

Abstract
Identification of novel mechanisms of apoptosis resistance of prostate cancer (PCa) cells has translational importance. Here, we discover that inhibition of tumor suppressor phosphatase PP2A by PME-1 inhibits anoikis (apoptosis in anchorage-independent conditions) in PTEN-deficient PCa cells. PME-1 physically associated with the nuclear lamina and regulated its deformability in PCa cells. In addition, PME-1 deficient cells, with highly deformable nuclear lamina, were particularly vulnerable to anoikis following cell detachment. As a molecular explanation for increased nuclear lamina deformability, PME-1 depletion induced dephosphorylation of nuclear lamina constituents, Lamin-A/C, Lamin-B1, Lamin-B2, LAP2A, LAP2B, and NUP98. PME-1 inhibition increased apoptosis also in an in ovo tumor model, and attenuated cell survival in zebrafish circulation. Clinically, PCa patients with inhibition of both PP2A and PTEN tumor suppressor phosphatases (PME-1high/PTENloss), have less than 50% 5-year secondary-therapy free patient survival, which is significantly shorter than survival of patients with only PTEN-deficient tumors.
In summary, we discover that PME-1 overexpression supports anoikis resistance in PTEN-deficient PCa cells. Further, increased nuclear lamina deformability was identified as plausible target mechanism sensitizing PME-1-depleted cells to anoikis. Clinically, the results identify PME-1 as a novel candidate biomarker for particularly aggressive PTEN-deficient PCa.
Clinical relevance While organ-confined PCa is mostly manageable, the local and distant metastatic progression of PCa remains a clinical challenge. Resistance to anoikis is critical for PCa progression towards aggressive CRPC. Our data show that PME-1 expression in human PCa cells protects the cells from apoptosis induction in anchorage-independent conditions both in vitro and in vivo. Clinically, our results identify PME-1 as a novel putative biomarker for extremely poor prognosis in PTEN-deficient PCa. Taken together, our results demonstrate novel post-translational regulation of key cancer progression mechanisms, with clear translational implications.
Introduction
Prostate cancer (PCa) is often detected early, and can remain non-aggressive, and non-metastatic, for years. One of the hallmarks of PCa progression towards an aggressive castration-resistant prostate cancer (CRPC) is that PCa cells acquire resistance towards specific type of programmed cell death, anoikis (1). Anoikis is induced in adherent cells by their detachment from other cells, or from surrounding extra-cellular matrix (ECM). Anoikis suppression is not only relevant for indolent PCa cells to acquire anchorage-independence, but also for survival of prostate cancer cells with metastatic potential in circulation (2, 3). Thereby, characterization of mechanisms supporting anoikis resistance of PCa cells could provide novel therapy opportunities for clinical management of PCa by preventing the progression to aggressive and metastatic disease.
Mechanistically anoikis resistance in PCa has been linked to changes in cell adhesion, cytoskeleton, as well as deregulated intracellular survival pathways (1). Tumor suppressor phosphatase PTEN is inactivated in a large fraction of high grade PCas (4), and prostate-specific PTEN deletion in a mouse model leads to metastatic PCa (5). Importantly, both PTEN deletion, and hyperactivity of PTEN downstream target AKT increases anoikis resistance (6–8). On the other hand, recent studies in other cancer types indicate for a very delicate balance between nuclear lamina stiffness and anchorage-independence, anoikis resistance, and cell migration. Whereas cell migration in three dimensional contexts requires a sufficiently deformable nuclear lamina to allow passage through physical restrictions (9–11), too deformable nuclei may limit cancer cell survival especially in anchorage-independent conditions (9, 12–15). Of nuclear lamina proteins, particularly inhibition of both Lamin-A/C and Lamin-B1 sensitizes cells to DNA damage and apoptosis (16, 17). Further, Lamin A/C deficiency reduces circulating tumor cell resistance to fluid shear stress (12), and to sensitize cells to anoikis (14). Together, this evidence highlights the emerging importance of nuclear lamina deformability in defining anoikis resistance. However, the post-translational regulation of nuclear lamina deformability, or its importance for PCa progression is poorly understood.
Protein phosphatase 2A (PP2A) is another tumor suppressor phosphatase, in addition to PTEN, that is commonly inactivated in PCa. PP2A functions as a trimeric protein complex composed of the scaffolding A-subunit, catalytic C-subunit, and a number of regulatory B-subunits (18, 19). In most human cancers, PP2A is inactivated by overexpression of PP2A inhibitor proteins such as CIP2A, SET or PME-1 (20–22), whereas in a small percentage of cancers it is inhibited by either loss of specific B-subunits or by inactivating mutations in A-subunits (18). Overexpression of PP2A inhibitor protein CIP2A clinically associates with CRPC (26), and inhibition of both CIP2A and SET inhibits malignant growth of PCa cells, including PTEN-deficient CRPC cells (26, 27). In contrast, role for PME-1 in PCa is currently unknown. Interestingly, PP2A, CIP2A and PME-1 all physically associate with Lamin-A/C (28–30), and PP2A B-subunit PPP2R2A promotes nuclear lamina reformation after mitosis (31). However, the specific phosphorylation sites regulated by PP2A on nuclear lamina constituents, and the oncogenic relevance of PP2A’s potential role on nuclear lamina regulation, remain to be identified.
In this study, we demonstrate that PP2A inhibitor protein PME-1 (21), that has not been previously implicated in PCa, has a critical role in anoikis suppression of PTEN-deficient PCa cells both in vitro and in vivo. PME-1 regulates phosphorylation of several nuclear lamina proteins. Consequently, PME-1-depleted PCa cells have significantly more deformable nuclei, and those cells are hypersensitized to cell death under anoikis-inducing conditions. Clinically, PCa tumors with inhibition of both tumor suppressor phosphatases PTEN (genetic deletion), and PP2A (PME-1 overexpression), had significantly shorter relapse-free survival. Finally, PME-1 overexpression suppressed anoikis and promoted Lamin-A/C phosphorylation in genetically defined PTEN-deficient cell model.
