disorganisation is observed upon the losses of Drosophila katanin (our unpublished results) or mammalian spastin (Denton et al., 2014; Fassier et al., 2013; Havlicek et al., 2014; Tarrade et al., 2006).
However, other studies of spastin, katanin and fidgetin led to contradictory findings, describing them as promoters rather than inhibitors of neurite growth and branching (Ahmad et al., 1999; Butler et al., 2010; Havlicek et al., 2014; Karabay et al., 2004; Riano et al., 2009; Stewart et al., 2012; Stone et al., 2012; Wood et al., 2006; Yu et al., 2008). Such stark, potentially context-dependent deviations seem to reflect the complex regulation of severing proteins. Thus, spastin, katanin and fidgetin are all members of the superfamily of AAA proteins (ATPases associated with diverse cellular activities; McNally and Roll-Mecak, 2018; Sharp and Ross, 2012; Zhang et al., 2007), but their severing activity is differentially regulated through their individual responses to (a) posttranslational MT modifications (in particular acetylation and poly-glutamylation; Bailey et al., 2015; Lacroix et al., 2010; Leo et al., 2015; Shin et al., 2019; Sudo and Baas, 2010; Valenstein and Roll-Mecak, 2016), (b) antagonistic MT shaft-binding proteins such as tau (Qiang et al., 2018; Qiang et al., 2006; Yu et al., 2008), or (c) spatial recruitment through specifically localised proteins such as CAMSAP (Jiang et al., 2018).
Through this precise spatiotemporal regulation of their activity, severing proteins are believed to either eliminate MTs or to break them up into stable fragments that serve as seeds for MT amplification (Baas et al., 2016; McNally and Roll-Mecak, 2018; Vemu et al., 2018). These properties of severing proteins could be used in different ways to prevent MT disorganisation: First, by targeting disorganised MTs for elimination, MT severing proteins could serve as quality control factors (‘6’ in Fig.3) that complement roles of cortical collapse factors (‘4’ in Fig.3). In agreement with this idea, katanin in plant cells was reported to localise and sever preferentially at MT cross-points, which can be used to take out non-aligned MTs (McNally and Roll-Mecak, 2018).
Second, MT shortening functions of katanin are required at MT minus ends. Thus, in both mammals and Drosophila, the minus-end capper CAMSAP/Patronin protects against MT disassembly, and recruits katanin to counterbalance against uncontrolled minus-end extension (‘13’ in Fig.3; Goodwin and Vale, 2010; Jiang et al., 2018; Nashchekin et al., 2016); such uncontrolled extension may cause MTs to go off-track or to buckle through extra forces produced.
Third, MT elimination functions could prevent MT bundle senescence. Thus, MTs suffer from damage through tear-and-wear (Dumont et al., 2015; Peet et al., 2018; Triclin et al., 2018; VanDelinder et al., 2016a), which might cause bundle aberration by abrogating interactions with MT-binding proteins (red cross at ‘16’ in Fig.3). On the one hand, this is addressed by immediate lattice repair involving katanin or spastin (Davis et al., 2002; Diaz-Valencia et al., 2011; Gasic and Mitchison, 2018; Triclin et al., 2018; Vemu et al., 2018). On the other hand, spastin and katanin could prevent senescence through selective elimination of aged MTs (as similarly suggested for kinesin-8 or −13; Gardner et al., 2011), followed by compensatory polymerisation (‘14’ in Fig.3). Such a mechanism might explain why spastin deficiency in the SpΔ mouse model causes a drop in MT polymerisation whilst triggering MT disorganisation (Fassier et al., 2013).
However, the MT phenotypes observed in the SpΔ mouse model could likewise be explained through the opposite role of spastin in MT multiplication, rather than elimination. Thus, without spastin, MT numbers might gradually decline and cause transport interruptions which, in turn, would affect MT bundle organisation and eventually cause systemic pathology (see section on motor proteins; Wali et al., 2018; Wali et al., 2016). In this way, not failing MT turn-over (causing senescence) but impaired homeostasis of MT numbers (causing transport defects) might lie at the root of the problem; curiously, axon swellings in this model were reduced with low doses of MT-stabilising or -destabilising drugs (Fassier et al., 2013), not favouring either of the explanations.
