

Abstract
Alternative splicing of pre-mRNA is a major mechanism to diversify protein functionality in metazoans from a limited number of genes. In the Drosophila Down Syndrome Cell Adhesion Molecule (Dscam) important for neuronal wiring up to 38,016 isoforms can be generated by mutually exclusive alternative splicing in four clusters of variable exons. However, it is not understood how a specific exon is chosen from the many variables and how variable exons are prevented from being spliced together. A main role in the regulation of Dscam alternative splicing has been attributed to RNA binding proteins, but how they impact on exon selection is not well understood. Serine-arginine-rich (SR) proteins and hnRNP proteins are the two main types of RNA binding proteins with major roles in exon definition and splice site selection. Here, we analyzed the role of SR and hnRNP proteins in Dscam exon 9 alternative splicing in mutant Drosophila embryos because of their essential function for development. Strikingly, Dscam alternative exon selection is very robust against loss or overexpression of canonical SR and hnRNP proteins even when multiple proteins are depleted together. Conversely, non-canonical SR protein Serine-arginine repetitive matrix 234 (Srrm234) is a main determinant of exon inclusion in Dscam exon 9 cluster. Since long-range base-pairings are absent in the exon 9 cluster, our data argue for a small complement of regulatory factors as main determinants of exon inclusion in the Dscam exon 9 cluster.
Introduction
During alternative splicing the order of exons can be varied to generate multiple different transcripts and proteins from one gene (Soller 2006; Nilsen and Graveley 2010; Fiszbein and Kornblihtt 2017). In humans, 95% of genes, and in Drosophila 63% of genes are alternatively spliced, respectively (Wang et al. 2008; Fu and Ares 2014). Among the genes where alternative splicing generates the greatest diversity of isoforms is the Drosophila homolog of human Down Syndrome Cell Adhesion Molecule (Dscam), which encodes a cell surface protein of the immunoglobulin superfamily. The Dscam gene comprises 95 alternatively spliced exons that are organized into four clusters, namely 4, 6, 9 and 17 which contain 12, 48, 33 and 2 variables, respectively. Hence, the Dscam gene can generate up to 38,016 different proteins (Schmucker et al. 2000; Neves et al. 2004; Hemani and Soller 2012; Sun et al. 2013). Dscam is functionally required for neuronal wiring in the nervous system, but also for phagocytosis of invading pathogens in the immune system (Schmucker et al. 2000; Watson et al. 2005). Interestingly, Dscam in mosquitos changes its splicing pattern to produce pathogen specific isoforms with higher binding affinity (Dong et al. 2006). However, despite intense research, relatively little is known about how Dscam alternative splicing is regulated in flies.
Pre-mRNA splicing is a multistep process catalyzed by the spliceosome sequentially assembled from five U snRNPs together with numerous proteins. Spliceosome assembly initiates by the recognition of the 5’ splice site by U1 snRNP and of the 3’ splice site by U2 snRNP together with U2AFs recognizing the branchpoint and the polypyrimidine tract and the AG of the 3’ splice site, respectively. Then the U4/5/6 tri-snRNP is recruited and upon several structural rearrangements where U4 snRNP leaves the spliceosome, catalysis takes place by two transesterification reactions (Luhrmann and Stark 2009).
Alternative splicing is to a large degree regulated at the level of splice site recognition involving base-pairing of U1 snRNP to the 5’ splice site YAG/GURAGU and U2 to the branchpoint WNCUAAU (W: A or U, Drosophila consensus(Lim and Burge 2001)) whereby splice sites closer to the consensus are preferably used (Soller 2006). Splice site selection is critically assisted by RNA binding proteins (RBPs) that support or inhibit recognition of splice sites.
Serine-arginine rich (SR) and heterogenous nuclear ribonucleoproteins (hnRNPs) are two prominent classes of RBPs involved in alternative splicing regulation (Busch and Hertel 2012; Fu and Ares 2014; Bradley et al. 2015). Humans have twelve and flies have eight SR proteins each having one or two RNA Recognition Motifs (RRM) and RS domain rich in serines and arginines (Busch and Hertel 2012). In addition, RS domains are present in some other splicing factors lacking RNA binding domains such as Tra2, SRRM1 (SRm160), and SRRM2 (SRm300), SRRM3, and SRRM4 (nSR100), which are termed non-canonical SR proteins (Blencowe et al. 1999; Long and Caceres 2009; Busch and Hertel 2012; Best et al. 2014). In contrast, hnRNP proteins are more diverse in their modular assembly containing RNA binding domains (e.g. RRM, KH or RGG domains) and various auxiliary domains (Geuens et al. 2016). In humans, the most prominent hnRNPs are the abundantly expressed hnRNP A and C family (Busch and Hertel 2012; Geuens et al. 2016).
SR proteins mostly bind to exonic splicing enhancers (ESEs) and recruit spliceosomal components to splice sites through their RS domains. SR protein binding sites are present in both alternatively spliced and constitutive exons to promote exon inclusion, but they can also repress inclusion of alternative exons (Black 2003; Shen et al. 2004; Wang et al. 2006; Chen and Manley 2009; Pandit et al. 2013). Although SR proteins recognize similar sequences, distinct functions have been shown either by binding distinct sites or when bound to the same site through differential regulation mediated by combinatorial interactions with other splicing regulators (Gabut et al. 2007; Anko et al. 2012; Pandit et al. 2013; Bradley et al. 2015).