Result
PME-1 promotes anchorage-independent growth of prostate cancer cells
Based on recent reports of potential clinical relevance of PP2A inhibition in human PCa (23–27, 32, 33), but lack of studies related to PME-1, we tested the impact of PME-1 depletion on colony growth of two PTEN-deficient PCa cells lines PC-3 (PTEN null) and DU-145 (PTEN heterozygous). PME-1 silencing by two independent siRNA sequences did not affect colony growth of either of the cell lines in 2D adherent culture conditions (Figure 1A). However, PME-1 silencing did decrease anchorage-independent growth on soft agar assay in both PC-3 and DU-145 cells (Figure 1B). To further assess whether PME-1 is particularly relevant for growth under low-attachment conditions, we performed spheroid growth assays on low attachment plates. PME-1 inhibition prevented the growth of floating PC-3 spheroids (Figure S1A), and reduced cell viability in these conditions (Figure 1C). Furthermore, we observed a strong synergy between PME-1 inhibition and low-attachment culture conditions in PARP cleavage (Figure 1D), indicating that apoptotic cell death contributes to decreased anchorage-independent growth of these cells. In order to rule out the possibility of siRNA off-target effects, we created a PC-3 PME-1 knock-out (KO) cell line by CRISPR/Cas9. Comparison of cleaved PARP expression between Cas9 overexpressing control cells with and without non-targeting gRNA (lanes 1-3), a pool of PME-1 gRNA transfected cells (lane 4), and a single cell subclone of PME-1 targeted cells (lane 5), demonstrated dose-dependent apoptosis induction upon PME-1 loss in cells following detachment (Figure 1E). PME-1 KO cells also displayed significantly reduced cell viability on low attachment plates (Figure S1B).

A. The effect of PME-1 depletion, utilizing two independent siRNAs, was characterized by colony formation assays (10 days of growth) in two PTEN-deficient PCa cells lines PC-3 and DU-145. The graphs show the quantified colony area, as mean fold change +/- SD compared to siScr. N=3.
B. PME-1 knock-down effect on anchorage-independent growth in soft agar assays (14 days of growth) in both PC-3 and DU-145 cells. The graphs show the quantified colony area, as mean +/- S.D. fold change compared to siScr. N =2, p-value: 2-sided t-test between colony areas in 15-20 image fields.
C. PME-1 inhibition effect on cell viability of PC-3 on low attachment plates, as measured by Cell Titer-GLO luminescence assay. The relative cell viability was calculated as compared to siScr (100%). N=2, Shown in mean + S.D, 2-sided t-test.
D. siScr and siPME-1 transfected PC-3 cells were plated 72h post-transfection on normal or low attachment plates for 24h, before collection and lysis, and subsequently analyzed by Western blot for cPARP induction.
E. Apoptosis induction, as measured by PARP cleavage, was studied by Western blot analysis in CRISPR/Cas9 generated PC-3 cells, with and without non-targeting gRNA (lanes 1-3), a pool of PME-1 gRNA transfected cells (lane 4), and a single cell subclone of PME-1 targeted cells (lane 5), after 24h detachment.
F. Mechanosensitive PARP cleavage in PME-1-depleted PC-3 cells. The cells were cultured on soft (0.5 kPa) or stiff (50 kPa) hydrogel for 24h before being scraped into PBS, spun down, lyzed and analyzed for protein expression. Results are representative of two independent experiments.
Previously, it was shown that non-transformed cells succumb to anoikis-type cell death when plated on soft substrates, whereas cancer cells do not die under same conditions (34). In order to test whether PME-1 expression is part of the apoptosis resistance mechanism also for cancer cells on low stiffness matrix, either control or PME-1 siRNA transfected PC-3 cells were plated on low- (0.5 kPa) or high (50 kPa)-stiffness hydrogels functionalized with ECM components (fibronectin and collagen I). Consistently with published results with other cancer cells (34), control PC-3 cells showed a very small increase in PARP cleavage on low stiffness (Figure 1F). Importantly, PME-1 depletion sensitized the cells to apoptosis induction selectively in low-stiffness conditions (Figure 1F).
Together these data demonstrate that PME-1 protects human PCa cells from apoptosis when cells are exposed to anchorage independence, or to low tissue stiffness.
PME-1 supports in vivo anoikis resistance and survival of prostate cancer cells in circulation
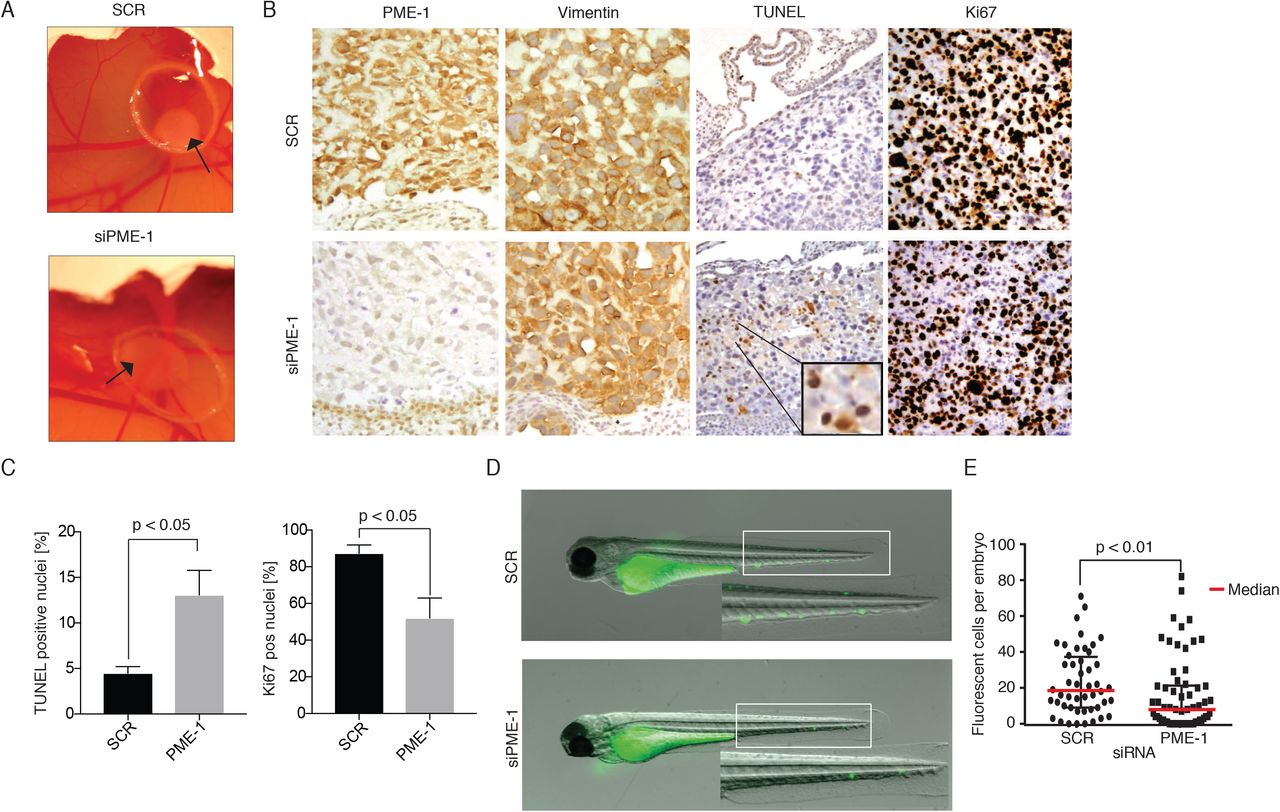
To test the in vivo relevance of PME-1-mediated inhibition of PCa cell anoikis, we used either scrambled or PME-1 siRNA transfected PC-3 cells in a chicken embryo chorioallantoic membrane (CAM) assay (Figure 2A). Tumors formed by PME-1 depleted cells were overall more translucent, suggesting decreased tumor growth (Figure S1C). In accordance with anoikis suppressing activity of PME-1, histological analyses of dissected tumors revealed increased TUNEL positivity in tumors derived from PME-1 siRNA transfected cells (Figure 2B,C). Tumor suppression was further confirmed by reduced number of Ki-67 positive cells in tumors derived from PME-1 siRNA transfected cells (Figure 2B,C).

