

SUMMARY
Mechanisms coupling the atypical PKC (aPKC) kinase activity to its subcellular localization are essential for regulating cell polarization but remain to be fully elucidated. Unlike other members of the PKC family, aPKC has no well-defined plasma membrane (PM) or calcium binding domains, leading to the assumption that its subcellular localization relies exclusively on protein-protein interactions. Here we show that in both Drosophila and mammalian cells the pseudosubstrate region (PSr) of aPKC acts as a polybasic domain sufficient to target aPKC to the PM via electrostatic binding to PM phosphoinositides. PM-targeting of aPKC requires the physical interaction between the N-terminal PB1 domains in both aPKC and Par-6, which not only allosterically exposes PSr to PM-binding, but also inhibits aPKC kinase activity. Such kinase inhibition requires the C-terminal domain of Par-6 and can be relieved through Par-6 interaction with apical polarity protein Crumbs. Our data suggest a mechanism in which allosteric regulation of polybasic PSr in aPKC by Par-6 couples the control of both aPKC subcellular localization and spatial activation of its kinase activity.
INTRODUCTION
For the majority of polarity proteins, achieving polarized plasma membrane (PM) / cortical localization is essential for their regulation of cell polarization (Hong, 2018; Rodriguez-Boulan and Macara, 2014). Mechanisms mediating the PM/cortical association of these polarity proteins had often been assumed to be based primarily on the intricate protein-protein interactions among polarity proteins and their regulators (Rodriguez-Boulan and Macara, 2014). One breakthrough in recent studies is the identification of so-called polybasic (i.e. basic-hydrophobic) domains in several polarity proteins such as Lgl, Numb and Miranda (Bailey and Prehoda, 2015; Dong et al., 2015). Polybasic domains are short but highly positively charged due to the enrichment of basic Arg/Lys residues. Polybasic domains can bind to PM specifically, as the inner surface of PM is the most negatively charged membrane surface due to membrane phosphatidylserine (Yeung et al., 2008) and its unique enrichment of polyphosphoinositides PI4P and PI(4,5)P2 (PIP2) (Hammond et al., 2012). Moreover, polybasic domains in Lgl, Numb and Miranda also contain conserved serine residues that can be phosphorylated by another key polarity protein atypical PKC (aPKC) (Bailey and Prehoda, 2015; Dong et al., 2015). Similar to the phosphorylation of the polybasic ED domain in MARCKS by PKC (Arbuzova et al., 2002), aPKC-phosphorylation neutralizes the positive charges in a polybasic domain therefore inhibits its electrostatic binding to phospholipids on the PM (Bailey and Prehoda, 2015; Dong et al., 2015). Such aPKC-dependent inhibition serves an elegant mechanism to polarize the PM targeting of polybasic polarity proteins, allowing apically localized aPKC to limit Lgl, Numb and Miranda to basolateral PM. Potential aPKC-phosphorylatable polybasic domains have been found in hundreds of proteins in metazoan genomes (Bailey and Prehoda, 2015)(Y.H. unpublished data), suggesting aPKC plays a critical role in regulating the PM targeting of many polybasic proteins.
However, to date the exact molecular mechanism governing the PM/cortical localization of aPKC itself remains unclear. Unlike conventional PKC (cPKC) and novel PKC (nPKC) that bind DAG (diacylglycerol), phospholipids and calcium for their kinase activation and PM targeting, aPKC has no well-defined PM or calcium binding domains and has not been demonstrated or proposed to directly bind the PM (Garg et al., 2013; Rosse et al., 2010). In fact, it is considered a unique feature of aPKC that its kinase activity and subcellular localizations appear to be exclusively regulated by protein-protein interactions other than lipids and/or calcium (Rosse et al., 2010). aPKC has a PB1 domain that binds to the PB1 domain in another polarity protein Par-6 which consistently colocalizes with aPKC in many polarized cell types such as epithelial cells and C. elegans one-cell embryos (Hong, 2018; Suzuki and Ohno, 2006). PM/cortical localization of aPKC/Par-6 complex has been assumed mainly based on protein interactions with other polarity proteins such as Baz (Izumi et al., 1998; Krahn et al., 2010; Morais-de-Sa et al., 2010), Crb (Sotillos et al., 2004), Sdt (Wang et al., 2004), Patj (Hurd et al., 2003) or Cdc42 (Joberty et al., 2000; Lin et al., 2000; Qiu et al., 2000). Recent studies have delineated some detail mechanisms by which Par-3 and Cdc42 coordinates the spatial and temprol control of aPKC kinase activity during the anterior-posterior (A-P) polarization of worm one-cell embryo (Dickinson et al., 2017; Rodriguez et al., 2017), but how aPKC PM localization and kinase activity are regulated during apical-basal (A-B) polarization is much less clear. Moreover, in vitro kinase assays yielded conflicting results regarding whether binding of Par-6 to aPKC inhibits or activates its kinase activity (Hong, 2018).
Here we report that the pseudosubstrate region (PSr) of aPKC is also a polybasic domain that directly binds to PM through electrostatic interaction with PM phosphoinositides PI4P and PIP2. Interestingly, PSr in aPKC appears to be non-phosphorylatable thus its binding to PM cannot be regulated by phosphorylations. Instead we show that protein interactions between aPKC and Par-6 appear to allosterically control the PM binding of polybasic PSr. Our data support a model that Par-6-dependent allosteric regulation of polybasic PSr in aPKC couples the PM targeting of aPKC and the spatially restricted activation of aPKC kinase during apical-basal polarization.
RESULTS
The conserved pseudosubstrate region (PSr) is a polybasic domain required for PM targeting of DaPKC in Drosophila epithelial cells
Our previous stuides showed that acute hypoxia induces loss of phosphoinositides in PM which in turn disrupts the PM localization of polybasic domain proteins such as Lgl and Numb in live fly tissues and cultured cells (Dong et al., 2015). Curiously, in the same studies we found that endogenous aPKC in Drosophila (DaPKC) also showed dramatic loss of PM localization in epithelial tissue under hypoxia (Dong et al., 2015), suggesting PM targeting of DaPKC may be based on direct binding to PM phospholipids in Drosophila epithelial cells. Similar to conventional PKC (cPKC) and novel PKC (nPKC), aPKC contains a pseudosubstrate region (PSr) that binds the kinase domain to induce autoinhibition (Rosse et al., 2010). We identified that the PSr in DaPKC is in fact highly polybasic (i.e. basic-hydrophobic) (Fig. 1A): the 17aa PSr contains eight Arg and Lys residues plus one Leu and one Trp residues, and the adjacent 13aa sequence in C1 domain contains additional four Arg and Lys residues plus two Phe residues (Heo et al., 2006). Besides the enriched Arg and Lys residues, the presence of Trp, Phe and Leu is highly indicative for a polybasic domain as the bulky or long hydrophobic side chains of these residues enhance the PM binding (Heo et al., 2006; McLaughlin and Murray, 2005; Yeung et al., 2006). The basic-hydrophobic index (Brzeska et al., 2010) of PSr in aPKC is 0.92, similar to polybasic domains in Lgl (1.00) and Numb (0.89). The PSr is extremely well conserved between DaPKC and mammalian aPKC isoforms PKCζ and PKCι (Fig. 1A).

(A) Alignment of the PSr (bold) and adjacent sequences in C1 domain from Drosophila and mammalian aPKC isoforms. Sequences are based on NCBI RefeSeq# NP_524892.2 (DaPKC), NP_002735.3 (PKCζ), and NP_002731.4 (PKCι), respectively. Lys/Arg residues mutated in PKCζKR8A are shown at the bottom. Same residues were mutated in DaPKCKR8Q or PKCζKR8Q.
(B) GST fusion of PSr from PKCζ (“GST-PSr”), but not the non-polybasic GST-PSr-KR8A, co-sedimented with PI4P- and PIP2-containing liposomes in vitro.
(C) DaPKC::GFP (“DaPKC”), but not non-polybasic DaPKCKR8Q::GFP (“KR8Q”), localized to PM in embryonic and larval disc epithelia.
(D) Follicular epithelial cells in ovaries from ubi-DaPKC::GFP or par-6::GFP adult females were imaged ex vivo under controlled oxygen environment. Cells are in cross-section view as indicated by the illustration at the far right. PM localization of DaPKC::GFP and Par-6::GFP were acutely inhibited by hypoxia (0.5% O2), but recovered after few minutes of reoxygenation by air (see also Movies S1 and S2). Kymographs on the right show the loss and recovery DaPKC::GFP and Par-6::GFP on PM during hypoxia and post-hypoxia reoxygenation treatment. White boxes indicate where kymographs were sampled.
