

Abstract
Herpesviruses attach to host cells by interacting with cell surface heparan sulfate (HS) proteoglycans prior to specific coreceptor engagement which culminates in virus-host membrane fusion and virus entry. Interfering with HS-herpesvirus interactions results in significant reduction in virus infectivity indicating that HS play important roles in initiating virus entry. In this study, we provide convincing evidence that specific sulfations as well as the degree of polymerization (dp) of HS govern human cytomegalovirus (CMV) infection and binding by following line of evidences. First, purified CMV extracellular virions preferentially bound to the sulfated longer chain of HS on a glycoarray compared to unsulfated glycosaminoglycans and shorter chain unsulfated HS. Second, the fraction of glycosaminoglycans (GAG) displaying higher dp and sulfation had a major impact on CMV infectivity and titers. Finally, cell lines knocked out for specific sulfotransferases Glucosaminyl 3-O-sulfotransferase (3-O-ST-1 and −4 and double −1/4) produced significantly reduced CMV titers compared to wild-type cells. Similarly, a peptide generated against sulfated-HS significantly reduced virus titers compared to the control peptide. Taken together, the above results highlight the significance of the chain length and sulfation patterns of HS in CMV binding and infectivity.
Importance The cell surface heparan sulfates (HS) are exploited by multiple viruses as they provide docking sites during cell entry and therefore are a promising target for the development of novel antivirals. In addition, the molecular diversity in HS chains generates unique binding sites for specific ligands and hence offers preferential binding for one virus over other. In the current study several HS mimics were analyzed for their ability to inhibit cytomegalovirus (CMV) infection. The results were corroborated by parallel studies in mutant mouse cells and virus binding to glycoarrays. Combined together, the data suggests that virus particles preferentially attach to specifically modified HS and thus the process is amenable to targeting by specifically designed HS mimics.
Introduction
The heparan sulfate (HS) proteoglycans are present on most cell types and function as primary cellular receptor for medically important viruses, including human immunodeficiency virus (HIV), hepatitis-C virus (HCV), human papillomavirus (HPV), and Dengue virus (DENV) 1-4. In addition, virtually all human herpesviruses, with the possible exception of Epstein Barr virus, use HS as an initial co-receptor for entry 5. The interaction between cell surface HS and virus envelope is the primary event in the complex process of virus entry. However, this binding is not sufficient for viral entry and requires fusion between the viral envelope and cell membrane 6.
Herpesviruses including other enveloped viruses enter the host cells using two distinct pathways: 1) A pH-intendent pathway which involves the fusion of the virus envelope with the plasma membrane; and 2) A pH-dependent pathway that involves endocytosis of the virus particle7. In cells, where binding of virus to cell surface receptors induces endocytosis, the usual consequence is the acidification of the endosome, which ultimately triggers fusion between the virus envelope and endosomal membrane 5. Interestingly, HCMV entry follows direct fusion at the cell surface in fibroblasts, while entry into other relevant cell types, such as endothelial cells, follows an endocytic route 8,9. In either case, HS functions as the primary attachment receptor. Since the presence of HS receptors are well documented in endosomal membranes it is likely that HS receptors also play a role in intracellular virus trafficking 10-13.
The herpesvirus envelope is a lipid bilayer derived from host cell membranes in which most cellular proteins have been displaced by viral membrane proteins. For human cytomegalovirus (HCMV), at least twenty three different viral glycoproteins have been found to be associated with purified virion preparations 14. For most herpesviruses, the conserved glycoprotein B (gB) is required for virus entry and binds to cell surface molecules, including HS, which is present not only as a constituent of cell surface proteoglycans but also as a component of the extracellular matrix and basement membranes in organized tissues 5,15. HCMV gB binds to HS resulting in virus attachment 16 similar to its counterparts in herpes simplex virus (HSV)-1 15,17 and varicella-zoster virus (VZV) 18. Corroborating this fact, treatment of cells with soluble form of gB inhibits HCMV entry 19. HCMV binding and infection are reduced by soluble heparin and HS, as well as in cells treated with heparinases or those unable to produce HS 20.