A. The effect of PME-1 effect on anchorage-independent growth of PC-3 cells in vivo was tested by Chicken embryo chorioallantoic membrane (CAM) assay. PC-3 cells were transiently transfected with either siScr or siPME-1, and 24h post-transfection placed on the CAM. Growth of tumors was followed for 3-5 days.
B-C. Immunohistological stainings of dissected tumors were conducted using antibodies for PME-1, VIM, TUNEL and Ki67, and subsequently the percentage of TUNEL- and Ki67-positive were quantified. Shown is mean + S.E.M, p-value; Mann Whitney test.
D-E. The survival of siScr and siPME-1 transfected PC-3 was studied by microinjecting them into the common cardinal vein of zebrafish embryos 72h post-transfection. After overnight incubation the embryos were imaged by fluorescence stereomicroscopy. The number of surviving fluorescent tumor cells per was counted manually from the images using FIJI and statistical analysis was performed using non-parametric Kruskal-Wallis test.
To test whether high PME-1 expression would suppress cell death also in response to flow-shear stress, and in disseminated PCa cells, we examined survival of control and PME-1 depleted PC-3 cells in zebrafish circulation using recently described experimental setting (35). In short, cell suspensions of scrambled and PME-1 siRNA transfected cells were microinjected into the common cardinal vein of the embryo using a glass microinjector and successfully transplanted embryos were selected to the experiment. After overnight incubation the embryos were imaged by fluorescence stereomicroscopy (Figure 2D). Quantitation of fluorescent tumor cells per zebrafish embryo demonstrated that PME-1 significantly supported survival of circulating PC-3 cells (Figure 2E).
Together, these results validate the in vivo relevance of PME-1-mediated anoikis resistance in PCa cells.
PME-1 associates with the nuclear lamina and regulates nuclear deformability
PME-1-mediated support of cancer cell survival in other cell types has been mostly attributed to increased AKT activity (36). However, we could not detect any consistent effects on AKT phosphorylation by PME-1 silencing in PC-3 cells either in vitro or in vivo (Figure S2A,B). As a reason for these discrepant results, it is possible that PTEN loss in PC-3 is alone sufficient for activation of AKT, and PME-1 has no further effect. Also, the levels of MYC, which is another oncogenic PP2A target relevant for PCa, were not affected by PME-1 silencing (Figure S2A,B).
To seek for alternative targets for PME-1-mediated anoikis suppression in prostate cancer cells, we focused on recently identified association of PME-1 with nuclear lamina. PME-1 was shown to interact with both Lamin-A/C, and with nuclear lamina-associated farnesylase FDPS (30, 37). Further, siRNA-mediated inhibition of either PME-1, or Lamin-A/C, or FDPS, resulted in comparable loss of viability phenotype in PC-3 cells (30). The rationale of nuclear lamina regulation being a potential mechanism protecting PME-1 expressing cells from anoikis is based on recent studies demonstrating that increased nuclear lamina deformability sensitizes cancer cells to apoptosis and anoikis (9, 12, 13). More specifically, inhibition of both Lamin-A/C and Lamin-B1 sensitizes cells to DNA damage and apoptosis (16, 17).
We confirmed the physical association of PME-1 with Lamin-A/C in PC-3 cells by proximity ligation assay (PLA) (Figure 3A). The specificity of PLA signal was confirmed by clear loss of signal in PME-1 depleted cells. To identify potential PME-1-regulated PP2A target phosphorylation sites on nuclear lamina, we re-analyzed recently reported LC-MS/MS phosphoproteome analysis data from PME-1 depleted cells (28). Notably, PME-1 inhibition was found to result in dephosphorylation of several nuclear lamina constituents. Specifically, PME-1 inhibition resulted in dephosphorylation of Lamin-A/C, Lamin-B1 and B2, NUP98, and of chromatin anchoring proteins LAP2A and LAP2B (Figure 3B), without altering cell cycle distribution (data not shown). As many of the regulated proteins impact nuclear deformability, the effects of PME-1 depletion on nuclear deformability was directly assessed by subjecting the control and PME-1 knock-out cells to a micropipette aspiration experiment as described previously (38). Indeed, PME-1 KO cells had significantly more deformable nuclei based on micropipette aspiration experiment (Figure 3C,D). Interestingly, both cell lines show a similar slope for the deformation over longer time periods and mostly vary in their initial deformation (Figure 3D), indicating differencies in their elastic properties.