Similar to the polybasic domain in Lgl (Dong et al., 2015), PSr in aPKC is also capable of direct binding to PI4P and PIP2 in vitro in liposome pull-down assays. Purified GST fusion of PSr from PKCζ bound PI4P- and PIP2-containing liposomes, while non-polybasic GST-PSr-KR8Q with all eight Arg/Lys residues in PSr mutated to Gln, did not (Fig. 1B). PSr is also required for PM targeting of DaPKC in vivo: while ubiquitously expressed aPKC::GFP localized to apical PM in both wild type and aPKC-/- epithelial cells, non-polybasic aPKCKR8Q::GFP stayed in the cytosol (Fig. 1C and Fig. S1A).
Similar to Lgl and Numb, our live imaging assays showed that hypoxia induced acute and reversible loss of DaPKC::GFP from PM in live Drosophila follicular and embryonic epithelial cells. (Fig. 1D, Fig. S1B and Movie S1), consistent with electrostatic binding to phosphoinositides being the primary mechanism targeting DaPKC to the PM in vivo. Furthermore, Par-6 is an essential regulatory partner of aPKC and forms a robust complex with aPKC (Hong, 2018; Suzuki and Ohno, 2006). Both proteins are mutually dependent on each other for proper localization and function during cell polarization processes. Consistently, hypoxia in live Drosophila follicular and embryonic epithelial cells also induced acute and reversible loss of PM targeting of Par-6::GFP (Fig. 1D, Fig. S1B and Movie S2). The loss of DaPKC/Par-6 complex from PM under hypoxia is not due to the concurrent loss of Lgl from PM. Par-6::GFP in lgl-/- mutant follicular cells showed expanded localization to basolateral PM but responded to hypoxia identically to Par-6::GFP in wild type cells (Fig. S1C).
Overall, our data suggest that polybasic PSr in DaPKC is essential for localizing DaPKC/Par-6 complex to the PM in Drosophila epithelia in vivo through electrostatic interaction with PM phosphoinositides such as PI4P and PIP2 (see below).
PM targeting of mammalian PKCζ depends on both PSr and Par-6 but not Cdc42
Given the well documented requirement of Par-6 in aPKC subcellular localization in many cell types (Chen et al., 2018; Hong, 2018; Suzuki and Ohno, 2006), we further investigated the role of Par-6 in polybasic PSr-mediated PM targeting of aPKC. For such purpose we took the over-expression approach in non-polarized HEK293 cells in which we can specifically reconstruct the potential interactions between aPKC and Par-6 or other exogenously introduced proteins. Mammalian aPKC family has two isoforms PKCζ and PKCι, and both contain PSr domains nearly identical to DaPKC’s (Fig. 1A). To test whether mammalian aPKC also require PSr for PM targeting, we expressed PKCζ::GFP in HEK293 cells and found PKCζ::GFP was surprisingly only cytosolic (Fig. 2A). However, although Par-6::RFP alone is also cytosolic in HEK293 cells, co-expression of PKCζ::GFP and Par-6::RFP results in strong and robust PM localization of both proteins (Fig. 2A). Such co-localization to PM requires direct physical interaction between Par-6 and PKCζ, as a C107Y mutant of PKCζ which specifically abolishes the physical interaction with Par-6 (Kim et al., 2009), failed to localize to PM when co-expressed with Par-6 (Fig. 2B,C). Furthermore, PM targeting of PKCζ/Par-6 complex requires the polybasic PSr in PKCζ. Co-expression of Par-6 with non-polybasic PKCζKR8Q (Fig. 2B) resulted in no PM localization of either protein, even though the physical interaction between Par-6 and PKCζKR8Q remained intact (Fig. 2C). The loss of PM targeting of PKCζKR8Q is not due to potential misfolding, as PKCζKR8Q phosphorylates mLgl similarly to PKCζ in HEK293 cells (Fig. 5C). We also confirmed that such results are not unique to HEK293 cells, as COS7 and MCF7 breast cancer cells also showed same results of Par-6-dependent PM targeting of PKCζ (Fig. S2A,B).

(A) PKCζ::GFP and Par-6::RFP were cytosolic when each expressed alone, but both became strongly PM localized when co-expressed.
(B) PKCζC107Y::GFP (“C107Y”), PKCζKR8A::GFP (“KR8A”), PKCζΔPSr::GFP (“ΔPSr”) did not localize to PM when coexpressed with Par-6::RFP.
(C) PKCζ::GFP and non-polybasic PKCζKR8A::GFP, but not PKCζC107Y::GFP, co-immunoprecipitated with FLAG-Par-6 in HEK293 cells by anti-GFP antibody.
(D) PKCζ::GFP was cytosolic when co-expressed with BFP::Cdc42CA. Par-6::RFP localized to PM in cells co-expressing of BFP::Cdc42CA. PKCζ and Pra-6 were PM localized in cells expressing BFP::Cdc42CA, PKCζ::GFP and Par-6::RFP or expressing BFP::Cdc42DN, PKCζ::RFP and Par-6::iRFP. Non-polybasic PKCζKR8Q::GFP was PM-localized when co-expressed with BFP::Cdc42CA and Par-6::RFP.
In all data plots boxes extend from 25 and 75 percentiles, with lines in the middle indicating the median and whiskers indicating 10 and 90 percentiles, respectively. Sample numbers are indicated in parentheses at the right edge of box plots. Red dashed lines in quantification figures indicate the PM localization index = 1 (see Materials and Methods). Measurements less than 1 indicate cytosolic localization, whereas larger than 1 indicate PM localization. Note that PM localization axes in all figures are in log2 scale.
Cdc42 has been shown to be essential for aPKC/Par-6’s localization to anterior PM in C. elegans one-cell embryo and has well characterized physical interactions with Par-6. We thus investigated whether Cdc42 is somehow required for aPKC/Par-6 PM targeting in HEK293 cells. In HEK293 cells expressing constitutively active Cdc42CA, both PKCζ and PKCζKR8Q can be localized to PM, but only in the presence of Par-6 (Fig. 2D). However, overexpression of dominant-negative Cdc42DN did not inhibit the PM targeting of aPKC/Par-6, suggesting that Cdc42 activity is unlikely required for PSr-dependent targeting of PKCζ/Par-6 (Fig. 2D). Furthermore, since Cdc42 interacts with the C-terminal CRIB domain in Par-6, we made two truncated proteins: PB1Par-6 which only contains the N-terminal PB1 domain of Par-6, and Par-6ΔPB1 which carries reciprocal truncation. We found that PB1Par-6 is fully capable of targeting PKCζ to PM, while Par-6ΔPB1 cannot (Fig. 4B). Given that PB1Par-6 cannot interact with Cdc42, PM targeting of PKCζ by PB1Par-6 could not be Cdc42-dependent in our cell-based assays.
In summary, our data show that PM targeting of PKCζ/Par-6 in HEK293 cells is both PSr and Par-6 dependent but Cdc42 independent, and physical interaction between the N-terminal PB1 domains in aPKC and Par-6 is sufficient to induce PSr-dependent PM targeting of aPKC.
PI4P and PIP2 act redundantly to target aPKC/Par-6 complex to PM
Phosphoinositides PI4P and PIP2 are uniquely enriched in PM and are considered the major negatively charged phospholipids responsible for electrostatically binding polybasic domains (Hammond, 2012). To confirm that PM targeting of aPKC/Par-6 complex in cells is indeed mediated by PI4P and PIP2, we used a well-established system to acutely and selectively deplete PI4P, PIP2 or both in HEK293. In this inducible system addition of rapamycin induces dimerization between FKBP-tagged phosphatases and FRB-tagged PM anchor protein Lyn11-FRB-CFP (Hammond, 2012), resulting in acute PM recruitment of phosphatase and concurrent depletion of target phospholipids. In particular, PM-recruitment of a chimeric lipid phosphatase PJ (pseudojanin) rapidly converts both PI4P and PIP2 in PM to phosphatidylinositol (PI) (Hammond, 2012) (Fig. 3A). In HEK293 cells expressing PKCζ::GFP, Par-6::iRFP together with RFP-FKPB-PJ and Lyn11-FRB-CFP, addition of rapamycin induced acute and concurrent loss of both PKCζ::GFP and Par-6::iRFP from PM (Fig. 3B). Depleting either PIP2 using FKBP-INPP5E or PI4P using FKBP-PJ-Sac induced very mild loss of PM targeting of both proteins (Fig. 3A,B).

(A) LEFT: INPP5E converts PIP2 to PI4P which can be further converted to PI by Sac, whereas PJ converts both PIP2 and PI4P to PI. RIGHT: mRFP-FKBP-PJ can be acutely recruited to PM through rapamycin (rapa)-induced heterodimmerization with PM-anchored Lyn11-FRB-CFP. PM-recruitment of PJ results in acute removal of both PI4P and PIP2.
(B) PM localization of PKCζ::GFP and Par-6::iRFP were quantified prior and after the rapamycin addition in HEK293 cells that were also expressing Lyn11-FRB-CFP and mCherry-FKBP-PJ or - Sac, -INPP5E, -PJ-dead (as a negative control). Representative time-lapse images of Lyn11- FRB-CFP (red), PKCζ::GFP (green), Par-6::iRFP (magenta) and mCherry-FKBP-PJ/Sac/INPP5E/PJ-dead (cyan) are shown under each quantification figure. For each quantification, means ± standard error of the mean from 20-30 cells pooled across three independent experiments were plotted.