Understanding spastin is important because it is by far the most prominent factor linking to spastic paraplegias worldwide (Koh et al., 2018; Schüle et al., 2016), and axonal swellings are a hallmark of the disease (Blackstone, 2018; Zempel and Mandelkow, 2015). Most SPG4-linked mutations lie within the AAA-ATPase domain (Shoukier et al., 2009), suggesting that MT severing is key to the disease pathology. However, point mutations might generate versions of spastin, which either act as dominant negative alleles (forming dysfunctional complexes that titrate out other spastin-interacting factors), or acquire gain-of-function qualities by diffusing away to perform very different roles. One such MT-independent role of spastin is the isoform-specific regulation of the endoplasmic reticulum (‘15’ in Fig.3), including its shape, interaction with the endosome or production of lipid droplets (Allison et al., 2017; Papadopoulos et al., 2015; Park et al., 2010; Solowska and Baas, 2015). Therefore, it is difficult to exclude that at least part of those mutations causes systemic rather than MT aberrations as the initial triggers of axon decay (‘3’ in Fig.4).
Structural support through MT-MT cross-linkage
MT-MT cross-linkage (‘12’ in Fig.3) is likely the oldest mechanistic concept put forward by neurobiologists to explain MT bundles (Lee and Brandt, 1992) and appears an obvious means of suppressing MT disorganisation. In physical terms, axons have been described as a “stiff spring in series with a viscoelastic (Voight) element composed of a less stiff spring in parallel with a fluid dashpot” (Heidemann et al., 1990), meaning that axons are under rest tension and combine elastic and viscous properties. A central structural component underpinning such properties is likely provided by networks of MT-MT cross-linkers (Fig.2), where each linker is able to detach upon super-threshold pull or compression, and re-attach thereafter (slip-bonds). Physical cross-linking strands between axonal MTs were observed decades ago (Hirokawa, 1982, 1986), and mathematical models support MT-MT cross-linkage as an important structural feature of axons (e.g. de Rooij and Kuhl, 2018; Lazarus et al., 2015; Li et al., 2018; Peter and Mofrad, 2012).
However, the underlying genetic factors remain surprisingly controversial to this day - to a degree that one cannot even fully exclude a model view where linkers do not attach MTs but rather separate them, whilst the corset of contractile cortical actin rings (Fig.1) constrains them into a dense bundle (Fan et al., 2017). This uncertainty is partly due to the surprisingly low number of publications reporting structural bundle defects upon loss of putative MT-MT cross-linkers (see below). Further doubt comes from the recognition that MT bundling observed upon neuronal linker expression in non-neuronal cells, might represent artefacts, because MT bundling can even be achieved through expression of isolated MT-binding domains, or the application of the MT-stabilising drug taxol which causes bundles with ultrastructural cross-bridges that are indistinguishable from those induced by tau or MAP2 (Chapin et al., 1991; DeBonis et al., 2015; Goriounov et al., 2003; Kader et al., 2017; Lee and Brandt, 1992). Another example is dynamin, which is linked to Charcot-Marie-Tooth disease, has been shown to bundle MTs in vitro, but in vivo seems to bind membranes instead (Scaife and Margolis, 1990; Shpetner and Vallee, 1989; Züchner et al., 2005).