A. Proximity ligation assay (PLA) was used to confirm the physical association of PME-1 with Lamin-A/C in PC-3 cells. Cleraly decreased PLA signal in PME-1-1 transfected cells confirm the specificity of PLA reaction. DNA was stained using DAPI
B. PME-1 depletion by transient siRNA transfection induced dephosphorylation of the indicated nuclear lamina constituents in HeLa cells. Shown is Log2 fold change in abundance of the indicated phosphopeptide 72 hours after transfection. Included are phosphopeptides detected with 1% FDR accuracy and for which FDR for reproducibility between 3 parallel samples was < 10%. The data was retrieved from (28).
C-D. CRISPR/Cas9 generated control and PME-1 KO cells were analyzed by micropipette aspiration, showing representative images (C) and quantification (D) of nuclear protrusions (um at different time-points).
E. The effect of PME-1 knock-down in PC-3 cells on the phosphorylation of Lamin-A/C S22 ans S392 was confirmed by Western blot analysis, after trypsin-induced detachment for the indicated time (0-210 minutes) prior to collection and lysis of samples. The signal was quantified as compared to total Lamin-A/C and GAPDH.
F. Quantification depicting the phosphorylation of Lamin-A/C S22 in PME-1-depleted and control PC-3 cells, 48h post-transfection and 24h after seeding on soft hydrogel (2 kPa PAG). After culture the cells were fixed, stained and analyzed by confocal microscopy. Mean +/- SD, Mann-Whitney test, n = 84-117 cells from two independent experiments.
G-H. PC-3 cells, in which the endogenous Lamin-A/C expression was suppressed by shRNA was analyzed by Western blot, and upon confirmation of Lamin-A/C suppression (E) transiently transfected with either wild-type Lamin-A/C (WT) or with a Lamin-A/C mutant with both serine 22 and 392 mutated to alanine (S22A/S392A). The effect of Lamin-A/C phosphorylation on cell viability was analysed, as measured by CellTiter-GLO assay. Shown in mean + S.D, 2-sided t-test, n=2.
Although the increased nuclear deformability in PME-1 negative cells potentially involves dephosphorylation of all identified nuclear lamina components (Figure 3B), Lamin-A was the only protein for which there were phosphoantibodies available for the PME-1-regulated sites. From the original PP2A LC-MS/MS phosphoproteome data (28), we identified 8 Lamin-A/C phosphopeptides, with 10 phosphosites, that were regulated negatively by PME-1 depletion, and positively by inhibition of the PP2A scaffold subunit PPP2R1A (Figure S3A). Out of these sites, currently there are phosphoantibodies available for Serine 22 (S22) and S392 and these were used to validate nuclear lamina phosphorylation regulation by PME-1 under anoikis-inducing conditions.
Recent studies using hematopoetic stem cells have demonstrated increase in S22 phosphorylation of Lamin-A/C in response to loss of cell adhesion (39). We could confirm this phenotype also in scrambled siRNA transfected PC-3 cells by trypsin-induced detachment (Figure 3E). Importantly, the cells in which PME-1 expression was inhibited, either by siRNA, or by knockout, showed clearly lower levels of Lamin-A/C serine 22 phosphorylation up to 210 minutes of follow-up after detachment (Figure 3E and S3B). Importantly, similar results were observed with serine 392 phosphorylation (Figure 3E). Further, inhibition of Lamin-A/C serine 22 phosphorylation was sustained for up to 24 hours in PME-1 depleted cells that were detached and re-plated on low stiffness hydrogel matrix (Figure 3F and S3C). To further strengthen the PP2A-dependence of Lamin-A/C phosphorylation regulation by PME-1, we carried out a rescue experiment by co-depletion of the PP2A B-subunit PPP2R2A. PPP2R2A mediates PME-1 effects in human glioma cell therapy resistance (40), and it is deleted in 8% of human prostate cancers (24, 25) (Figure S3D). Reassuringly, co-depletion of PPP2R2A prevented inhibition of Lamin-A/C phosphorylation in detached PME-1 depleted cells (Figure S3E,F).
Given that Lamin-A/C is the major determinant of nuclear lamina elasticity (14), and that PME-1 regulated several Lamin-A/C phosphorylation sites, we used Lamin-A/C phosphorylation mutants to study the functional relevance of this phosphorylation regulation for anoikis resistance. To this end we used PC-3 cells in which endogenous Lamin-A/C expression was suppressed by shRNA targeting the 3’UTR of Lamin-A/C gene (Figure 3G), and the cells were transiently transfected with either wild-type Lamin-A/C or with a Lamin-A/C mutant with both serine 22 and 392 mutated to alanine (Figure S3G). Supportive of the importance of Lamin-A/C S22 and S392 phosphorylation for survival in low attachment conditions, re-expression of phospho-deficient Lamin-A/C mutants reduced the survival of cells with low endogenous Lamin-A/C levels, whereas overexpression of the WT Lamin-A/C resulted in a modest increase in survival (Figure 3H).
These results identify nuclear deformability as a novel PP2A-regulated phenotype in cancer cells. Together with data linking nuclear lamina deregulation to increased apoptosis susceptibility (9, 12, 13, 16, 17), PME-1-mediated regulation of nuclear lamina is a plausible mechanism by which PME-1 negative prostate cancer cells are protected from anoikis (Fig. 4H).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
A. PME-1 protein expression in PCa tissue microarray material from 358 patients treated primarily with radical prostatectomy in the Helsinki University Hospital between 1983 and 1998 was assessed by immunohistochemical (IHC) stainings. PME-1 protein content was scored using 4-tier scale; negative (not shown), low, intermediate and strong expression.
B. Assessment of the activity status of PTEN and PP2A in the patient populations by using genetic deletion of PTEN and PME-1 overexpression as surrogate markers
C-E. Kaplan-Meyer analysis of time to secondary therapies after primary treatment based on PME-1 status alone (C) and in combinations with PTEN deletion (D) and AR expression (E).
F) Effect of stable PME-1 overexpression in anoikis in mouse embryo fibroblasts (MEFs) from a PTEN/p53 KO mouse model after 24h incubation in low attachment conditions. Apoptosis induction was assessed by Western blot utilizing antibodies for cleaved PARP.
G. Effect of stable PME-1 overexpression in S22 phosphorylation of Lamin-A/C in PTEN/p53 KO MEFs.
H. Schematic representation of prostate cell properties, such as nuclear lamina stiffness, invasion and anoikis sensitivity, in relation to PME-1 protein levels and cell malignancy. Based on the results and existing literature, we propose that during cellular transformation nuclear lamina becomes softened to allow cell migration, whereas PME-1 overexpression prevents excess softening of the lamina to the levels that would threaten the viability of these cells.