Thus, complete inhibition of PKCζ/Par-6 PM targeting requires depleting both PIP2 and PI4P, suggesting that PI4P and PIP2 act redundantly to bind PKCζ/Par-6 complex to PM. This is in contrast to Lgl which appears to rely more on PIP2 for its PM targeting (Dong et al., 2015),. Consistently, when we used the similar rapamycin-inducible system to acutely deplete PIP2 in Drosophila follicular epithelial cells, Par-6::GFP remained on the PM (Fig. S3) whereas Lgl::GFP showed significant loss from PM in PIP2-depleted cells (Dong et al., 2015). We could not carry out assays by inducible depletion of PI4P or both PI4P and PIP2 as corresponding genetic tools are currently unavailable in Drosophila.
PM-binding of PSr in aPKC is allosterically regulated by Par-6
Previous studies suggested that the PSr in PKCζ binds the kinase domain (KD) and autoinhibits its kinase activity, and that binding of Par-6 likely causes an allosteric conformation change in PKCζ that displaces the PSr from the kinase domain (Graybill et al., 2012). We postulate that in unbound aPKC its PSr is occluded by the kinase domain from binding to the PM but is allosterically exposed once Par-6 binds aPKC. To test this hypothesis, we generated two KD-deletion mutants PKCζ-ΔKD and PKCζKR8Q-ΔKD. Indeed, in contrast to full length PKCζ, PKCζ-ΔKD localizes to PM in the absence of Par-6 and its Par-6-independent PM localization still requires the positive charges of PSr region, as non-polybasic PKCKR8Q-ΔKD is cytosolic regardless the presence of Par-6 (Fig. 4A). Further supporting our allosteric regulation model, while interaction between PKCζ and Par-6 PB1 domain alone is sufficient to induce PKCζ PM localization (Fig. 4B), merely reducing the interaction between PSr and kinase domain is not sufficient to make PSr accessible to PM binding. A PKCζA119D mutant, in which Ala119 in PSr is mutated to phosphomimetic Glu to reduce its autoinhibition on the kinase domain, was still cytosolic and only localized to PM when coexpressed with Par-6 (Fig. 4A).
Although the lack of full protein structures of aPKC and Par-6 makes it difficult to conduct comprehensive structure based experiments to further confirm the allosteric regulation of PSr region by Par-6, our data strongly support a model that PB1/PB1 interaction between aPKC and Par-6 is both sufficient and necessary to allosterically displace the polybasic PSr in aPKC from kinase domain, exposing it to electrostatically bind the PM.

(A) PKCζ-ΔKD::GFP (“ΔKD”), in which kinase domain (KD) is deleted, localized to PM when either expressed alone or co-expressed with Par-6::RFP. Non-polybasic PKCζ-ΔKDKR8Q::GFP (“ΔKD-KR8Q”) failed to localize to PM with or without the co-expression of Par-6::RFP. PM targeting of PKCζA119D::GFP (“A119D”) remained Par-6-dependent.
(B) Both PKCζ::RFP and Par-6ΔPB1::iRFP remained in cytosol when co-expressed. PB1Par-6::iRFP was cytosolic when expressed alone but when co-expressed with PKCζ::RFP both became PM-localized. Experiments were carried out in HEK293 cells.
Par-6-dependent PM targeting inhibits PKCζ kinase activity
Binding of Par-6 is considered an essential step in regulating the kinase activity of aPKC, although whether Par-6 activates or inhibits aPKC remains unsettled and may well depend on additional regulators presented in different cell types (Hong, 2018). To investigate how Par-6 regulates PKCζ activity in vivo, we established aPKC kinase activity assays in cultured cells based on the loss of Lgl PM localization that serves as a sensitive, robust and quantifiable readout for measuring aPKC-phosphorylation of Lgl in live cells. When expressed alone in HEK293 cells mammalian mLgl::GFP showed consistently strong and robust PM localization (Fig. 5A) which was strongly reduced in cells co-expressing PKCζ but not kinase-dead PKCζK281W (Fig. 5A). An anti-phospho-mLgl antibody confirmed phosphorylation of mLgl in HEK293 cells coexpressing PKCζ (Fig. 5B), suggesting that overexpressed PKCζ alone contains basal kinase activity sufficient to phosphorylate Lgl, consistent with the fact that in vitro purified aPKC has ∼10% of activated kinase activity (Zhang et al., 2014). To further confirm that the loss of PM localization of mLgl is due to phosphorylation by PKCζ, we generated a non-phosphorylatable mLglS6A::GFP in which all six conserved phospho-serines were mutated to alanine. As expected, mLglS6A::GFP remained on the PM when co-expressed with PKCζ (Fig. 5A).
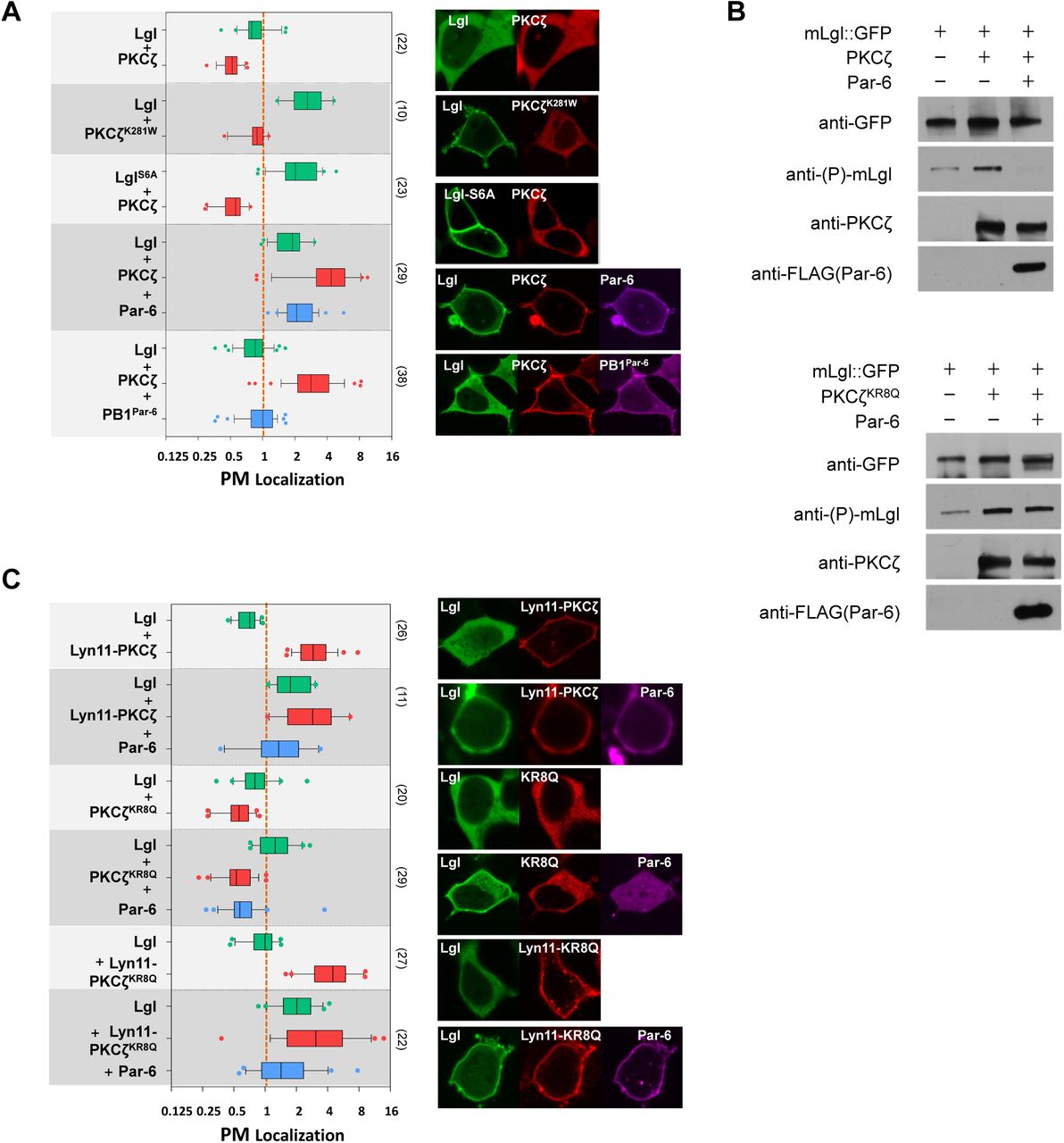
(A) PM localization of Lgl::GFP was strongly reduced in cells co-expressing PKCζ::RFP but not kinase dead PKCζK281W::RFP. PM localization of non-phosphorylatable LglS6A::GFP was resistant to PKCζ::RFP co-expression. Lgl::GFP showed strong PM-localization in cells co-expressing PKCζ::RFP and Par-6::iRFP, but not in cells expressing PKCζ::RFP and PB1Par-6::iRFP.