In contrast to SR proteins, hnRNP proteins mostly bind to intronic splicing silencers (ISSs) and repress inclusion of alternative exons (House and Lynch 2006; Wang et al. 2008). In addition, they can act antagonistically to SR proteins by binding to exonic splicing silencers (ESSs) and compete with SR proteins for binding (Soller 2006; Long and Caceres 2009). However, a more comprehensive analysis revealed that SR and hnRNP proteins can also act coordinately in many instances in exon inclusion or repression (Brooks et al. 2015).
With regard to the alternative splicing in the Dscam gene a model has been proposed for the exon 6 cluster whereby long-range base-pairing brings variable exons into the proximity of the proximal constant flanking exon amid a conserved docking sequence in the first intron of the exon 6 cluster and complementary selector sequences in front of each variable exon (Graveley 2005). This model also requires that the entire cluster is maintained in a repressed state and variable exons are selected under the control of RBPs. However, the architecture in the exon 4 an 9 cluster is different and conserved “docking site” sequences are found at the end of the exon 4 and 9 clusters, but the support for this model based on evolutionary sequence conservation is weak (Yang et al. 2011; Haussmann et al. 2019).
RNAi knock-down of RBPs in cell culture revealed little changes for most variable exons in the Dscam exon 4 cluster, but this result could be due to residual protein left (Park et al. 2004). Hence, we wanted to investigate the role of SR and hnRNP proteins in Drosophila Dscam alternative splicing more comprehensively at an organismal level using knock-out mutants and overexpression. Most SR and hnRNP proteins are essential for development to adult flies, but embryonic development proceeds such that Dscam alternative splicing could be analysed in late stage embryos in SR and hnRNP mutants or when over-expressed. Unexpectedly, we find that inclusion of Dscam exon 9 variables is little affected in loss or gain of function conditions. Likewise, splicing is robust even upon removal even of multiple factors. However, the non-canonical SR protein Srrm234 is required for inclusion of most exon 9 variables. We further find that long-range base-pairing is not supported as a general model. Hence, our results argue that a small complement of RBPs are main regulators of Dscam exon 9 alternative splicing.
Results
Analysis of Dscam exon 9 alternative splicing by restriction digests
All 33 exons in the Drosophila melanogaster Dscam exon 9 variable exon cluster have about the same length and run as a single band on an agarose gel (Figure 1A and B). The sequences in variable exons, however, differ enough such that a complement of restriction enzymes can digest the complex mix of PCR products to identify a majority of isoforms on sequencing type gels using one 32P labelled primer after reverse transcription of the mRNA (Figure 1C and D) (Haussmann et al. 2019). Using a combination of SacII, ClaI, PshAI, HaeIII, MseI, BsrI and BstNI yields 26 fragments of unique size identifying 20 variable exons (Fig 1D).