PME-1 overexpression in prostate cancer associates with PTEN loss and with therapy relapse
To evaluate the clinical relevance of PME-1 in human PCas, PME-1 protein expression, and its clinicopathological associations, were evaluated in PCa tissue microarray (TMA) material consisting of 358 patients treated primarily with radical prostatectomy in the Helsinki University Hospital between 1983 and 1998. The clinical cohort has been previously described in detail (41, 42) (see also Table S1 for demographics).
The specificity of the PME-1 antibody for immunohistochemical (IHC) stainings has been validated previously (43). PME-1 expression was scored using 4-tier scale of negative, low, intermediate and strong expression (Figure 4A). As each patient had three cancerous cores in the TMA, the maximum values of PME-1 scores were used and each patient was dichotomized as either high or low (strong vs. negative to intermediate staining). In the correlation analysis with clinical variables, strong PME-1 protein expression correlated with higher Grade group and advanced stage (Table 2). We also correlated PME-1 status with previously assessed PTEN, ERG and AR status. Notably, high PME-1 expression statistically significantly associated with complete PTEN loss, but also with ERG positivity and high AR expression status (Figure S4A). Further assessment of patient populations based on this data, and regarding predicted activity status of PTEN and PP2A, defined a subcohort of approximately 8% of patients with impaired activities of both tumor suppressors (Figure 4B).
Demographics of the radical prostatectomy patient cohort.
Univariate Analysis of PME IHC and clinical variables.
Survival analysis showed that the patients with high PME-1 expression have shortened disease-specific survival, although this was not statistically significant (Figure S4B). However, patients with high PME-1 expression had statistically significantly shorter time to secondary therapies after primary treatment (i.e. relapse free survival) (Figure 4C). Most importantly, the patients with both complete loss of PTEN and high PME-1 expression defined a remarkably aggressive patient population with less than 50% 5-year secondary-therapy free survival (Figure 4D). Similar, albeit less prominent cooperative effects were observed with PME-1 overexpression and with either ERG or AR expression (Figure 4E and S4C).
Together with functional data these observations indicate clinical relevance for PME-1-mediated anoikis suppression in PTEN-deficient PCa. To confirm this functional association of two tumor suppressor phosphatases in a more genetically defined model system, PME-1 cDNA construct was stably transfected to PTEN/p53 knock-out mouse embryonic fibroblasts (MEFs). In accordance with siRNA and Crispr/Cas results, overexpression of PME-1 in PTEN/p53 knock-out MEFs prevented PARP cleavage upon 24h incubation in low attachment conditions (Figure 4F). Moreover, PME-1 overexpression increased Lamin-A/C phosphorylation in PTEN/p53 knock-out MEFs consistently with other results (Figure 4G).
Discussion
During tumor growth, and particularly during invasion and metastatic spread, cancer cells are exposed to varying levels of stress. Particularly detachment of cells from other cells, and from ECM, makes cells vulnerable to anoikis induction. Thereby, mechanisms that inhibit cancer cell anoikis facilitate local tumor spreading, and eventually metastasis. Here we report high expression of PP2A inhibitor PME-1 as a anoikis resistance mechanism in PTEN-deficient PCa cells. The clinical relevance of anoikis suppression by PME-1 is supported by association of patient population with PTENloss/PME-1high tumors with remarkably short time to secondary therapies.
Although anoikis suppression is a generally relevant mechanisms for tumor progression, it may have particular clinical importance in slowly progressing cancers such as PCa where cancers can be diagnosed in indolent phase, and there is a strong need to be able to both predict and inhibit the likelihood of disease progression (4). Previously anoikis resistance in PCa cancer has been linked mechanistically to regulation of cell adhesion, cytoskeleton (1), PTEN inhibition (6), and AR activity (44). In addition to remarkable clinical connection between PTEN and PME-1 (Figure 4D), we found that low PME-1 and AR expression tumor expression was associated with significantly prolonged therapy-free survival (Figure 4E). These results are particularly interesting given that pharmacological PP2A reactivation leads to AR degradation, and to significant PCa tumor regression (32), corroborating situation seen in the less aggressive tumors with high PP2A activity (PME-1low) and low AR expression (Figure 4E).
PTEN and PP2A have both been identified independently as PCa tumor suppressor phosphatases (4, 23, 27, 33), but the clinical relevance of their co-operation has not been studied thus far. Here, we demonstrate an important role for PME-1-mediated PP2A inhibition in anoikis suppression in various PTEN-deficient models. Clinically, simultaneous inhibition of both tumor suppressor phosphatases was associated with very aggressive disease with high propensity for early disease relapse (Figure 4D). As PTEN deficiency has also been shown to promote anoikis resistance of PCa cells (6), the tumor cells with inhibition of both PP2A and PTEN are assumed to be particularly well protected from anoikis, and we hypothesize that this is likely to contribute to observed clinical aggressiveness of these cancers. Mechanistically PTEN-mediated anoikis resistance is mediated by AKT signaling (6, 7), whereas we showed that PME-1 depletion had no effect on AKT phosphorylation (Figure S3A,B). The non-overlapping downstream mechanisms for PTEN and for PME-1 in anoikis suppression further explains their synergetic actions. As PTEN genetic status can be routinely evaluated in current clinical PCa diagnostic practice (4), our results indicate diagnostic utility of assessment of PME-1 status on patients with complete PTEN loss. Although further studies are clearly needed to validate these conclusions, our results indicate that patients with PTENloss/PME-1high tumors might benefit from more intensive follow-up, and/or from more aggressive therapies as the first, and second line treatments.