For example, MTLC1 and MAP1B (Futsch in Drosophila) appear ideal cross-linkers, because they both possess an N-and a C-terminal MT-binding domain, and were shown to induce bundles upon expression in non-neuronal cells (although MAP1B appears a weak bundler; Kader et al., 2017; Penazzi et al., 2016; Satake et al., 2017). However, there are only isolated reports that axonal bundle defects occur in their absence (Bettencourt da Cruz et al., 2005; Satake et al., 2017). Instead, the long history of MAP1B research is mostly dedicated to aspects of axon development (Villarroel-Campos and Gonzalez-Billault, 2014). Its fly homologue Futsch promotes MT spools at synaptic terminals (Roos et al., 2000), but its axonal defects comprise merely growth aberrations but no MT disorganisation (Hummel et al., 2000; our unpublished results). Therefore, existing data for MAP1B/Futsch only vaguely support roles in axonal MT bundling.
The other conserved linker candidate tau, has only one central MT-binding region and achieves physical MT-MT linkage in vitro through N-terminal dimerisation (Chung et al., 2016; Méphon-Gaspard et al., 2016; Rosenberg et al., 2008). However, its dwell time on MTs seems very short (Janning et al., 2014; Samsonov et al., 2004), and reported tau-deficient phenotypes hardly ever comprise bundle aberration, but developmental neuronal defects instead (Krieg et al., 2017; Penazzi et al., 2016).
Pinpointing MT-MT cross-linkage through tau is further complicated by the fact that normal tau has been associated with a whole array of further molecular functions relevant for MT dynamics. For example, tau can protect MTs from severing by katanin (Qiang et al., 2006), bind tubulin hetero-dimers (Shin et al., 2018), switch between bundled and single MT states (Prezel et al., 2018), cross-link MTs with actin or the cortex (Biswas and Kalil, 2018; Cabrales Fontela et al., 2017; Maas et al., 2000), stabilise MTs during axon initiation (Brandt, 1998), maintain labile domains along MT shafts (Qiang et al., 2018), regulate End-binding proteins (Sayas et al., 2015), compete with kinesins (Trinczek et al., 1999), and promote MT nucleation and polymerisation (Penazzi et al., 2016). A similarly broad pleiotropy has been reported for MAP1B (Villarroel-Campos and Gonzalez-Billault, 2014).
A further complicating factor for pinpointing relevant MT-MT cross-linking activities are the obvious functional redundancies in the system. Thus, MAP1B and tau have enhanced growth phenotypes when both mutant conditions are combined in double-mutant mouse neurons (Takei et al., 2000). Complementary to this, co-expression of Futsch and Tau causes enhanced phenotypes in the Drosophila CNS (Hummel et al., 2000). Such functional redundancy likely extends to further potential cross-linkers. For example, Kinesin-5 (KIF11), kinesin-6 (KIF23, Pavarotti in Drosophila) and kinesin-12 (KIF15) are best known for their ability to slide anti-parallel MTs in the mitotic spindle (Baas, 1999); however, in axons where MTs are arranged in parallel these kinesins seem to inhibit sliding (Dong et al., 2019; Lin et al., 2012; Liu et al., 2010; Lu et al., 2013; Myers and Baas, 2007; Nadar et al., 2012) - and this can be considered a form of MT-MT cross-linkage. Notably, we observe that loss of Drosophila Pavarotti causes axonal MT disorganisation (our unpublished data), providing a readout for studying its potential linker functions. By incorporating redundancies of linker candidates into experimental approaches, there might be new opportunities to decipher the true molecular nature of MT-MT cross-linkage in axons.
Are MT bundles anchored to the axonal cortex?
Apart from cross-linkage between MTs, another bundle-stabilising aspect could be anchorage to the wall of the axonal tube. For example, polymerisation occurs all along axon shafts, and it has been proposed from studies in developing vertebrate and Drosophila neurons, that the MT mass generated in the axonal shaft during axon growth gradually shifts anterogradely (Miller and Sheetz, 2006; O’Toole et al., 2008; Reinsch et al., 1991; Roossien et al., 2013). Contributing forces were suggested to be generated by pulling forces in the rear of growth cones (O’Toole et al., 2015), by MT polymerisation, by thermal motion of MT-MT cross-linkers (Lansky et al., 2015), by kinesins actively sliding MTs along other MTs (‘B’ in Fig.3; Lu and Gelfand, 2017) or by dyneins sliding MTs along cortical F-actin (‘10’ in Fig.3; Ahmad et al., 2006; He et al., 2005; Myers et al., 2006; Roossien et al., 2014).