(B) Cells expressing Lgl::GFP only, expressing both Lgl::GFP and PKCζ::RFP (or PKCζKR8Q::RFP), or expressing Lgl::GFP together with PKCζ::RFP (or PKCζKR8Q::RFP) and FLAG::Par6 were directly lysed in SDS-loading buffer and analyzed by western blot. Anti-phospho-Lgl antibody detected a low level phosphorylation of Lgl::GFP expressed alone in HEK293 cells. Lgl::GFP phosphorylation was much increased in cells expressing PKCζ::RFP or PKCζKR8Q::RFP. Lgl::GFP phosphorylation was strongly inhibited in cells co-expressing PKCζ::RFP and FLAG::Par-6, but not in cells expressing non-polybasic PKCζKR8Q::RFP and FLAG::Par-6.
(C) PM localization of Lgl::GFP was also strongly reduced in cells expressing Lyn11- PKCζ::RFP, PKCζKR8Q::RFP (“KR8Q”) and Lyn11-PKCζKR8Q::RFP (“Lyn11-KR8Q”), respectively. In all three cases, co-expression of Par-6::iRFP resulted in increased PM localization of Lgl::GFP.
All experiments were carried out in HEK293 cells.
We then tested the kinase activity of PM targeted aPKC/Par-6 complex. Strikingly, in HEK293 cells co-expressing mLgl::GFP, PKCζ::RFP and Par-6::iRFP, all three proteins were strongly PM localized (Fig. 5A) and anti-pLgl antibody failed to detect phosphorylation on mLgl, suggesting that binding of Par-6 not only targets PKCζ to PM, but also strongly inhibits its kinase activity (Fig. 5B). Such apparent inhibition of PKCζ kinase activity by Par-6 is not due to PM localization of PKCζ alone, as a constitutively PM-bound Lyn11-PKCζ delocalized mLgl::GFP to the cytosol and was efficiently inhibited by Par-6 (Fig. 5C). Interestingly although both cytosolic PKCζKR8Q and Lyn11- PKCζKR8Q reduced the PM localization of Lgl (Fig. 5C), Par-6 apparently inhibited Lyn11-PKCζKR8Q more efficiently than PKCζKR8Q (Fig. 5C), suggesting that PM localization of PKCζ may enhance the inhibition of kinase activity by Par-6.
Thus, our data suggest that binding of Par-6 inhibits PKCζ kinase activity in live cells and such inhibition is enhanced by the PM targeting of PKCζ/Par-6 complex.
Lgl does not form robust inhibitory complex with aPKC/Par-6 on PM
Lgl antagonizes aPKC activity genetically and is considered a potential inhibitor of aPKC (Lee et al., 2006; Rolls et al., 2003; Wirtz-Peitz et al., 2008), therefore inhibited Lgl phosphorylation by aPKC/Par-6 observed in HEK293 cells could be due to Lgl forming a stable inhibitory complex with aPKC/Par-6 on PM. While Lgl certainly forms robust complex with aPKC and Par-6 in solution as evidenced by co-immunoprecipitation experiments (Betschinger et al., 2003; Plant et al., 2003; Yamanaka et al., 2003), whether Lgl forms stable complexes with aPKC/Par-6 on PM in live cells has not been investigated. Taking advantage of the fact that both Lgl and aPKC/Par-6 require PM PI4P and PIP2 for targeting, we made a Lyn11-Lgl::GFP so its PM localization will be independent of PI4P and PIP2. If aPKC/Par-6 form a stable complex with Lgl, in the presence of Lyn11-Lgl their PM localization will also become resistant to the loss of PI4P and PIP2.
In COS7 cells, Lyn11-Lgl, aPKC and Par-6 were all localized to PM when co-expressed (Fig. 6A). We then used a well-established PI4K kinase inhibitor PAO (phenylarsine oxide) to deplete PI4P and PIP2 in PM. While Lyn11-Lgl remained on PM under PAO treatment, aPKC and Par-6 were readily lost from PM (Fig. 6A). Conversely, in COS7 cells co-expressing Lgl, Lyn11-aPKC and Par-6 undergoing PAO treatment, only Lgl became lost from PM while Lyn11- aPKC and Par-6 remained PM localized (Fig. 6A). Our results support a stable PM complex of aPKC and Par-6, but argue against that Lgl forming a stable inhibitory complex with aPKC and Par-6 on the PM.

(A) COS7 cells expressing Lyn11-Lgl::GFP, PKCζ::RFP and Par-6::iRFP and cells expressing Lgl::GFP, Lyn11-PKCζ::RFP and Par-6::iRFP were treated with PAO (20µM). (-): before PAO addition. (+): 20 minutes after PAO addition. (A’) Quantification of PM localization of Lgl, PKCζ and Par-6 in cells treated with PAO. PM localization data at 0min of all samples were normalized to 2.
(B) PKCζ::RFP localized to PM in HEK293 cells co-expressing Lgl::GFP with either PB1Par-6::iRFP or Par-6ΔPDZ::iRFP. Lgl::GFP was strongly reduced from PM in these cells.
(C) In HEK293 cells expressing PKCζ::RFP and Par-6::iRFP, PM localization of Lgl::GFP was strongly reduced when BFP::Crbintra, but not BFP::Crbintra-ΔERLI or BFP::Cdc42CA, was co-expressed. LglS6A::GFP remained on PM in HEK293 cells expressing PKCζ::RFP, Par-6::iRFP and BFP::Crbintra. Lgl::GFP remained on PM in HEK293 cells expressing BFP::Crbintra, Par-6::iRFP and kinase-dead PKCζK281W::RFP.
(D, D’) Drosophila lgl::GFP or lglS6A::GFP follicular epithelial cells overexpressing Crb were immunostained for Lgl::GFP (green), DaPKC (red) and Crb (magenta). Images in D are in tangential view and were sectioned below the apical surface of follicular cells where Crb and aPKC normally are absent. Images in D’ are in cross-section view of follicular epithelial cells. Overexpressed Crb expanded into the lateral PM and apparently also mislocalized DaPKC to lateral PM. Cells overexpressing Crb are highlighted by asterisks in green channel images.
Crb activates aPKC/Par-6 kinase activity to phosphorylate Lgl
The PB1 domain of Par-6 alone (PB1Par-6) was still capable of targeting PKCζ to PM, so did Par-6ΔPDZ in which the C-terminal PDZ domain was deleted (Fig. 4B). However, neither protein inhibited the phosphorylation of mLgl::GFP by PKCζ (Fig. 6B), suggesting the PDZ domain in Par-6 could be specifically required for inhibiting PKCζ kinase activity on Lgl (Fig. 6B). The C-terminus of Par-6 interacts with multiple proteins, including activated Cdc42 which binds the Par-6 CRIB domain and moderately activates kinase activity of aPKC/Par-6 complex in vitro (Yamanaka et al., 2001). However, in HEK293 cells co-expression of Cdc42CA did not reduce PM localization of mLgl::GFP in the presence of aPKC/Par-6 (Fig. 6C), suggesting that activated Cdc42 is not sufficient to activate aPKC in our cell-based assays. Our results are consistent with genetic evidences that Cdc42 is not required for aPKC to phosphorylate Lgl (Hutterer et al., 2004) and Baz (Walther and Pichaud, 2010) in vivo.
Besides Cdc42, apical polarity protein Crb also interacts with Par-6. Crb is a transmembrane protein and the C-terminus of its intracellular domain is a PDZ-binding motif (PBD) that can bind the PDZ domain in Par-6 (Kempkens et al., 2006). Overexpression of Crb in Drosophila follicular cells and embryonic epithelial cells expanded Crb and DaPKC into basolateral PM, and completely delocalized Lgl, but not non-phosphorylatable Lgl-S5A, from basolateral PM (Fig. 6D, S4A). In contrast, in Drosophila crb-/- embryos aPKC also extended to the basolateral PM but Lgl remained on PM (Fig. S4B). Notably, overexpression of a supposedly constitutively active and PM-bound DaPKC-CAAX only caused partial loss of PM localization of Lgl (Fig.S4C). Such data suggest that Crb promotes DaPKC-phosphorylation on Lgl in epithelial cells.