PME-1 expression protected PCa cells from anoikis in various anchorage-independent conditions (soft agar, trypsin-detachment), or under conditions with low ECM-cell mechanotransduction (low adhesion plates, low stiffness). We also demonstrate for the first time that PP2A modulation regulates nuclear deformability. Our results are fully consistent with the published model that different levels of nuclear lamina deformability support different cellular functions (Figure 4H). Whereas a stiff lamina prevents cell migration through physical restrictions, a highly deformable lamina renders cells vulnerable to apoptosis induction by low tissue tension, and fluid stress (9, 12, 15, 17, 45). Further, a recent study indicates that aggressive prostate cancer cells such as PC-3 and DU-145 have softer nuclei than immortalized benign prostate epithelial cells (46). As we demonstrate an important role for PME-1 in preventing further softening of the nuclear lamina in PC-3 cells, and link this to the anoikis suppression, we speculate that during cellular transformation nuclear lamina becomes softened to allow cell migration, whereas PME-1 overexpression in cancerous cells prevents softening of their nuclear lamina to the levels that would make them sensitized to anoikis (Figure 4H). Although alternative mechanisms downstream of PME-1 may contribute to its anoikis suppressing function, we emphasize that nuclear lamina deformability regulation may have been overlooked as a critical mechanism supporting viability of PCa cells during their progression towards full malignancy. Importantly, although Lamin-A/C phosphor-antibody analysis was used to validate PP2A-mediated regulation of nuclear lamin proteins, it is very likely that PP2A-mediated dephosphorylation of other detected nuclear lamina proteins are important for both the observed nuclear deformability phenotype. In addition to phosphotargets of PME-1, PME-1 also interacts with a FDPS(30) which contributes to farnesylation of Lamin-A/C, B1 and B2(37). Therefore also this nuclear lamina-associated PME-1 protien interaction may contribute to the observed nuclear deformability regulation. The molecular mechanisms by which too deformable nuclei sensitizes apoptosis is currently under debate. As nuclear chromatin is tightly connected with lamina (47), and DNA arrangements sensitize to apoptosis, it is possible that dephosphorylation of chromatin arranging LAP2A/B proteins may contribute to anoikis induction in PME-1 negative cells under low attachment conditions.
Together these results identify anoikis resistance as a candidate mechanism by which PME-1-mediated PP2A inhibition promotes malignant progression of prostate cancer. Together with emerging orally bioavailable PP2A reactivating compounds with profound antitumor activity in in vivo PCa models (32), these results clearly emphasize future importance of comprehensive understanding of PP2A biology for management of aggressive prostate cancer patients. The results also identify PME-1 as a potential biomarker for improved stratification and better therapy response among PCa patients with complete PTEN loss. Together with other findings, results further indicate that diagnostic PME-1 evaluation from PTEN negative tumors may help to identify patients that would benefit from more intensive follow-up after therapy.
Material and Methods
Cell lines
PC-3 and DU-145 prostate cancer cell line were obtained from ATCC and cultured in RPMI. PtenΔ/Δ; Trp53Δ/Δ; lsl-tdTomato mouse embryonic fibroblasts were provided by Lloyd Trotmann and cultured in DMEM. All growth media were supplemented with 10% heat-inactivated FBS (Biowest), 2 mmol/L l-glutamine, and penicillin (50 units/mL)/streptomycin (50 μg/mL; pen/strep). Cells were confirmed to be mycoplasma-free and grown at 37° in a humidified atmosphere of 5% CO2.
siRNAs
Scrambled (SCR.2): CGU ACG CGG AAU ACU UCG A
PME-1.1 (PME-1.3): GGA AGU GAG UCU AUA AGC A
PME-1.2 (PME-1 hr2): GAA UGA AAC UGG CAA GGA U
PP2R2A: CUG CAG AUG AUU UGC GGA U
Validated PP2A B-subunit siRNA was purchased from Qiagen, PME-1 siRNAs from Eurofins Genomics (MWG).
LMNA: ON-TARGETplus human siRNA SMARTpool (L-004978-00-0005)
Antibodies
The following antibodies were used at the indicated dilutions: PME-1: Santa Cruz Biotechnology sc-20086 (H-226) Western blotting 1:1000; PME-1: Santa Cruz Biotechnology sc-25278 (B-12) Immunohistochemistry 1:1000, Immunofluorescence 1:100; PPP2R2A: Cell Signalling Technology #5689 Western blotting 1:1000; cleaved PARP: Abcam ab32064 [E51] Western blotting 1:1000; GAPDH: Hytest 5G4-6C5 Western blotting 1:500.000 c-MYC: Abcam ab32072 [Y69] Western blotting 1:1000; p-Myc S62:Abcam ab78318 Western blotting 1:1000; Akt1/2/3: Cell signalling #9272 Western blotting 1:2000; p-Akt S473: Cell Signalling Technology #4060 Western blotting 1:1000; p-Akt T308: Cell Signalling Technology #13038 Western blotting 1:1000; Lamin A/C: Santa Cruz Biotechnology sc-20681 (H-110) Western blotting 1.10,000; Lamin A/C: Santa Cruz Biotechnology sc-6215 (N-18) Western blotting 1:1000 Immunofluorescence 1:100; Lamin A/C: Santa Cruz Biotechnology sc-7292 (636) Immunofluorescence 1:250; p-Lamin S22: Cell Signalling Technology #2016 Western blotting: 1:500 Immunofluorescence 1:50 or 1:100; p-Lamin S392: Abcam ab58528 Western blotting 1:5000, Immunofluorescence 1:200
siRNA/plasmid transfection
For siRNA transfections Lipofectamine RNAi MAX (Thermo Fisher Scientific) was used following the manufacturer’s instructions. In most cases 2.5×105 cells were seeded on a 6-well plate one day before transfection to reach 60-70% confluency. Cells were then transfected with 25pmol siRNA, 7.5µl siRNA MAX per 6-well and assayed 48-72h after transfection unless otherwise stated.
For plasmid transfections Lipofectamine 3000 (Thermo Fisher Scientific) was used following the manufacturer’s instructions. In most cases 3-4 x105 cells were seeded on a 6-well plate one day before transfection to reach 80-90% confluency. Cells were then transfected with 2.5µg DNA, 5µl P3000 reagent, 3.75µl Lipofectamine 3000 per 6-well and assayed 24-48h after transfection unless otherwise stated.
Colony formation assay
Cells were reseeded at low confluency (1-4×103) on a 12-well dish 24h after siRNA transfection and grown for around 10 days to allow formation of cell colonies. Colonies were then fixed with ice-cold methanol and stained with 0.2% crystal violet solution (made in 10% ethanol) for 10 minutes at room temperature each. Excess stain was removed by repeated washing with PBS. Plates were dried and scanned with Epson perfection V700 scanner. Quantifications were performed with ColonyArea ImageJ plugin (48), and graphs were plotted using the area % values.