Potential MT sliding along cortical actin would be a form of tethering MT bundles to the axonal surface and raises the fundamental question as to whether MT bundles are free-flowing within the axonal tube, or anchored to it. Potential anchorage is suggested by observed co-drift of the axolemma with the axon core (Lamoureux et al., 2010; Popov et al., 1993; Zheng et al., 1991). Such anchorage would not have to be static, but might involve an interface of slip-bonds, as similarly suggested for back-flowing actin networks at stable focal adhesion sites (Case and Waterman, 2015). Furthermore, anchorage of MTs would not have to be restricted to the rigid cortical actin networks (Fig.1; ‘2’ in Fig.3; Xu, 2013 #6895}, but could also involve links to adhesion factors (‘3’ in Fig.3), thus forming mechano-sensing modules (Yap et al., 2018) that can respond to local shear forces generated between MT bundles and the axonal environment (Fig.1). Such adhesion-mediated local mechano-sensing could explain local regulation phenomena: for example, net rates of fast mitochondrial transport are regulated in a way that they gradually decrease towards distal axon segments, indirectly proportional to the slow local MT drift rate along the axon (which increased towards the distal end; Miller and Sheetz, 2006). This proximo-distal differences of fast mitochondrial transport could therefore be determined by mechano-sensing as a function of the local MT drift rate relative to the outer axonal environment.
Apart from dynein (see above), spectraplakins are candidate anchoring factors. This is suggested by Drosophila neurons lacking the spectraplakin Shot and treated with the MT-stabilising drug taxol: in these neurons, axonal MTs shifted distally, often leaving tubulin-free zones in the proximal axon shaft (Voelzmann et al., 2017). However, this same effect was not observed when applying taxol whilst also removing actin (Sánchez-Soriano et al., 2010), suggesting that Shot does more than simply anchoring to cortical actin in this experimental context.
This conclusion is in agreement with the breadth of axonal functions proposed for spectraplakins, making them the most conspicuous regulators of the axonal cytoskeleton in any species investigated so far (Voelzmann et al., 2017). For example, the role of Shot in preventing MT shift may involve a combination of molecular functions including MT-MT cross-linkage (‘11’ in Fig.3), actin-MT cross-linkage (‘2’ and ‘5’ in Fig.3), as well as two other potential anchoring mechanisms: Firstly, Shot is able to compartmentalise the localisation of the cell adhesion molecule Fasciclin 2 (Bottenberg et al., 2009; Prokop et al., 1998), suggesting that it might be able to link to membrane-associated proteins like its mammalian homologue dystonin (‘3’ in Fig.3; Voelzmann et al., 2017). Secondly, work on non-neuronal cells of fly and mammals revealed that spectraplakins can interact with the MT minus end-stabilising factor CAMSAP/Patronin (a factor known to be relevant for neuronal morphology; Yau et al., 2014), thus anchoring MT minus ends to the cell cortex (‘1’ in Fig.3; Nashchekin et al., 2016; Ning et al., 2016; Noordstra et al., 2016).
Taken together, axonal MT bundles are likely tethered to the cortex, with dynein and/or spectraplakins as potential anchors. But further factors have to be considered. For example, several MT lattice-binding and/or -cross-linking proteins were similarly reported to bind to actin or to the cortex, and these include tau, MAP1B, APC and dynamin (Biswas and Kalil, 2018; Blanchoin and Michelot, 2012; Brandt et al., 1995; Elie et al., 2015; Gu et al., 2010; Maas et al., 2000; Mohan and John, 2015; Villarroel-Campos and Gonzalez-Billault, 2014). They could cross-link MTs to actin at the cortex (‘2’ in Fig.3) or to central longitudinal actin trails (Fig.1; Leterrier et al., 2017), thus further contributing to the intricate cross-linking networks expected to stabilise MT bundles. Cross-linking MT bundles internally and anchoring them to the axonal tube would constitute a firm structure, able to prevent MT buckling and bundle deformation caused by the enormous forces imposed by axonal cargo transport.