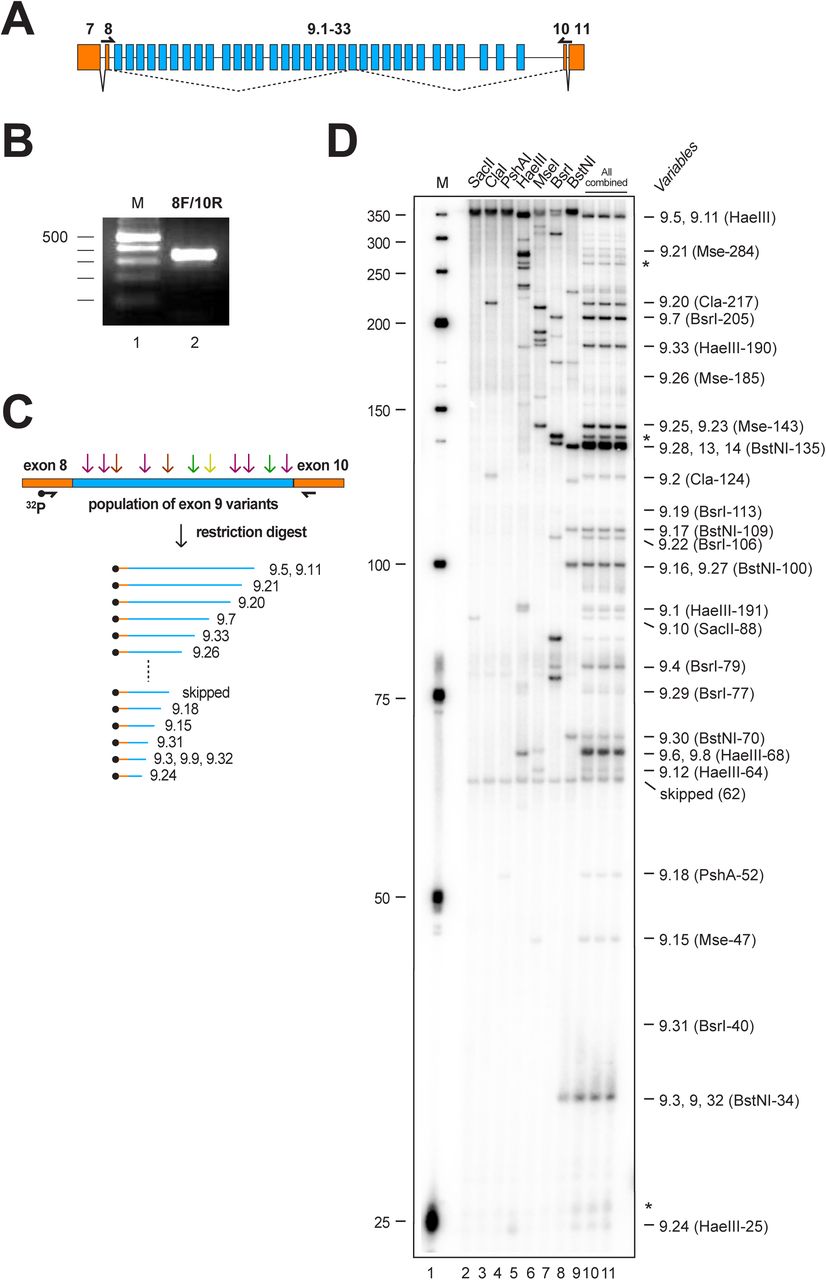
(A) Schematic of the Dscam exon 9 variable cluster gene region. Constitutive exons are shown in orange and variable exons in blue. Primers to amplify the variable part are shown on top of the exons. (B) RT-PCR product for variable exon cluster 9 shown on a 3% agarose gel. (C) Schematic of the method used to resolve inclusion levels of variable exons using a 32P labelled forward primer in combination with a set of restriction enzymes followed by separation of a denaturing polyacrylamide gel. (D)Denaturing acrylamide gel (6%) showing a restriction digest (SacII, ClaI, PshAI, HaeIII, MseI, BsrI, BstNI) of Dscam exon 9 variables amplified with a 32P labelled forward primer from 14-18 h Drosophila embryos. Single enzyme reference digests are shown on the left (lanes 2-8) and the combination of all enzymes on the right (lanes 9-11).
Alteration of SR proteins minimally impacts on Dscam exon 9 alternative splicing
SR proteins are organized into five families and representative orthologues are present in Drosophila (Fig 2) (Busch and Hertel 2012). To determine the role of SR proteins in Drosophila Dscam exon 9 alternative splicing we obtained mutants and over-expression lines for most of the canonical SR proteins as well as for general splicing factor SF1 and non-canonical SR protein Srrm1 (SRm160) and Srrm234 (SRm300, CG7971) (Fig 2A). For loss of function (LOF) alleles of SR genes, five from the ten analysed, which are X16 GS1678, SF2GS22325, B5229, Srrm1 B103 and Srrm234ΔN, are required to reach adulthood (Fig 2A and B). Gain of function (GOF) conditions by pan-neural elavGAL mediated over-expression via UAS was lethal in larval instars for UAS GFP-X16, UAS RSF1, UAS GFP-SC35, UAS GFP-SF2 and UAS GFP-B52, while over-expression of UAS SF1 and UAS Srrm1 from EP lines did not result in a phenotype (Fig 2A and C).

(A) Evolutionary relationship of Drosophila SR proteins with human homologues is shown on the left and the domain structure is indicated by coloured boxes. Arginine-serine-rich domain (RS, green), RNA recognition domain (RRM, light blue), hnRNP K homology domain (KH, purple), zinc finger domain (ochre), Proline-Tryptophan-Isoleucine domain (PWI, yellow) and CWF21 domain (red). The type of allele obtained, and the loss (LOF) and gain of function (GOF) phenotypes are indicated on the right. (B) Viability was determined from stocks that harbour a zygotically expressing GFP marked balancer chromosome, which contains a set of inversions to supress recombination and a recessive lethal mutation. If a gene is essential, only heterozygous flies will survive. Homozygous mutant embryos were identified in the progeny of these stocks, by the lack of GFP and advanced development as homozygous balancer carrying embryos die early before GFP expression. (C) To obtain embryos overexpressing SR proteins flies carrying the yeast GAL4 transcription factor under the control of the neuronal elav promoter were crossed with lines harbouring SR proteins under the control of the yeast UAS promoter.