The synthesis of HS is a complex process involving multiple specialized enzymes and is initiated from a tetrasaccharide (GlcA-Gal-Gal-Xyl) that is attached to the core protein (Fig 1). HS polymerase is responsible for building the polysaccharide backbone with a repeating unit of -GlcA-GlcNAc- (Fig 2). The backbone is then modified by N-deacetylase/N-sulfotransferase (NDST) responsible for N-deacetylation and N-sulfation of selected glucosamine residues, C5-epimerase responsible for epimerization of selected glucuronic moieties to iduronic acid, 2-O-sulfotransferase (Hs2st; 2-O-ST) responsible for 2-O-sulfation of selected iduronic acid residues, 6-O-sulfotransferase (H6st; 6-O-ST) for 6-O-sulfation and finally (but rarely) 3-O-sulfotransferases (Hs3st; 3-O-ST) responsible for 3-O-sulfation 21,22. The substrate specificities of these biosynthetic enzymes dictate the structures of HS product, including sulfation levels, the contents of IdoA units and the size of the polysaccharides 21. The location of the sulfo groups and IdoA in turn play a crucial role in determining the binding and functions of HS.

HS is a linear polysaccharide composed of repeating uronic acid [D-glucuronic acid (GlcA) or L-iduronic acid (IdoA)] and D-glucosamine (GlcN) disaccharide subunits. Synthesized chain of HS represents assembly of the tetrasaccharide linker region (GlcA-Gal-Gal- Xyl) at reducing end on serine residues of the protein core followed by the addition of alternating GlcA and GlcNAc residues. The chain extension is also accompanied by a series of modifications, which include 6-O, 3-O sulfations on GlcN and the 2-O sulfation on GlcA. The arrow shows the 3-O position of the GlcN where sulfation is important for herpesvirus binding13,23.

Heparan sulfate chains are initially synthesized as repeating disaccharide units of N-acetylated glucosamine and glucuronic acid. HS can then be modified by a series of enzymatic reactions, including N-deacetylation and N-sulfation of N-acetylated glucosamine converting it to N-sulfo-glucosamine, C5 epimerization of glucuronic acid to iduronic acid, and O-sulfation at the 2-OH, 6-OH, and 3-OH positions. Among sulfations, first is 2-O-sulfation of iduronic acid and glucuronic acid, followed by 6-O-sulfation of N-acetylated glucosamine and N-sulfo-glucosamine units, and finally 3-O-sulfation of glucosamine residues 26,47.
The enzymatic modification of HS chain is known to generate unique binding sites for viral ligands. For example, 3-O-sulfation modification in HS chain generates fusion receptor for HSV glycoprotein D (gD) promoting viral entry and spread 23. The 3-O-S HS is a product of enzymatic modification at C3 position of glucosamine residue, which is relatively rare in comparison to other HS modifications (Fig 2). Expression of Hs3st can make normally resistant Chinese hamster ovary (CHO-K1) cells susceptible to HSV-1 infection 24. Studies in clinically relevant primary human corneal fibroblasts have also shown 3-O-S HS as a primary receptor for HSV entry 25. Interestingly, both HSV-1 and HSV-2 use HS as an attachment receptor but HSV-1 binds to distinct modification sites on HS that HSV-2 is unable to, which could explain some of the differences in cell tropism exhibited by these two viruses 26. For example, while N-sulfation and carboxyl groups are required for both HSV-1 and HSV-2 binding, only HSV-1 is able to bind the specific modification sites generated by 2-O, 6-O, and 3-O-sulfations 27. The O-desulfated heparins have little or no inhibitory effect on HSV-1 infection but inhibit HSV-2 infection. This susceptibility to O-desulfated heparins can be transferred to HSV-1 by recombinant transfer of the gene for glycoprotein C (gC-2) from HSV-2 27. It was recently established that 3-O-S HS are important for HCMV entry in human iris stromal (HIS) cells 28. The expression of Hs3st in HIS cells promoted HCMV internalization, while pretreatment of HIS cells with heparinase enzyme or treatment with anti-3-O-S HS (G2) peptide significantly reduced HCMV plaques/foci formation. In addition, co-culture of the HCMV-infected HIS cells with CHO-K1 cells expressing 3-O-S HS significantly enhanced cell fusion. A similar trend of enhanced fusion was observed with cells expressing HCMV glycoproteins (gB, gO, and gH-gL) co-cultured with 3-O-S HS cells. These results highlight the role of 3-O-S HS during HCMV entry.
Owing to their inherent structural features, certain sulfated glycans can exert therapeutic effects against infections caused by pathogenic microorganisms. A study by Pomin et al., laid the proof-of-concept by administering sulfated glycans to disrupt the pathogen protein-host glycosaminoglycan (GAG) complex formation causing impairment of microbial binding onto host cells 29. Similarly, sulfated GAG, glycosphingolipids and lectins have been shown to inhibit DENV entry.30 Heparan sulfate mimics, such as suramin, pentosan polysulfate, and PI-88, SPGG 31,32 have been reported to be effective against multiple viruses including herpesviruses 2,33,34. The inhibitory activity of HS mimics, including these compounds, is believed to be due to their association with GAG binding sites of the putative receptor-binding domain on the viral protein 2,35. Thus, HS mimics can inhibit virus adsorption and entry.
In the current study, we investigated the impact of specific sulfations as well as degree of polymerization (dp) in HS chain on both human and mouse CMV infection and binding. Purified CMV extracellular virions preferentially bound strongly to the longer chain sulfated HS but not to the shorter chain unsulfated HS on a glycoarray. Glycosaminoglycans of different dp were derivatized from enoxaparin (a low molecular weight heparin) and tested for their ability to inhibit CMV infection in cell culture. The results show that longer glycan chains are more efficient at reducing CMV titers in cells compared to shorter chain glycans. Finally, the cell lines defective in 3-O-ST −1 and −4 expression had reduced CMV replication. Moreover, a peptide generated against the sulfated-HS significantly reduced HCMV titers compared to control peptide. Overall, these results indicate that CMV binding to cell surface glycans is dependent on branch length and sulfation pattern of HS.
Materials and Methods
Preparation of Glycosaminoglycans (GAGs) oligosaccharides
Glycosaminoglycans of different dp were fractionated from enoxaparin (a low molecular weight heparin) by Bio-Gel P-10 chromatography as previously described 36. Briefly, 30 mg/2 mL Enoxaparin Sodium derived from porcine intestinal mucosa (Sanofi-Aventis U.S., Bridgewater, NJ) was applied to a Bio-Gel P-10 column (2.5×120 cm, Bio-Rad, Hercules CA) and eluted with 0.2 M NH4HCO3 at a flow rate of 14 mL/h. Elution of oligosaccharides was monitored by absorbance at 232 nm. NH4HCO3 was removed by heating in oven at 50°C for 24 h.
Preparation of the 6-O desulfated Arixtra with MTSTFA
A detailed procedure on the preparation of 6-O desulfated Arixtra was published previously 37. Briefly, 4 mg of Arixtra was added to 10 volumes (w/w) of N-Methy-N-(trimethylsilyl)-trifluoroacetamide (MTSTFA, Sigma, ≥98.5%) and 100 volumes (v/w) of pyridine. The mixture was heated at 100 ℃ for 30 min, then quickly cooled in an ice-bath, followed by extensive dialysis and freeze-drying. The sample was resuspended in 50% acetonitrile/water at a concentration of 30 µM for later LC-MS/MS analysis.