We then investigated whether Crb directly activates aPKC kinase activity through its interaction with Par-6 PDZ domain. Par-6 became localized to the PM in HEK293 cells expressing membrane-bound Crb intracellular domain (Crb-intra), but not Crb-intraΔPBD in which the C-terminal PBD was deleted, confirming that Par-6 binds to the PBD of Crb-intra (Fig. S4D). More importantly, in HEK293 cells expressing Crb-intra, but not Crb-intraΔPBD, Lgl::GFP was strongly delocalized from the PM in the presence of aPKC and Par-6 (Fig. 6C). The loss of Lgl::GFP from PM in cells expressing Crb-intra, aPKC and Par-6 is phosphorylation-dependent, as Lgl::GFP remained on the PM in cells expressing Crb-intra, Par-6 and the kinase-dead aPKCK281W (Fig. 6C). In addition, non-phosphorylatable LglS6A::GFP remained on PM in cells expressing Crb-intra, aPKC and Par-6 (Fig. 6C). These results support a model of direct interactions between Crb-intra and Par-6 in a PM-bound aPKC/Par-6 complex that activates aPKC kinase activity and thus Lgl phosphorylation in vivo (Fig. 7).

(A) Free cytosolic aPKC in auto-inhibited conformation has polybasic PSr blocked by the kinase domain from binding to PM. Binding of Par-6 to aPKC induces conformation changes that expose the PSr in aPKC, as well as allows the C-terminus of Par-6 to simultaneously inhibit the aPKC kinase domain.
(B) Polybasic PSr in aPKC/Par-6 complex binds to PM via electrostatic interaction with PI4P and PIP2 which are uniquely enriched on PM.
(C) Intracellular domain of apical polarity protein Crb interacts with the C-terminal PDZ domain of Par-6 and release its inhibition on aPKC kinase domain. Interaction with Crb could also facilitate the apical-enrichment of PM-bound aPKC/Par-6.
(D) Activated aPKC phosphorylates Lgl to prevent it from binding to apical PM.
Illustration is based on Drosophila epithelial cells. AJ: adherens junction. PM: plasma membrane.
DISCUSSION
Electrostatic binding to phosphoinositides by polybasic PSr is sufficient to target aPKC to PM
In this report we show that the pseudosubstrate region (PSr) in aPKC is a typical polybasic domain capable of directly targeting aPKC to PM through its electrostatic interaction with negatively charged phosphoinositides PI4P and PIP2 in PM. This is in contrast to the current assumption that protein-protein interactions are solely responsible for localizing aPKC to PM or cell cortex. In addition, different from phosphorylatable polybasic domains in Lgl, Numb and Miranda, polybasic PSr in aPKC does not appear to be phosphorylatable therefore its PM binding cannot be inhibited by charge-neutralization. Instead we showed here a novel example that a polybasic domain can also be allosterically controlled on its binding to the PM. Our data support a model that the polybasic PSr in unbound aPKC is occluded by the kinase domain from binding PM, and the binding of Par-6 to aPKC via PB1 domains induces conformational changes in aPKC that make the polybasic PSr accessible to PM-binding (Fig. 7).
Consistent with Cdc42 playing an important role in mediating the PM targeting of aPKC/Par-6 in certain cell types such as C. elegans one-cell embryos, we found that activated Cdc42 binds Par-6 which in turn can recruit non-polybasic aPKCKR8Q to the PM. However, our studies showed that electrostatic binding to PI4P and PIP2 alone is sufficient to localize aPKC/Par-6 to the PM without interacting with additional proteins such as Cdc42. Our results are consistent with findings that removing Cdc42 in C. elegans late embryos or in Drosophila pupal epithelial cells does not severely disrupt aPKC and Par-6 localization (Georgiou et al., 2008; Zilberman et al., 2017). Although mechanisms governing the aPKC/Par-6 subcellular localization appear to be heavily cell-type specific and multi-layered, our results highlight that the electrostatic PM-binding property of aPKC needs to be taken into consideration when studying the protein regulators of aPKC/Par-6 subcellular localization. In fact, our hypoxia assay suggest that at least in Drosophila follicular and embryonic epithelial cells, binding to PI4P and PIP2 is likely the primary mechanism localizing aPKC/Par-6 to the PM in vivo. In these cells the Par-6-dependent electrostatic binding to PM by PSr likely functions as the first step to localize aPKC/Par-6 to the PM, followed by further recruitment to specific membrane domains such as apical PM through protein-protein interactions with, for instance, apical Crb complex.
Our studies also suggest potential new negative regulators that function to block this electrostatic binding step to prevent aPKC from binding to the PM, by mechanisms such as masking the PSr, inhibiting the allosteric changes induced by Par-6 or sequestering the phospholipids in the PM. Moreover, it is notable that a PI4P- and PIP2-dependent mechanism also makes aPKC PM targeting vulnerable to stress conditions such as hypoxia and ATP depletion that diminish levels of these phospholipids in the PM (Dong et al., 2015). The acute and reversible loss of PM targeting of polybasic polarity proteins like Lgl and aPKC (in complex with Par-6) may have profound implications in epithelial cells for maintaining and restoring their apical-basal polarity when undergoing hypoxia, ischemia and reperfusion.
Par-6 controls both aPKC PM targeting and kinase activity
Mechanisms spatially coupling the aPKC kinase activity with its subcellular localization are essential for aPKC to phosphorylate targets at the right place and right time (Hong, 2018), but molecular details about these mechanism remain largely unknow. Recent studies began to reveal exciting details on how aPKC/Par-6 kinase activity and subcellular localization maybe regulated by the clustering of Par-3(Baz) and diffusive interaction with Cdc42 (Dickinson et al., 2017; Rodriguez et al., 2017), but these studies so far have been limited to the process of anterior-posterior (A-P) polarization in worm one-cell embryos. Here we show that, in Drosophila epithelial cells and cultured mammalian cells, the electrostatic binding of aPKC to PM provides an elegant mechanism enabling Par-6 to play a pivotal role coupling the PM targeting and control of kinase activity of aPKC (Fig.7). It is possible that binding of Par-6 to aPKC induces conformational changes that not only expose PSr to electrostatically binding to the PM but also allow the C-terminus of Par-6 to simultaneously inhibit aPKC’s kinase domain.
Our allosteric model is consistent with previous studies that also suggested the displacement of PSr from aPKC upon binding of Par-6 (Graybill et al., 2012). However, in that study Par-6 appears to activate aPKC in vitro and in Drosophila S2 cells. Such discrepancy could be due to the fact that S2 cells already express proteins capable of releasing the inhibition of Par-6 on aPKC. It should be noted that studies from multiple labs yielded conflicting results on whether Par-6 inhibits or activates aPKC kinase activity (Atwood et al., 2007; Chabu and Doe, 2008; Graybill et al., 2012; Lin et al., 2000; Yamanaka et al., 2001). Most of these studies relied on immunoprecipitated or reconstituted aPKC/Par-6 complex to measure the kinase activity in solution in vitro. Given that the majority of aPKC substrates are PM-bound polybasic domain-containing proteins, it should be considered critical to assay aPKC/Par-6 kinase activity in its PM-bound form in live cells.
In addition, it needs to be noted that binding to PM should shield the polybasic domain in target proteins from being accessible to aPKC, as suggested by the increased resistance of membrane-bound Lgl to aPKC phosphorylation in vitro (Visco et al., 2016). This is consistent with our finding that PM-bound Lgl does not form robust complex with aPKC/Par-6 on PM, suggesting that, whereas Lgl forms robust complex with aPKC and Par-6 in solutions, Lgl/aPKC/Par-6 complex formation is inhibited when both Lgl and aPKC/Par-6 complex are electrostatically attached to PM. On the other hand, the transient and dynamic nature of lipid-binding by polybasic domains (Hammond et al., 2009) could also effectively enrich a local cytosolic pool of target proteins near PM, which works in favor of PM-bound aPKC for efficiently interacting with its substrates. Such intricate relationships between PM-targeting and aPKC-phosphorylation of polybasic polarity proteins remain to be further explored.
PM targeting and spatial control of aPKC kinase activation
Similar to cPKC and nPKC, the electrostatic binding of PSr to PM could potentiate the kinase activity of aPKC/Par-6 complex by preventing PSr from autoinhibiting the kinase domain (Garg et al., 2013; Rosse et al., 2010), as supported by in vitro studies that Par-6 binding to aPKC both inhibits its kinase activity but potentiates its activation upon subsequent molecular events (Yamanaka et al., 2001). PM-bound aPKC/Par-6 complexes with inhibited but potentiated kinase activity are ideal targets for additional activation to fine-tune aPKC localization and kinase activity. Curiously, we found that Cdc42 is not capable of activating PM-bound aPKC/Par-6 to phosphorylate Lgl in our cultured cell assays, but interaction between Par-6 and apical transmembrane protein Crumbs could release the Par-6’s inhibition on aPKC kinase activity. Supporting the role of Crb in enriching and activating aPKC/Par-6, Crb colocalizes with aPKC/Par-6 complex and is required for enriching aPKC/Par-6 complex to the apical PM in fly embryonic epithelial cells and in many specialized membranes in Drosophila including apical membranes photoreceptors (Hong et al., 2003; Pellikka et al., 2002) and lumens in scolopale cell in chondotoral organs (Y.H., data not shown). However, Crb is not required for apical enrichment of aPKC in follicular cells (Sherrard and Fehon, 2015), therefore additional proteins besides Crb must responsible for the apical enrichment and activation of aPKC/Par-6 in different types of epithelial cells.
Our studies were primarily based on cultured cells and limited types of Drosophila epithelial cells, and likely present a simplified model regarding the relationship between Crb and aPKC/Par-6. Whereas Crb certainly is not the only regulator activating aPKC/Par-6, our studies highlight that aPKC/Par-6 activity can be regulated by multiple regulators such as Cdc42 or Crb under different cellular and polarity contexts. Extrapolating our study to further understanding the role of aPKC in regulating the cell polarity requires experiments to take into account the more complicated and variable cellular contexts in different cell types.
COMPETING INTERESTS
The authors declare no competing or financial interests.
AUTHOR CONTRIBUTIONS
Conceptualization: Y.H., W.D., G.R.H.; Investigation: W.D., J.L., X.Z., Y.W., K.L., G.R.H. and Y.H.; Writing - Review &Editing: Y.H., W.D., G.R.H.; Funding acquisition: Y.H., G.R.H.; Supervision: Y.H.
FUNDING
This work was supported by grants NIH- NCRR R21RR024869 (Y.H.), NIH-NIGMS R01GM086423 and R01GM121534 (Y.H.), NIH 1R35GM119412-01 (G.R.H.). University of Pittsburgh Medical School Center for Biologic Imaging is supported by grant 1S10OD019973-01 from NIH.
SUPPLEMNTARY INFORMATION
1. SUPPLEMENTARY FIGURES
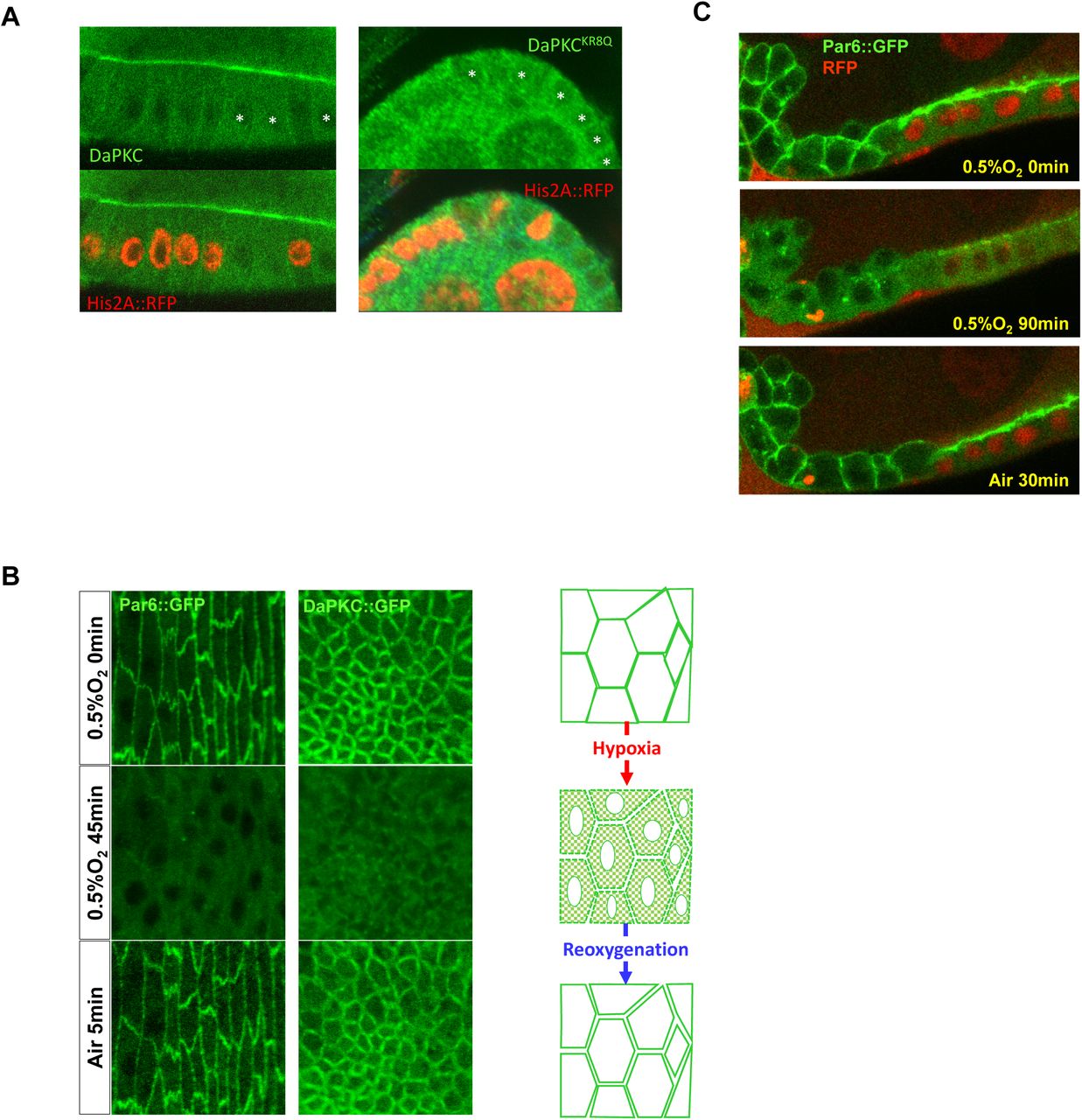
(A) PM localization of wild type DaPKC::GFP and non-polybasic DaPKCKR8Q::GFP in Drosophila wild type and DaPKC-/- mutant follicular epithelial cells. Asterisks indicate aPKC-/- mutant cells identified by the loss of Histon2A::RFP (His2A::RFP). Images are in cross section view.
(B) In Drosophila embryonic epithelial cells, PM localization of Par-6::GFP or aPKC::GFP was lost under hypoxia (0.5% O2) but recovered after post-hypoxia reoxygenation. Images are in tangential view of the apical surface of embryonic epithelia.
(C) Par-6::GFP showed acute and reversible loss of PM targeting under hypoxia in both wild type and lgl-/- mutant (marked by the loss of nuclear RFP) follicular epithelial cells. Note that in lgl-/- mutant cells, Par-6 was no longer restricted to apical PM but localized to both apical and lateral PM. Images are in cross section view.

(A-B) Representative images showing that in MCF7 and COS7 cells, PKCζ::GFP and Par-6::RFP were cytosolic when each expressed alone, but both became PM localized when co-expressed.

Cells overexpressing mRFP-FKBP-INPP5E and PM-bound Lck-FRB::CFP (not imaged) (asterisked in GFP images) are labeled by nuclear RFP. Rapamycin treatment induced strong PM localization of mRFP-FKBP-INPP5E, but Par-6::GFP remained largely on apical PM (A, cross-section view) while showed expansion to lateral PM (B, tangential view at lateral PM position).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Wild type lgl::GFP embryos and embryos of lgl::GFP UAS-Crb/Mat-Gal4 or lglS5A::GFP UAS-Crb/Mat-Gal4 were immunostained for GFP (green), Crb (red) and aPKC (magenta). Embryonic epithelial cells were in cross-section view. Note the loss of Lgl::GFP, but not LglS5A::GFP from the PM, under Crb over-expression (driven by Mat-Gal4, see Materials and Methods).
(B) Wild type lgl::GFP embryos and lgl::GFP; crb-/- mutant embryos were immnuostained for GFP (green), Crb (red) and aPKC (magenta). Crb signal was enhanced in crb-/- embryos to confirm no detectable expression of Crb. In crb-/- embryonic epithelial cells both Lgl and DaPKC became localized all around PM.
(C) Lgl::GFP showed moderate reduction from PM in Drosophila follicular cells overexpressing DaPKC-CAAX (marked by the expression of nuclear RFP). Asterisks indicate wild type cells that did not express DaPKC-CAAX.
(D) Par-6::iRFP localized to PM in HEK293 cells expressing BFP::Crb-intra, but not in cells expressing BFP::Crb-intraΔERLI. Non-polybasic PKCζKR8Q::RFP became PM-localized in cell expressing Par-6::iRFP and BFP::Crb-intra, but not in cells expressing Par-6::iRFP and BFP::Crb-intraΔERLI.
2. SUPPLEMENTARY MOVIES
Movie S1. Acute and reversible loss of PM aPKC::GFP under hypoxia.
Ovaries from a 3-day old ubi-DaPKC::GFP female were dissected and imaged live in an environment-controlled micro chamber. Hypoxic (0.5% O2) gas was flashed into the chamber starting 0 minute. Normal air was flashed into chamber starting 42min for reoxygenation. Time intervals are 3 minutes during hypoxia and 10 seconds during reoxygenation.
Movie S2. Acute and reversible loss of PM Par-6::GFP under hypoxia.
Ovaries from a 3-day old genomically rescued par-6::GFP par-6Δ226 female were dissected and imaged live similarly as samples in Movie S1. Reoxygenation starts from 33min. Time intervals are 3 minutes during hypoxia and 10 seconds during reoxygenation.
Movie S3. PM localization of Lyn11-Lgl::GFP, PKCζ::RFP and Par-6::iRFP in PAO-treated COS7 cells.
COS7 cells triple transfected with Lyn11-Lgl::GFP, PKCζ::RFP and Par-6::iRFP were cultured for 24 hours. Cells were treated with 20µM PAO and images were captured every 30 seconds using a laser-scanning confocal microscope (Nikon A1; Nikon Corp.).
Movie S4. PM localization of Lgl::GFP, Lyn11-PKCζ::RFP and Par-6::iRFP in PAO-treated COS7 cells.
COS7 cells triple transfected with Lgl::GFP, Lyn11-PKCζ::RFP and Par-6::iRFP were cultured for 24 hours. Cells were treated with 20µM PAO and images were captured every 30 seconds using a laser-scanning confocal microscope (Nikon A1; Nikon Corp.).
MATERIALS AND METHODS
Fly stocks and genetics
Drosophila stocks: Knock-out alleles of lglGX (“lglKO”) and crbGX (“crbKO”) and knock-in alleles of lgl::GFP and lglS5A::GFP were previously described (Dong et al., 2015; Huang et al., 2009).
w; UAS-Crb; (gift from Dr. Eli Knust, Dresdon, Germany),
w par-6Δ226 FRT9-2/FM6 (a gift from Dr. Jurgern Knoblich, IMP, Vienna, Austria),
y w; DaPKCk06403/CyO (BL#10622),
w; His2Av::mRFP (BL#23651),
y w ubi-GFPNLS FRT9-2 (BL#5154),
w; Act5C(FRT.CD2)-Gal4, UAS-RFP/TM3, Sb (BL#30558),
w; UAS-PLCδ-PH::GFP (BL#39693),
w; UAS-GRP1-PH::GFP(tGPH) (BL#8163),
w; FRTG13 DaPKCk06403/CyO; ubi-DaPKC::GFP/TM6;
w; FRTG13 DaPKCk06403/CyO; ubi-DaPKCKR8Q::GFP/TM6; w par-6::GFP hs-FLP par-6Δ226 FRT9-2/FM7C;
w; lgl::GFP hs-FLP38; Act5C(FRT.CD2)-Gal4, UAS-RFP/TM3, Sb;
w; lgl::GFP UAS-Crb/CyO; w, lglS5A::GFP UAS-Crb/CyO;
w; lglS5A::GFP/CyO; hs-FLP Act5C(FRT.CD2)-Gal4, UAS-RFP/TM3; w hs-FLP; FRTG13 His2Av::mRFP;
w par-6::GFP hs-FLP par-6Δ226 FRT9-2/FM7C; lglGX FRT40A/CyO;
w par-6::GFP hs-FLP par-6Δ226 FRT9-2/FM7C; ubi-RFPNLS FRT40A/CyO;
w; lgl::GFP UAS-aPKC-CAAX (UAS-aPKC-CAAX is a gift from Dr. Sonsoles Campuzano, Univerisidad Autonoma de Madrid, Spain);
w UASp>mRFP::FKBP-5Ptase (“FKBP-INPP5E”) and w;; UASp>Lck-FRB::CFP are gifts from Dr. De Renzis, EMBL Heidelberg, Germany (Reversi et al., 2014).
w; αTub67C-Gal4V2H; αTub67C-Gal4V37 (“Mat-Gal4”, a gift from Dr. Mark Peifer, University of North Carolina at Chapel Hill, USA) (Schaefer et al., 2018).
Transgenic flies of ubi-DaPKC::GFP, ubi-aPKCKR8Q::GFP and par-6::GFP were generated by phiC31-mediated integration protocol (Huang et al., 2009). attPVK00033 (BL#24871) stock was used to integrate ubi-DaPKC::GFP and ubi-DaPKCKR8Q::GFP constructs into the 3rd chromosome, while attPZH2A (BL#24480) stock was used to integrate par-6::GFP constructs into the X chromosome. par-6::GFP was further recombined with par-6Δ226 null allele to generate w par-6::GFP par-6Δ226 of which homozygote females and hemizygote males are fully viable and fertile, indicating a complete rescue of par-6Δ226 by par-6::GFP.
Drosophila cultures and genetic crosses are carried out at 25°C. Detail information about the strains from Bloomington stock center can be found in FlyBase.
Molecular cloning
To make ubi-aPKC::GFP, ubiquitin promoter (1872bp) was PCR from plasmid pWUM6 (a gift from Dr. Jeff Sekelsky, University of North Carolina at Chapel Hill) using primers 5-AGTGTC GAATTC CGCGCAGATC GCCGATGGGC and 5-CTGGAC GCGGCCGCGGTGGATTATTCTGCGGG and inserted into pGE-attB vector (Huang et al., 2009) to generate vector pGU. DNA fragments encoding aPKC::GFP and aPKCKR8Q::GFP were then inserted into pGU vector.
To make par-6::GFP, a 4.3kb par-6 genome DNA including 1Kbp upstream and 250bp downstream sequences was PCR-amplified from Drosophila genomic DNA using primers 5- ATGCGGCCGC GCTCTTCGGC TCTCGGATAG TCG and 5-GACGCGTGAT TAAGGCCCGGCTAATG, subcloned into pGE-attB vector. AvrII enzyme site was added before stop code for GFP insertion.
More details about DNA constructs used in this report are listed in Supplementary Table S1. NCBI RefSeq ID: Par-6 (human): NP_058644.1, Lgl (mouse): NP_001152877.1. Plasmids containing PKCζ and PKCι coding sequences were gifts from Dr. Jane Wang (U of Pittsburgh)
Live imaging and hypoxia treatment of aPKC::GFP and Par-6::GFP in Drosophila - epithelial cells
Embryos and dissected ovaries were imaged according to previously described protocol (Dong et al., 2015; Huang et al., 2011). Embryos were collected over night at 25°C. Ovaries from adult females of several days old were dissected in halocarbon oil (#95). Dechorinated embryos or dissected ovaries were mounted in halocarbon oil on an air-permeable membrane (YSI Membrane Model #5793, YSI Inc, Yellow Springs, OH) that is sealed by vacuum grease on a custom-made plastic slide over a 10mmx10mm cut-through window. After placing the coverslip on top, membrane at the bottom ensures the sufficient air exchange to samples during the imaging session. The slide was then mounted in an air-tight micro chamber (custom made) for live imaging under confocal microscope. Oxygen levels inside the chamber were controlled by flow of either air or custom O2/N2 gas at the rate of approximately 1-5 cc/sec. Images were captured at room temperature (25°C) on a Leica TCS-NT confocal microscope (PL APO 40x oil objective, NA=1.25) by Leica TCS-NT software, or an Olympus FV1000 confocal microscope (40x Uplan FL N oil objective, NA=1.3) by Olympus FV10-ASW software, or a Nikon A1 confocal microscope (Plan Fluo 40x oil objective, NA=1.3) by NIS-Elements AR software. Images were further processed in ImageJ and Adobe Photoshop.
Liposome pull-down assays
Bacteria BL21 was used to express and purify GST and GST fusion proteins as previously described (Dong et al., 2015). Liposomal binding assays were carried out as described (Kim et al., 2008). To prepare liposomes, lipid mixture of 37.5% PC (Cat#840051C), 10% PS (Cat#840032C), 37.5% PE (Cat#840021C), 10% Cholesterol (Cat#700000P) and 5% PI(4,5)P2 (Cat#840046X) or PI4P (Cat#840045X, all lipids were purchased from Avanti Polar Lipids Inc) was dried and resuspended to a final concentration of 1 mg/ml of total phospholipids in HEPES buffer. After 30 min sonication, formed liposomes were harvested at 16,000 × g for 10 min and resuspended in binding buffer (HEPES, 20 mM, 7.4, KCl 120 mM, NaCl 20mM, EGTA 1mM, MgCl 1mM BSA 1mg/ml). In each liposome-binding assay, approximately 0.1 μg of purified protein was mixed with 50μl of liposome suspension. After 15 min incubation at room temperature, liposomes were pelleted at 16,000 × g for 10 min and were analyzed by western blot to detect co-sediment of target proteins.
Cell culture and imaging
HEK293 cells were cultured in glass bottom dishes (In Vitro Scientific) and were transfected with DNA using X-treme Gene 9 DNA transfection reagent (Sigma Cat# 6365787001). After 24 to 40 hours of transfection cells were mounted and imaged on an Olympus FV1000 confocal microscope (40x Uplan FL N oil objective, NA=1.3) by Olympus FV10-ASW software, or a Nikon A1 confocal microscope (Plan Fluo 40x oil objective, NA=1.3) by NIS-Elements AR software. For images to be used for quantification, parameters were carefully adjusted to ensure no or minimum over-exposure. In addition, when appropriate a fluorescent PM dye (CellMask DeepRed Plasma Membrane Stain, ThermoFisher, Cat#C10046) was added the cell culture prior to live imaging to help tracing the PM for later quantifications.