Conclusions and future perspectives
Here we have presented a conceptual view by describing a functional interactome that integrates the enormous complexity of cross-regulatory networks acting at the local level in axons. We propose that there has to be a fine balance between damaging effects inflicted by life-sustaining ‘associated’ motor movements (‘A-E’ in Fig.3) and the activities that maintain the highways required for this movement (MT-‘taming’ mechanisms; ‘1-15’ in Fig.3), fine-tuned through a number of cross-regulatory mechanisms (‘a-e’ in Fig.3).
Our model integrates a broad range of findings from the literature and speculative conclusions drawn. But its original foundations are derived from our own work in Drosophila neurons as one consistent model. Like other genetic invertebrate models, Drosophila provides a cost-effective and fast system to unravel the functional overlap and interface of different genetic factors - ideal to dissect complex machinery and deliver data that then often apply to axons of higher animals (Beaven et al., 2015; Prokop, 2018; Prokop et al., 2013).
This strategy offers one feasible path towards solving the daunting task of disentangling the enormous regulatory complexity; the model of local axon homeostasis could provide the basis on which to formulate helpful working hypotheses. A good starting point might be to break down the local axon homeostasis machinery into classifiable sub-machineries, such as those discussed in the different sections of this review.
In our view, we urgently require a shift away from prioritising molecular mechanisms and should rather dedicate to exploring their integration into wider regulatory networks - a type of research that is too often disqualified as ‘incremental science’ (Cohen, 2017). In our opinion, this would be a much needed paradigm shift with a realistic chance of providing us with far better understanding of axonal cell biology at the organisational level at which axonopathies become manifest.
Such an integrative approach has to take into consideration that knowledge derived from non-neuronal cells might not apply in neurons (Beaven et al., 2015). Furthermore, the interactome shown in Fig.3 makes clear that we will need quantitative approaches: we know increasingly well how factors bind to MTs and partly understand how they might compete with each other. But how crowded can a single MT be, how many molecules are there at any time in its surrounding, and how much dynamic exchange is taking place? Computational modelling will be an unavoidable means to make sense of existing data and make reasonable predictions to inform experimentation (Cohen, 2004; Gunawardena, 2014).
Integrated understanding of axon biology will also improve our knowledge of the next higher level of complexity, i.e. the mechanisms that orchestrate axon homeostasis, and can help to maintain balance even during phases of change (when switching from growth to differentiation, or during stress, injury, regeneration) - or tip the balance inducing degeneration. Obviously, signalling networks or dynamic changes of systemic factors such as second messengers or the ‘tubulin code’ will be key players to this end (Baas et al., 2016; Park and Roll-Mecak, 2018; Schelski and Bradke, 2017; Wilson and Gonzalez-Billault, 2015) - and glial cells will likely act as important external influencers of such processes (Pan and Chan, 2017).
Finally, MTs have been recognised as promising therapeutic targets (Baas and Ahmad, 2013; Eira et al., 2016; Zempel and Mandelkow, 2015), and urgently needed advance on this translational path will be facilitated by a better understanding of the axonal MT homeostasis system.
Acknowledgements
Work underpinning this article was made possible through support by the BBSRC to A.P (BB/I002448/1, BB/P020151/1, BB/L000717/1, BB/M007553/1), by parents to Y.-T.L., by the Leverhulme Trust to I.H. (ECF-2017-247). The Manchester Bioimaging Facility microscopes used in this study were purchased with grants from the BBSRC, The Wellcome Trust and The University of Manchester Strategic Fund. The Fly Facility has been supported by funds from The University of Manchester and the Wellcome Trust (087742/Z/08/Z). We thank Anthony Brown for introducing A.P. to the arguments that support a role of intermediate filaments in axon diameter.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵