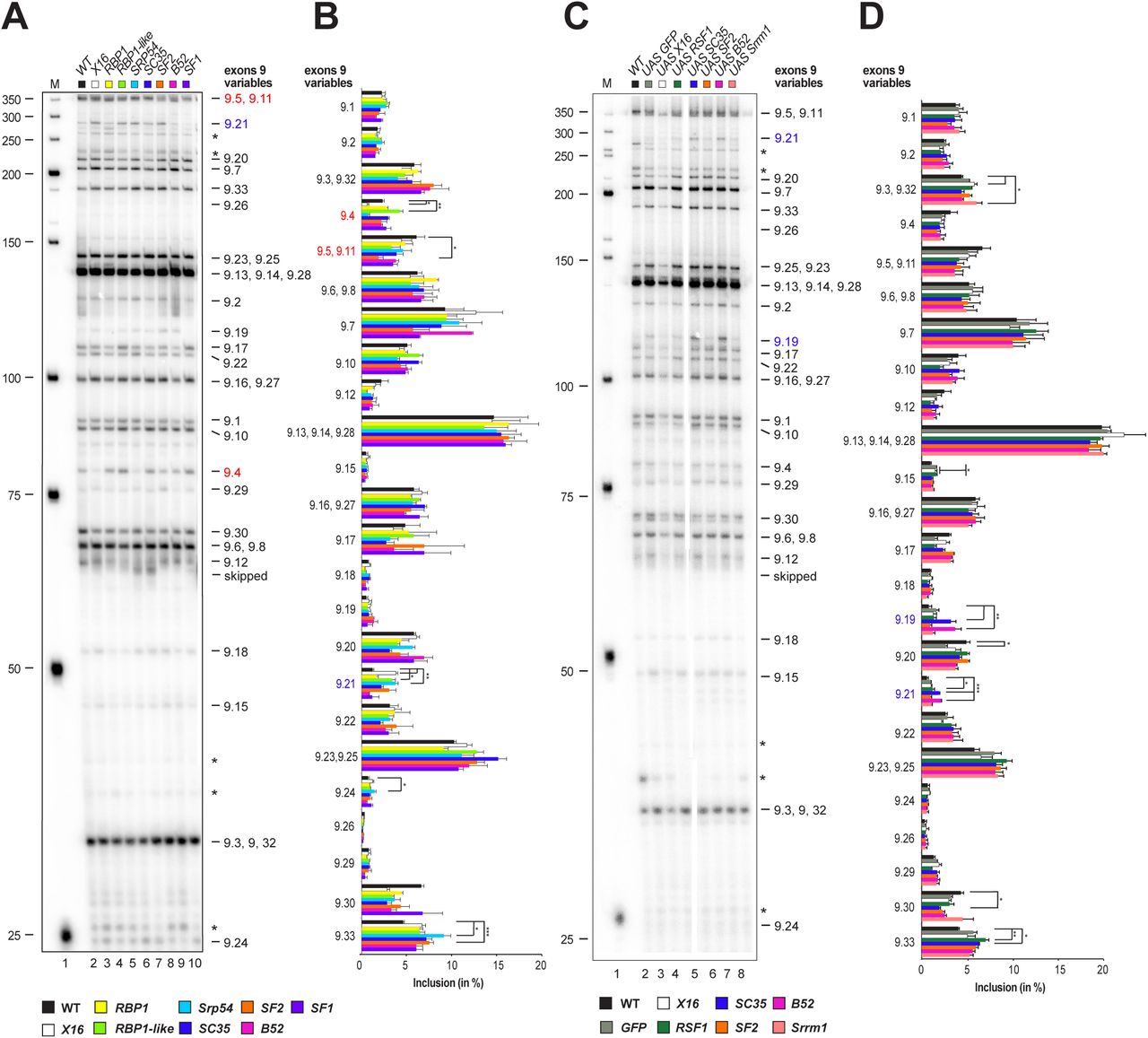
Next, we analysed inclusion levels of exon 9 variables in embryos for LOF and GOF conditions of canonical SR proteins and SF1. For LOF alleles X16 GS1678, RBP1 HP37044, RBP1-like NP0295, Srp54GS15334, SC35KG02986, SF2GS22325, B5228 and SF1G14313 the Dscam exon 9 splicing pattern unexpectedly remained largely unchanged and significant changes are prominent in exon 9.4, 9.21 and 9.5/9.11 for mutants compared to wild type (marked in red for decrease and blue for increase, Figure 3A and B). Similar results were obtained for over-expression of UAS GFP-X16, UAS RSF1, UAS GFP-SC35, UAS GFP-SF2, UAS GFP-B52 and UAS-Srrm1 with significant changes only prominent in exon 9.19 and 9.21 for GOF conditions compared to wild type (marked in red for decrease and blue for increase, Fig 3C and D).

(A, C) Denaturing acrylamide gel showing restriction digests of Dscam exon 9 variables amplified with a 32P labelled forward primer from 14-18 h Drosophila embryos for canonical SR protein LOF (A) and elavGAL4 UAS GOF mutants (C). Quantification of inclusion levels are shown as means with standard error from three experiments for canonical SR protein LOF (B) and GOF mutants (D). Prominent changes in inclusion levels in mutants compared to wild type are marked with red letters for a decrease and in blue for an increase, and statistically significant differences are indicated by asterisks (***p≤ 0.001, **p≤ 0.01, *p≤ 0.05).
Non-canonical SR protein Srrm234 is required for selection of Dscam exon 9 variables
The canonical SR proteins contain an RRM and bind to RNA. In contrast, the large Srrm1 and Srrm234 proteins contain RS domains, but seem not to bind RNA and exert splicing enhancing functions through association with proteins bound to ESEs (Fig 2) (Blencowe et al. 1998; Eldridge et al. 1999; Blencowe et al. 2000; Szymczyna et al. 2003).
We obtained null mutants for both genes, Srrm1 B103 and Srrm234ΔN, which are late embryonic lethal (Fan et al. 2014). While loss of Srrm1 had little effect on Dscam exon splicing, loss of Srrm234 resulted in significant reduction in inclusion for many variable exons (marked in red, 9.4, 9.7, 9.10, 9.16/9.27, 9.17, 9.18, 9.20, 9.23/9.25, 9.29 and 9.33) that is compensated by increased inclusion of a few variable exons (marked in blue, 9.3/9.32, 9.6/9.8, 9.12 and 9.30) (Figure 4A and B).

(A) Denaturing acrylamide gel showing restriction digests of Dscam exon 9 variables amplified with a 32P labelled forward primer from 14-18 h Drosophila embryos for Srrm1 and Srrm234 protein LOF mutants. Quantification of inclusion levels are shown as means with standard error from three experiments (B). Red arrows point towards exons with reduced inclusion levels in the Srrm234ΔN mutant compared to wild type. Prominent changes in inclusion levels are marked with red letters for a decrease and in blue for an increase in the Srrm234ΔNmutant compared to wild type. Statistically significant differences are indicated by asterisks (***p≤ 0.001, **p≤ 0.01, *p≤ 0.05).
Alteration of hnRNP proteins minimally impacts on Dscam exon 9 alternative splicing
Since alterations of individual canonical SR proteins had little impact on selection of Dscam exon 9 variables, we focused on hnRNP proteins as candidates for repressing inclusion of exon 9 variables (Olson et al. 2007; Chen and Manley 2009; Fu and Ares 2014). Drosophila has four members of the highly expressed hnRNP A family (Hrp36, Hrp38, Rb97D and Hrp48) and one member of the hnRNP C, although the Drosophila orthologue is considerably longer (Fig 5)(Appocher et al. 2017). Other highly expressed hnRNP proteins are Hrp40 (hnRNP D), Glorund (hnRNP F/H), Hrb57A (hnRNP K) and Hrb59 (hnRNP M).
To determine the role of hnRNP proteins in Drosophila Dscam exon 9 alternative splicing we could obtain null-mutants for all major hnRNP proteins (Hrp36BG02743, Hrp38MI1059, RB97D1, Hrp48 GS14498, Hrp40 GS18188, glo f02674, Hrb57 G13574 and Hrp59GS6029) and gene-switch or EP over-expression lines for most (Hrp36 GS15926, Hrp38 GS12795, Hrp48 EY12571, and Hrp59 GS6029, Fig 5). Half of the tested major hnRNPs are required for viability (Rb97D, Hrp40, glo and Hrp59), while only over-expression of Hrp36 was lethal (Fig 5).

Evolutionary relationship of Drosophila hnRNP proteins with human homologues is shown on the left and the domain structure is indicated by coloured boxes. RNA recognition domain (RRM, light blue) and hnRNP K homology domain (KH, purple). The type of allele obtained, and the loss (LOF) and gain of function (GOF) phenotypes are indicated on the right.
Then, we analysed Dscam exon 9 inclusion levels for LOF and GOF of hnRNPs. For LOF alleles, the Dscam exon 9 splicing pattern also unexpectedly remained largely unchanged and significant changes are prominent in exon 9.17, 9.19 and 9.21 for mutants compared to wild type (marked in red for decrease and blue for increase, Fig 6A and B). Similar results were obtained for GOF conditions with significant changes only prominent in exon 9.2, 9.4, 9.17, 9.19, 9.21, and 9.30 (marked in red for decrease and blue for increase, Fig 6C and D).

(A, C) Denaturing acrylamide gel showing restriction digests of Dscam exon 9 variables amplified with a 32P labelled forward primer from 14-18 h Drosophila embryos for general hnRNP protein LOF (A) and elavGAL4 UAS GOF mutants (C). Quantification of inclusion levels are shown as means with standard error from three experiments for canonical SR protein LOF (B) and GOF mutants (D). Prominent changes in inclusion levels are marked with red letters for a decrease and in blue for an increase, and statistically significant differences are indicated by asterisks (***p≤ 0.001, **p≤ 0.01, *p≤ 0.05).
Selection of Dscam exon 9 variables is robust against removal of multiple SR and hnRNP proteins
hnRNP36 and SR protein B52 genes lie next to each other and potentially could cross-regulate to compensate for each other (Fig 7A). Therefore, we generated a double knock-out of hnRNP36 and B52 genes (hnRNP36/B52Δ1). In addition, we also combined this double knock-out with the hnRNP38 mutant as hnRNP36 and hnRNP38 are closely related and since they are highly expressed, they could act redundant.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Schematic of the genomic region of Hrp36 and B52 genes. Transposon inserts are shown with triangles and the deletion Hrp36/B52Δ1 is indicated at the bottom. (B) Denaturing acrylamide gel showing restriction digests of Dscam exon 9 variables amplified with a 32P labelled forward primer from 14-18 h Drosophila embryos for Hrp36BG02743, Hrp36/B52Δ1, Hrp38d05172 and Hrp36/B52Δ1 Hrp38 d05172 mutants. (C) Quantification of inclusion levels are shown as means with standard error from three experiments. Prominent changes in inclusion levels are marked with red letters for a decrease and in blue for an increase, statistically significant differences are indicated by asterisks (***p≤ 0.001, **p≤ 0.01, *p≤ 0.05).
Surprisingly, even in hnRNP36/B52Δ1hnRNP38d05172 triple mutants, Dscam exon 9 was robustly spliced with only differences in inclusion levels of variables 9.4, 9.16/9.27, 9.17, 9.18, 9.19, and 9.21 compared to controls (marked in red for decrease and blue for increase, Fig 7B and C.
Variable exon selection is not explained by long-range base-pairing in Dscam exon 9
In the exon 4 cluster, we could not detect conserved sequences adjacent to every variable exon that could mediate long-range base-pairing arguing that such a mechanism is not involved in variable exon 4 selection (Haussmann et al. 2019). A sequence alignment between D. melanogaster and D. virilis showed strong conservation in the coding sequences and the architecture of the exon 9 cluster with only few insertions and deletions of exons (Supplementary Figure 1). There are conserved sequence elements in the intron before constant exon 10 that potentially could serve as a docking site (Yang et al. 2011), but we did not find conserved sequence elements between every variable exon by manual inspection (Supplementary Figure 2). A systematic bioinformatics comparison of this potential docking site with arbitrary sampled sequences of the same length and sequence complexity further did not reveal a special propensity of the docking site to form more or stronger complementary alignments within Dscam intronic sequences in the variable exon 9 cluster. The same picture arises when comparing predicted energies of the best secondary duplex structures formed by the reverse intronic sequence, the docking site, and sampled sequences (Supplementary Figure 3). In addition, we didn’t find other instances of the docking site in any other gene of D. melanogaster (except in the anti-sense RNA CR45129 of Dscam), indicating that the sequence is not recurrent in other genes.
Discussion
Although the sequence determinants that direct the spliceosome to its correct position are very degenerate, splicing needs to occur precisely and with high accuracy to prevent disease (Cooper et al. 2009; Zaharieva et al. 2012). It is therefore thought that RNA binding proteins play key roles in localizing functional splice sites. In particular, the abundant SR and hnRNP proteins have been attributed key roles in this process by forming RNA-protein complexes co-transcriptionally to recruit early splicosomal components for defining splice sites. Although SR proteins were initially viewed as binding ESEs to activate splicing, and hnRNP proteins to bind ISSs for antagonizing SR proteins, a number of genome-wide studies draw a more complex picture for both SR and hnRNP binding and function (Blanchette et al. 2009; Anko et al. 2010; Anko et al. 2012; Huelga et al. 2012; Pandit et al. 2013; Brooks et al. 2015). In fact, both SR and hnRNP proteins can have very specific functions in one context, but also redundant functions in another context.
In this regard, we hypothesised that an array of similar exons as found in the Drosophila Dscam gene would provide a platform for SR and hnRNP proteins to evolve exon specific functions to regulate their inclusion. Surprisingly, however, Dscam exon 9 alternative splicing is exactly the opposite and the splicing pattern is very robustly maintained when SR and hnRNP proteins were either removed or overexpressed. In particular, the advances of Drosophila genetics allowed us to use complete knock-outs of most canonical SR and general hnRNP proteins and thus avoid the ambiguity of RNAi that would leave residual protein. Hence, despite complete loss of individual SR and hnRNP proteins, or combinations thereof, the Dscam splicing pattern is robustly maintained.
Two explanations are possible for this scenario. First, SR and hnRNP proteins act redundantly at the very extreme such that fluctuations in many would need to occur to impact on Dscam alternative splicing. However, whether this model applies will be difficult to test as removal of many general splicing factors will likely lead to global perturbations affecting many genes.
A second scenario could be that Dscam alternative splicing is fairly independent of general splicing factors. This would imply a more specific mechanism. Initially, it has been though that long-range base-pairing would provide such a mechanism as conserved sequences have been found in the Dscam exon 6 cluster (Graveley 2005). Our previous analysis of the exon 4 cluster, and the in depth analysis of the exon 9 cluster in this paper, however, rule out such mechanism in these two clusters (Haussmann et al. 2019). Hence, the question remains whether two independent mechanisms arose to regulate mutually exclusive alternative splicing in Dscam variable clusters. Potentially, the conserved sequences present in the variable clusters could provide binding sites for RBPs that have adopted cluster specific roles. In this context it is interesting to note that deletion of the docking site in exon 6 leads to inclusion of mostly the first exon in the cluster (May et al. 2011). This is unexpected as removal of the splicing activating mechanism should result in skipping of the entire variable cluster, because the proposed repressor hnRNP36 would still be present. Accordingly, the docking site in the exon 6 cluster also exerts a repressive role in maintaining the entire cluster in a repressed state.
Likewise, our finding that non-canonical Srrm234 regulates inclusion of many variables in the Dscam exon 9 cluster suggests a mechanism in Dscam mutually exclusive alternative splicing that differs from more general splicing rules directed by canonical SR and general hnRNP proteins. One of the human homologs of Srrm234, SRRM4 has key roles in the regulation of microexons (Irimia et al. 2014). Due to their small size, microexons cannot be defined through standard mechanism of exon definition. Hence, a distinct mechanism must apply, that can accurately direct splicing of microexons. Because microexons are often found in large introns, a robust process must underlie their selection and involves a newly described enhancer of microexons domain (eMIC), present in human SRRM3 and SRRM4. Intriguingly, in vertebrates, the ancestral pro-homologue Srrm234 has duplicated into three genes to adopt distinct functions through dedicated protein domains, but these features are maintained by alternative mRNA processing in Drosophila Srrm234 to include the eMIC at the C-terminus of the protein in neuronal tissue (Torres-Mendez et al. 2019).
Dscam alternative exons comply with the general average length of exons and thus the mechanism of their regulation is likely distinct from microexons. Dscam exon 9 cluster regulation by Srrm234 seems to involve its Cwf21 domain, which is not required for microexon inclusion (Torres-Mendez et al. 2019). Transposon inserts in the middle of the Srrm234 gene resulting in a truncated protein, that contains the Cwf21 domain do not affect exon 9 diversity (Mi{ET1}CG7971MB07314 and Mi(Liang et al.)CG7971MI04068, data not shown). The Cwf21 domain, which is homologous to the yeast Cwc21 domain, has been attributed key roles in splicing due to co-purification of the human ortholog SRRM2 with active spliceosomes and its localization in the catalytic centre of the spliceosome (Bessonov et al. 2008). Furthermore, the Cwc21 domain interacts with the U5 snRNP core components Snu114 and Prp8 involved in key structural rearrangements in the spliceosome during catalysis (Grainger et al. 2009; Gautam et al. 2015). Interestingly, Cwc21 has been attributed roles in splicing of meiotic genes which are regulated differently of general intron containing genes (Gautam et al. 2015). Srrm2 forms a complex with Srrm1 to promote alternative splicing of Drosophila doublesex required for sex determination, but the mechanism in Dscam is different as loss of Srrm1 does not impact on alternative splicing in the exon 9 cluster (Blencowe et al. 1998; Eldridge et al. 1999).
A Dscam cluster-specific role has been suggested for Hrp36 for exon 6 splicing in S2 cells, but whether the results are the same in flies remains to be tested (Olson et al. 2007).
Taken together, Dscam exon 9 mutually exclusive alternative splicing is robust against fluctuations of in canonical SR and general hnRNP proteins arguing for a specific mechanism regulating inclusion levels of variable exons. Indeed, non-canonical SR protein Srrm234 plays a key role increasing inclusion of many exon 9 variables. However, since Srrm234 does not have not have one of the classic RNA binding domains, additional RNA binding proteins likely connect Srrm234 to Dscam exon 9. Hence, our data obtained from knock-outs of general splicing factors indicate that a small complement of RNA binding proteins are likely key regulators of Dscam mutually exclusive alternative splicing.
The gene structure of invertebtrate Dscam harbouring arrays of variable exons for mutually exclusive splicing is at the extreme, but arrays of several alternative exons are common to many genes in metazoans. Likely, a yet to be discovered feature of the spliceosome has been exploited in mutually exclusive alternative splicing of Dscam such that only on exon is chosen. Likewise, since sequences that look like splice site are common in large introns, such a mechanism could be broadly relevant for robust select of isolated exons.
Materials and Methods
Fly genetics
Flies were maintained on standard cornmeal agar food as described (Haussmann et al. 2013). CantonS was used as a wild type control. The following loss of function mutants were used: X16 (P{GSV6}Hrb27CGS1678, DGRC Kyoto #206763), RBP1 (P{EPg}mRpL37HP37044, BDSC #22011), RBP1-like (P{GawB}Rbp1-likeNP0295, DGRC Kyoto #103580), Srp54 (P{GSV6}Srp54GS15334,, DGRC Kyoto #206174), SC35 (P{SUPor-P}SC35KG02986, BDSC #12904), SF2 (P{GSV7}SF2GS22325, DGRC Kyoto #203903), B5228(Gabut et al. 2007), SF1 (P{EP}SF1G14313 BDSC #30203), Hrp36 (P{GT1}Hrb87FBG02743, BDSC #12869), Hrp38 (Mi(Liang et al.)Hrb98DEMI1059, BDSC #55509), Rb97D (P{PZ}Rb97D1, BDSC #11782), Hrp48 (P{GSV6}Hrb27CGS14498, DGRC Kyoto #205836), Hrp40 (P{GSV6}sqdGS18188, DGRC Kyoto #201020), Glo (PBac{WH}glof02674, BDSC #18576), Hrb57A (P{EP}HnRNP-KG13574, BDSC #29672), Hrp59 (P{GSV3}rumpGS6029, DGRC Kyoto #200852), Srrm1 B103 (SRm160 B103, (Fan et al. 2014) and Srrm2340ΔN. Whether lethality of mutants mapped to the locus was tested by crossing to chromosomal deficiencies.
The null-allele Srrm234ΔN (CG7971) was generated by GenetiVision CRISPR gene targeting services. The 3.2kb deletion at the N-terminus of the gene was generated using sgRNAs AGTCTGCTGGGGACACTGCT and CGCCGCAGGACATATAACAG together with donor template harbouring two homology arms flanking a loxP 3xP3-GFP loxP cassette. Left and right homology arms of the donor were amplified using primers CG7971-LAF1 (GTTCCGGTCTCTTAGCCCTGCAGCAGCTTCTGCTTG) and CG7971-LAR1 (TCCAAGGTCTCACAGTTTATATGTCCTGCGGCGCTGC), and CG7971-RAF2 (GTTCCGGTCTCTGTCAGCTGGGAGCCGGCAGTGC) and CG7971-RAR2 (TCCAAGGTCTCAATCGAGTGGAGAACCCATACGTACTTAGATCC), respectively. Successful deletion and integration of the cassette was validated by PCR and Sanger sequencing using primers CG7971-outF1 (CATCGATTGTGTTGCATGAAGTTCAC) and CG7971-outR2 (GGGGAGTATCTGTGAGCAGTTGTATC), and LA-cassette-R (AAGTCGCCATGTTGGATCGACT) and Cassette-RA-F (CCTGGGCATGGATGAGCTGT), respectively. For the analysis of Dscam alternative splicing the 3xP3-GFP marker was removed by Cre mediated recombination using an insert on a third chromosome balancer (TM6B, P{w[+mC]=Crew}DH2, Tb, BDSC #1501) and the resulting chromosome was rebalanced with a zygotically YFP-expressing balancer (TM6B, P{Dfd-EYFP}3 Sb, Tb, BDSC #8704) to collect the embryonic lethal homozygous mutants.