Quantification of PM localization
PM localization were measured in Image J. In a given image, only cells expressing all transfected fluorescent proteins were measured. PM marker or proteins showing clear PM localization were selected for tracing PM. For each cell, a portion of PM and a spot in cytosol were manually traced and measured using line and area measurement tools in Image J. In each image, three separated spots in blank areas were also manually traced and measured for background levels. Fluorescence signals in each channel were quantified. The PM localization for each fluorescent protein were then calculated as the ratio of [PM - background]/[cytosol - background]. A custom ImageJ macro was used for semi-automatic batch measuring multiple cells in an image. Data were processed in Excel, visualized and analyzed in Graphpad Prism 7.
Biochemistry
For Lgl-phosphorylation assay, HEK293 cells were cultured in DMEM supplemented with 10% FBS. 24 hours after transient transfection, cells were directly lysed in SDS loading buffer and equal volumes of cell lysates were resolved in 12% SDS-PAGE. Proteins were detected by western blot using antibody chicken anti-GFP, 1:5,000 (Aves Lab, Cat# GFP-1020), rabbit anti-phospho-mLgl, 1:1000(Abgent, Cat# AP2198a), rabbit anti-PKC, 1:5000 (Santa Cruz, Cat# Sc-216) or mouse anti-Flag 1:5,000 (Sigma, Cat# F3165). For immunoprecipitation experiments, cells were lysed in RIPA buffer (25mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Triton X-100). Protein G-Sepharose beads were incubated with home-made and affinity-purified rabbit anti-GFP antibody (Huang et al., 2009) for 1 hour followed by incubation with the equal amount of each lysate for 1 hour. The beads were washed and boiled with SDS loading buffer. Supernatants were detected by western blot.
Drug treatments in mammalian cells
For PAO treatment, COS7 cells were seeded in glass bottom dish and transfected with target DNA plasmids. 24 hours after transfection, cells were imaged live under a Nikon A1 confocal microscope (Plan Fluo 40x oil objective, NA=1.3) by NIS-Elements AR software at room temperature. Around five to ten cells with satisfying expression were registered and multi-position time-lapse imaging started immediately after bath addition of PAO (10mM stock concentration in DMSO) to the final concentration of 20μM.
Induction of FKBP12-phosphatase and Lyn11-FRB::CFP dimerization in HEK293 cells
The procedure has been described in detail previously (Hammond et al., 2014). In brief, HEK293 cells cultured in 35 mm glass bottom dishes (In Vitro Scientific) were transiently transfected with 1 µg total DNA which included the Lyn11-FRB::CFP recruiter, FKBP12- phosphatases (Hammond et al., 2012), PKCζ::GFP and Par-6::iRFP as indicated. After 22-26 hours, cells were imaged in Fluoro-Brite medium (Life Technologies) using a Nikon A1R confocal laser scanning microscope though a 100x, NA/1.45 plan apochromatic objective lens. Time lapse imaging started 2 minutes prior to bath addition of 1 µM rapamycin. CFP::Lyn11-FRB image images were used to generate binary masks to define PM. Plasma membrane localization of each reporter was then calculated from the ratio of fluorescence within the PM to the whole cell, and is expressed relative to the average before rapamycin addition (Hammond et al., 2014).
Induction of mRFP::FKBP-5Ptase and Lck-FRB::CFP dimmerization in live Drosophila follicular cells
Young females of w UASp>mRFP::FKBP-5Ptase / w par-6::GFP par-6Δ226; hs-FLP Act5C(FRT.CD2)-Gal4 UAS-RFP/UASp>Lck-FRB::CFP or w UASp>mRFP::FKBP-5Ptase / +; lgl::GFP hs-FLP /+; Act5C(FRT.CD2)-Gal4 UAS-RFP / UASp>Lck-FRB::CFP were heat-shocked at 37°C for 1hr. Ovaries were dissected 4 days later in 1xPBS prior, mounted in a drop of 20μl Schneider’s medium containing 10μM rapamycin on a gas-permeable slide and imaged live, as previously described (Huang et al., 2011) (Dong et al., 2015). Treated lgl::GFP ovaries served as a positive control (Dong et al., 2015).
Generation of mitotic mutant clones in Drosophila follicular epithelia
Mutant follicular cell clones of lglKO or aPKCk06403 were generated by the routine FLP/FRT technique. Young females were heat-shocked at 37°C for 1 hour and their ovaries were dissected 3 days later.
Immunostaining and confocal imaging
Immunostaining of embryos and adult ovaries were carried out as described (Huang et al., 2009). Primary antibodies: rabbit anti-GFP (Huang et al., 2009) 1:1500; chicken anti-GFP (Aves Lab) 1:1000; rabbit anti-Lgl (d-300, Santa Cruz) 1:200; rabbit anti-aPKC (Santa Cruz) 1:1000. Secondary antibodies: Cy2-, Cy3 or Cy5-conjugated goat anti-rabbit IgG, anti-chicken IgG, goat anti-rat IgG, goat anti-mouse IgG and goat anti-guinea pig IgG (The Jackson ImmunoResearch Lab), all at 1:400. Images were collected on an Olympus FV1000 confocal microscope and processed in Adobe Photoshop and ImageJ.
Genotypes of Drosophila Samples in Figures
(C) w; FRTG13 DaPKCk06403/CyO; ubi-DaPKC::GFP/TM6; w; FRTG13 DaPKCk06403/CyO; ubi-DaPKCKR8Q::GFP/TM6.
(D) w; FRTG13 DaPKCk06403/CyO; ubi-DaPKC::GFP/TM6;
w par-6::GFP hs-FLP par-6Δ226 FRT9-2.
(D) w; lgl::GFP hs-FLP/ lgl::GFP UAS-crb; Act5C(FRT.CD2)-Gal4 UAS-RFP/+;
w; lglS5A::GFP UAS-crb/+; hs-FLP Act5C(FRT.CD2)-Gal4 UAS-RFP/+.
(A) w/w hs-FLP; FRTG13 DaPKCk06403/ FRTG13 His2Av::mRFP; ubi-DaPKC::GFP/+;
w/w hs-FLP; FRTG13 DaPKCk06403/ FRTG13 His2Av::mRFP; ubi-DaPKCKR8Q::GFP/+.
(B) w par-6::GFP hs-FLP par-6Δ226 FRT9-2/FM7C;
w; FRTG13 DaPKCk06403/CyO; ubi-aPKC::GFP/TM6.
(C) w par-6::GFP hs-FLP par-6Δ226 FRT9-2; lglKO FRT40A/ubi-RFPNLS FRT40A.
(A, B) w UAS-mRFP-FKBP-5’Ptas / par-6::GFP hs-FLP par-6Δ226 FRT9-2; +/+; Act5C(FRT.CD2)- UAS-RFPNLS/ UAS-lck-FRB::CFP.
(A) w; lgl::GFP UAS-crb /+; +/+;
w; lgl::GFP UAS-crb / αTub67C-Gal4V2H; αTub67C-Gal4V37 +;
w; lglS5A::GFP UAS-crb/ αTub67C-Gal4V2H; αTub67C-Gal4V37 /+.
(B) w; lgl::GFP; crbKO/ crbKO.
(C) w; lgl::GFP hs-FLP38/ lgl::GFP UAS-DaPKCCAAX; Act5C(FRT.CD2)-GAL4, UAS-RFP/+.
Online Supplementary Material
Figure S1 shows that the polybasic PSr was required for PM targeting of Drosophila aPKC and both Par-6::GFP and DaPKC::GFP showed hypoxia-sensitive PM localization in embryonic epithelia. In addition, hypoxia-sensitive PM localization of Par-6::GFP was not affected by the loss of Lgl.
Figure S2 shows that in MCF7 and COS7 cells PM targeting of PKCζ required co-expression of Par-6.
Figure S3 shows that acute depletion of PIP2 in Drosophila follicular cells does not strongly inhibits the PM localization of Par-6::GFP.
Figure S4 shows that in embryonic epithelial cells overexpression of Crb but not loss of Crb inhibited Lgl::GFP PM localization. In addition, Crb-intra was capable of recruiting Par-6 to PM in HEK293 cells.
ACKNOWLEDGEMENTS
We are grateful to Drs. Jane Wang, Gerald Apodoca, Mark Peifer, Eli Knust and Thomas Weide for reagents and fly stocks, Lu Jiang for technical assistances, Dr. Simon Watkins and University of Pittsburgh Medical School Center for Biologic Imaging for generous imaging and microscopy support, Bloomington Stock Center for fly stocks, and Developmental Studies Hybridoma Bank (DSHB) for antibodies.
REFERENCES