Anchorage-independent colony formation assay
For the anchorage-independent colony formation assay, which correlates typically with in vivo tumorigenicity, 2×104 cells were resuspended in 1.5ml growth medium containing 0.4% agarose (4% Agarose Gel, Termo Fisher Scientific Gibco; top layer) and plated on 1ml bottom layer containing growth medium and 1.2% agarose in a 12-well plate. After 14 days of growth, colonies were stained over night with 1mg/ml Nitro blue tetrazolium chloride (NBT; Molecular Probes) in PBS. Colonies were imaged using a Zeiss SteREO Lumar V12 stereomicroscope. Analysis was done using the ImageJ software. First, the background was subtracted using the rolling ball function with a radius of 50μm, then auto-thresholding was applied to separate the colonies. Area percentage was calculated using the ImageJ built-in function ‘Analyze Particles’ with exclusion of particles smaller than 500μm2 that are not considered colonies.
Cell viability assay on low attachment plates
Cell viability on 96 Well Clear Black Round Bottom Ultra Low Attachment Spheroid Microplates (Corning) was determined by using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega). For this purpose, a small number of cells (250-1000/well) were seeded on the plates and grown for the indicated number of days. Cells were then lysed in the CellTiter-Glo® reagent by vigorous pipetting and luminescence was read with a Synergy H1 reader.
Western blotting
Western blot protein lysates were prepared in 1x RIPA buffer (150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS and 50 mM Tris, pH 7.5) containing PhosSTOP™ phosphatase and cOmplete™, EDTA-free protease inhibitors (Roche). The DNA in the protein samples was sheared by sonication and the protein amount was estimated using the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Lysates were usually separated on 4–20% Mini-PROTEAN® TGX™ Gels (Biorad) and transferred by wet blotting to PVDF membranes (Millipore). Unspecific antibody binding was blocked with 5% non-fat dry milk in TBS-T. Incubation for most of the primary antibodies was performed overnight at 4°C in either 5% non-fat dry milk or 5% BSA for most of the phospho-specific antibodies. For detection, HRP-labelled secondary antibodies (DAKO) followed by incubation with Pierce™ECL Western Blotting Substrate (Thermo Fisher Scientific) was used.
Generation of PtenΔ/Δ; Trp53Δ/Δ mouse embryonic fibroblasts
PtenΔ/Δ; Trp53Δ/Δ; lsl-tdTomato MEFs were generated as previously described (49). To stably overexpress PME-1 in those cells, PME-1 cDNA was cloned into pCSCIW-2 lentiviral construct which was then packaged into lentivirus. Following transduction, GFP positive cells were sorted and overexpression of PME-1 was confirmed by western blotting.
CRISPR/Cas9 mediated PME-1 knock-out
To generate PME-1 knock-out cells, PC-3 cells were transduced with lentivirus containing lentiCas9-Blast construct (Addgene, Feng Zhang lab) and selected with growth medium containing 4µg/ml blasticidin for about one week. In a second round, cells were infected with lentivirus sgRNA against PME-1 exon 3 (sequence: ACTTTTCGAGTCTACAAGAGTGG) cloned into pKLV-flipedU6gRNA_PB_BbsI_PGKpuro2ABFP construct. After puromycin selection (2µg/ml in growth medium), PME-1 KO on the protein level was confirmed by western blotting. Single cell clones were then obtained by single-cell sorting and the knock-out was confirmed by sequencing (Primers. Forward CACCGCTTTTCGAGTCTACAAGAGGT; reverse TAAAACGAAGATCTGTCTGCAGAAAC). Cas9, non-targeting sgRNA expressing cells served as a negative control in the functional assays.
Chick chorioallantoic membrane (CAM) assay
To start chick embryonic development for the chorioallantoic membrane assay, fertilized eggs were kept rotating in an incubator at 37°C and 50-60% humidity for four days. After that initial incubation time, a small hole was introduced at the sharp edge of the eggs and sealed with parafilm. Following four more days of incubation on stationary racks, the hole was enlarged and a plastic ring was placed on top of blood vessels in the chorioallantoic membrane. Next, 1×106 PC-3 cells in 20µl volume of a 1:1 mixture of ice-cold PBS and matrigel were pipetted inside the ring on the membrane. The hole was then covered with parafilm and the eggs were incubated for three more days. At day twelve of embryonic development, the animals were sacrificed by freezing the eggs for 15min and the tumor cell mass was dissected from the membrane and processed for further analysis.
Immunohistochemistry
Hematoxylin/eosin staining and immunohistochemistry were performed on 3-μm-thick sections of 4% paraformaldehyde-fixed and paraffin-embedded tissues. Following rehydration, endogenous peroxidase was blocked by incubation in 50% MetOH, 1% H2O2. Subsequent antigen retrieval was performed with the 2100 Retriever (Aptum) in R-Universal Buffer. Unspecific antibody binding was blocked with 10% goat serum in 2% BSA/PBS prior to overnight incubation at 4°C with the primary antibody. For detection, the DAKO EnVision peroxidase system, followed by incubation with 0.01% diaminobenzidine (Sigma-Aldrich) was used.
Zebrafish in vivo dissemination assay
The xenotransplantation of zebrafish embryos was performed as described in detail in (35) with some modifications. PC-3 cells were washed with PBS, and stained with CellTracker Green CMFDA dye (5uM, Thermo Fisher Scientific) and detacher and detached with trypsin-EDTA in a single incubation step at +37°C. Subsequently, cells were pelleted by centrifugation and washed with PBS twice. This was followed by filtration through 40µM mesh into FACS tube (BD Falcon, 352235) and pelleting cells by centrifugation. Finally, cells were resuspended into 30µl of injection buffer (2%PVP in PBS) and kept on ice until transplanted.
Zebrafish (Danio rerio) of pigmentless casper strain (roy-/-; mitfa-/-)(50) was used in the experiments under licence no. MMM/465/712-93 (issued by Finnish Ministry of Forestry and Agriculture) and following legislation: the European Convention for the Protection of Vertebrate Animals used for Experimental and other Scientific Purposes and the Statutes 1076/85 and 62/2006 of The Animal Protection Law in Finland and EU Directive 86/609. The embryos were obtained through natural spawning, and embryos were cultured in E3-medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) at +33°C. At 2 or 3 days post-fertilization, the embryos were anesthesized with 200mg/ml Tricaine and embedded in 0.7% low-melting point agarose. Subsequently, the cell suspension was microinjected into common cardinal vein (duct of Cuvier) of the embryo using glass capillaries (Transfertip), CellTramVario microinjector and InjectMan micromanipulator (all from Eppendorf). Embryos were liberated from the agarose gel using forceps and successfully transplanted embryos were selected to the experiment. After overnight incubation at +33°C, the embryos were anesthesized again with Tricaine and imaged using Zeiss StereoLumar V12 fluorescence stereomicroscope. The number of surviving cells was counted manually from the images using FIJI and statistical analysis was performed using non-parametric Kruskal-Wallis test (GraphPad Prism 6.05 software, GraphPad Software, Inc.).