For gain of function experiments the following UAS lines, gene switch vector inserts and EP lines were used: UAS GFP control line (Gabut et al. 2007), UAS GFP-X16 (Gabut et al. 2007), UAS RSF1 (Labourier et al. 1999), UAS GFP-SC35 (Gabut et al. 2007), UAS GFP-SF2 (Gabut et al. 2007), Srrm1 (P{EP}Srrm1G18603, BDSC #26938), UAS GFP-B52 (Gabut et al. 2007), Hrp36 (P{GSV6}GS15926, DGRC Kyoto #206416), Hrp38 (P{GSV6}GS12795, DGRC Kyoto #204283), Hrp48 ({EPgy2}Hrb27CEY12571, BDSC #20758), Hrp59 (P{GSV3}GS6029, DGRC Kyoto #200852).
Hrp36 and B52 genes lie next to each other. The Hrp36/B52Δ1 double mutant was generated by FRT/FLP mediated recombination using PBac{RB}e01378 and PBac{WH}f01884 transposon insertions as described (Zaharieva et al. 2015). The lethal Hrp36/B52Δ1 allele was balanced and validated using primers e01378 Rev (GCCACATTTAGATGATTCAGCATTAT), f01884 Rev (GATTCCAATAGATCCCAACCGTTTCG) and RB 3’ MINUS (TCCAAGCGGCGACTGAGATG).
Lethal lines were rebalanced with balancers expressing YFP zygotically, but not maternally under a Dfd promoter (CyO, P{Dfd-EYFP}2, BDSC #8578, TM6B, P{Dfd-EYFP}3 Sb, Tb, BDSC #8704) to allow for selection of homozygous lethal mutants. Non-GFP expressing 14-18 h embryos were further selected according to the morphology of the auto-fluorescing gut to distinguish them from homozygous balancer carrying animals, which die before they express GFP (Haussmann et al. 2008). For over-expression a third chromosomal elavGAL4 insert was used (P{w[+mmC]=GAL4-elav.L}3, BDSC #8760).
RNA extraction, RT-PCR, restriction digestion, denaturing acrylamide gels
Total RNA was extracted using Tri-reagent (SIGMA) and reverse transcription was done with Superscript II (Invitrogen) as described (Koushika et al. 1999) using primer Dscam 11RT1 (CGGAGCCTATTCCATTGATAGCCTCGCACAG, 1 pmol/ 20 µl reaction). PCR to amplify Dscam exon 9 cluster was done using primers 8F1 (GATCTCTGGAAGTGCAAGTCATGG) and 10R1ΔST (GGCCTTATCGGTGGGCACGAGGTTCCATCTGGGAGGTA) for 37 cycles with 1 µl of cDNA. Primers were labeled with 32P gamma-ATP (6000 Ci/ mmol, 25 mM, Perkin Elmer) with PNK (NEB) to saturation and diluted as appropriate. From a standard PCR reaction with a 32P labelled forward primer, 10–20% were sequentially digested with a mix of restriction enzymes (NEB) according to their buffer requirements and temperatures. PCR reaction and restriction digests were phenol/CHCl3 extracted, ethanol precipitated in the presence of glycogen (Roche) and analyzed on standard 6% sequencing type denaturing polyacrylamide gels. After exposure to a phosphoimager (BioRad), individual bands were quantified using ImageQuant (BioRad) and inclusion levels for individual variable exons were calculated from the summed up total of all variables. Statistical analysis was done by one-way ANOVA followed by Tukey– Kramer post-hoc analysis using Graphpad prism. Percent inclusion levels of exon 9 variables of embryos were calculated from the total sum of variables.
Sequence analysis
Vista alignments were generated as described (Haussmann et al. 2011). The exon 9 docking site was scanned against all gene sequences (as downloaded from FlyBase on the 26th of February 2019) using Blat (Version 35, parameters: -stepSize=1 tileSize=6 -minScore=0 -minIdentity=0, filtering for hits of at least bit-score of 30 and a coverage over the docking sequence of at least 20 residues) (Kent 2002).
In order to compare the docking site’s propensity to form potential long-range base-pairing with other intronic Dscam sequences of the same length, we sampled 100 strictly intronic sequenced of the same or higher sequence complexity as determined by Shannon entropy. We then used the BioPython Bio.pairwise2 local alignment implementation to get pairwise alignments against all (excluding the sampled sequenced and the docking sequence) reverse and reverse-complement sub-sequences of the same length in any Dscam intron, using a custom substitution matrix to allow for G-U pairings (scoring scheme: G->U 0.8, A->U: 1, G->C: 1.2, gap opening penalty 0.1, gap extension penalty 0.1) (Cock et al. 2009). We then retained all hits with less than 3 gaps and normalized the alignment scores by each queries’ potential highest score. To assess the best potential secondary structure formed by the docking sequence with any intronic sequence, we run the same sampled sequenced against the concatenated reverse and reverse complement intronic sequences of Dscam (masking each query) using RNAduplex from the Vienna package and obtained for each sequence the predicted lowest energy secondary structure prediction (Lorenz et al. 2011).
Author contributions
M.S. and P.U. designed and directed the project. P.U., M.S., I.H., H.L. performed experiments. A. T-M and M.I. generated and validated the Srrm234ΔN allele. R.A. performed bioinformatic analysis. P.U. and M.S. wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgments
We thank Bloomington, Kyoto and Harvard stock centres, and J. Tazi and L. Rabinow for fly lines, and P. Grzechnik for comments on the manuscript. We acknowledge funding from the Sukran Sinan Memory Fund to P.U, BBSRC and the European Research Council (ERC-StG-LS2-637591) to M.I.
Footnotes
↵‡ Zhongkai University of Agriculture and Engineering, Haizhu District, Guangzhou, China
References