Proximity ligation assay (PLA)
Cells were grown to about 80% confluency on sterilized coverslips, fixed for 10min in 4% PFA and permeabilized for 10min with 0.5% Triton X-100 in TBS for proximity ligation assay (PLA). The following steps were completed following the manufacturer’s instructions (Sigma-Aldrich). Briefly, unspecific antibody binding was blocked with the provided blocking solution for 30min. The slides were then incubated with the primary antibodies overnight at 4°C, with the mouse and rabbit probes for one hour at 37°C, with the ligation mix for 30min at 37°C and the amplification mix for 100min at 37°C. The washing steps in-between the individual steps were carried out with Buffer A (Sigma), the final washing step with Buffer B (Sigma). Slides were mounted in mowiol mounting medium and imaged with a Zeiss LSM780 confocal microscope.
Immunofluorescence
For immunofluorescence cells were fixed for 10min with 4% PFA and permeabilized for 10min with 0.5% Triton X-100 in TBS. Unspecific antibody binding was blocked by incubation with 10% serum from the host of the secondary antibody in 2% BSA/PBS for 30min. Primary antibodies were diluted in 2% BSA/PBS and cells were usually incubated with the primary antibody over-night at 4°C. As secondary antibodies appropriate Alexa-Fluor conjugates (Thermo Fisher Scientific) were used at a 1:2000 dilution in PBS for 30min. Nuclei were stained with DAPI (4’,6-diamidino-2-phenylindole).
Polyacrylamide hydrogels
35 mm glass bottom dishes (MatTek Corporation, P35G-1.0-14-C) were treated with 100 µl of Bind-silane solution [7.14% Bind-silane (Sigma, M6514) and 7.14% acetic acid in absolute ethanol] for 15 min, washed twice with absolute ethanol and left to dry completely. A pre-polymer mix comprising 5.6% acrylamide (Sigma) and 0.078% N,N′-methylenebisacrylamide (Sigma) in PBS was prepared to obtain hydrogels with an elastic modulus of 1.8-2 kPa. Polymerization was initiated by adding 2.5 µl of 20% ammonium persulfate (Bio-Rad) and 1 µl of N,N,N′,N′-tetramethylethylenediamine (Sigma). The solution was vortexed, 13 µl was added on top of the glass bottom dish, a 13 mm glass coverslip was placed on the drop and the gel was left to polymerize for 1 h at room temperature. After polymerization, the dish was filled with PBS and the coverslip was carefully removed. Hydrogels were made permissive for protein binding by incubating them in 500 µl of Sulfo-SANPAH solution [0.2 mg/ml Sulfo-SANPAH (Sigma, 803332) and 2 mg/ml N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (Sigma, 03450) in 50 mM HEPES] for 30 min on slow agitation, followed by a 10 min UV exposure (∼30 mW/cm2, 253.7 nm). Activated gels were washed three times with PBS to get rid of residual Sulfo-SANPAH.
Alternatively, pre-activated polyacrylamide hydrogels of variable stiffness were ordered from Matrigen Life Technologies. 35 mm glass bottom dishes (SV3510-EC-0.5, SV3510-EC-50) were used for imaging and 6-well plates (SW6-EC-0.5, SW6-EC-50) for growing cells for lysis. All hydrogels and corresponding plastic controls were functionalized with bovine plasma fibronectin (Merck-Millipore, 341631) and collagen type I (Sigma, C8919) by incubating the dishes in 5 µg/ml of each protein for 1-2 h at +37 °C, or overnight at +4 °C, before use.
For Western blot analysis from cells in hydrogels, 0.5 and 50 kPa hydrogel-coated 6-well plates were immersed in cell culture medium for 30 min and seeded with 300,000 PME-1 or control siRNA-treated PC-3 cells per well. The cells were incubated on the gels for 24 h. Once the cultures reached 80-90% confluency, the cells were washed once and scraped into cold PBS, spun down and lysed in radioimmunoprecipitation assay (RIPA) buffer.
For immunofluorescence experiments, PME-1 or control siRNA transfected cells were seeded on 2 kPa hydrogel-coated glass bottom dishes and incubated for an additional 24 h before fixing. Cells were fixed with 4% PFA for 10 min at room temperature, and simultaneously permeabilized and blocked with 0.3% Triton in 10% horse serum (Gibco) for 15 min at room temperature. All samples were incubated in primary antibodies overnight at +4 °C, and stained with secondary antibodies for 1-2 h at room temperature on the following day. The antibodies were diluted in 10% horse serum before use.
Microscopy and image analysis
Fluorescent specimens were imaged using a laser scanning confocal microscope LSM780, controlled by Zen 2010 (Zeiss), and the objective used was a 40x/1.2 W C-Apochromat objective (Zeiss). Images were analyzed using ImageJ (National Institutes of Health) and CellProfiler (Broad Institute) softwares.
Acknowlegdements
We thank The Proteomics and The Cell Imaging Core at Turku Centre for Biotechnology supported by the University of Turku and Biocenter Finland, for technical assistance. Taina Kalevo-Mattila is thanked for excellent technical help. This work was supported, in part, by grants from Academy of Finland (J.W, J.I), Academy of Finland CoE for Translational Cancer Research (J.I), Sigrid Juselius Foundation (J.W, J.I.), and the Finnish Cancer Organization (J.W, J.I., P.T.), the National Institutes of Health [R01HL082792 and U54CA210184 to JL], the Department of Defense Breast Cancer Research Program [Breakthrough Award BC150580 to JL], and the National Science Foundation [CAREER Award CBET-1254846 to JL]. This work was performed in part at Cornell NanoScale Facility (CNF), an NNCI member supported by NSF Grant NNCI-1542081. A.I. has been supported by the University of Turku Doctoral Programme in Molecular Life Sciences (DPMLS).
References