LC-MS/MS Analysis
The 6-O-desulfated Arixtra (30 µM) was analyzed on a Thermo Orbitrap Fusion Tribrid (Thermo Fisher Scientific) coupled with an Ultimate 3000 Nano LC system (Dionex) using direct infusion. The flow rate was set to 1 µL/min. Mobile-phase was 50% acetonitrile. Nanoelectrospray voltage was set to 2.0 kV in negative ion mode. Full MS scan range was set to 200-2000 m/z at a resolution of 60,000, RF lens was 6%, and the automatic gain control (AGC) target was set to 2.0 × 105. For the MS/MS scans, the resolution was set to 50,000, the precursor isolation width was 3 m/z units, and ions were fragmented by collision-induced dissociation (CID) at a normalized collision energy of 80%.
Cells
Mouse embryonic fibroblasts (MEF) and human foreskin fibroblasts (HFF) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Cellgro, Manassas, VA) containing 4.5 g/ml glucose, 10% fetal bovine serum (SAFC, Lenexa, KS), 1 mM sodium pyruvate, 2 mM L-glutamine, and 100 U/ml penicillin-streptomycin (Cellgro, Manassas, VA) at 37°C with 5% CO2. Mutant mouse lung endothelial cells (WT, H3st1- knockout, H3st4-knockout, H3st1/4-double-knockout) were obtained from Wang laboratory at University of South Florida and maintained as described earlier 38. Cells were split upon confluency.
Virus
MCMV (strain K181) was grown in MEF cells, while HCMV (Towne strain) was grown on HFF cells. Virus stock was prepared in 3X autoclaved milk, sonicated 3 times and stored at −80°C. During infection, media was removed from the wells of cell culture plates and appropriately diluted virus stock was absorbed onto the cells in raw DMEM. Cells were incubated for 1 hour with gentle shaking every 10 mins followed by washing 3X with PBS. Fresh complete medium was added and cells were incubated until the end point. For extracellular virus (ECV) purification, HFF were seeded in roller bottles, grown to confluency and infected with HCMV (Towne strain) at MOI of 0.01. Two days after 100% cytopathic effect was observed, infected cell medium was collected and centrifuged at low speed to pellet cellular debris, and the supernatant was transferred to new tubes and centrifuged at 20,000 g for 1 hour to pellet the ECV. This ECV pellet was re-suspended in phosphate buffer, sonicated to eliminate any aggregates, loaded over 15-50% continuous sucrose gradients and centrifuged in a SW-41 rotor at 39,000 RPM for 20 min. ECV bands were visualized in incandescent light and harvested by puncturing the sides of the centrifuge tubes. These bands were washed once with phosphate buffer, spun again and the final pellet resuspended in low salt phosphate buffer. An aliquot of the sample was used for assessment of initial quality of ECV by negative staining and transmission electron microscopy. Purified ECV were shipped on ice to Z biotech (Aurora, CO) for glycoarray binding analysis.
Cell Viability Assay
Cells plated in 12 well tissue culture plates were grown to confluency and pretreated for 1h with 10 µM concentration of candidate HS and then infected with HCMV (Towne strain) at a multiplicity of infection (MOI) of 3.0 or mock- infected. Five hundred µl of fresh complete medium was added to the wells on day 3 and day 6. At the designated time points, media was removed and cells were harvested by trypsinization. Cell viability was determined using trypan blue exclusion on TC20 automated cell counter (BioRad Laboratories, Hercules, CA) following manufacturer’s protocol.
Virus Titers
Infected or mock-infected samples were harvested within the medium at the designated end points and stored at −80°C before titration. In some experiments, media and cells were separated by low-speed (< 1000 × g) centrifugation and viral loads in supernatant and cells were quantified by titering on wild-type cells. Titers were performed as described earlier 39 with some modifications. In brief, monolayers of fibroblasts grown in 12 well plates and serial dilutions of sonicated samples were absorbed onto them for 1 h, followed by 3X washing with PBS. Carboxymethylcellulose (CMC) (Catalog No. 217274, EMD Millipore Corp., Billerica, MA) overlay with complete DMEM media (1-part autoclaved CMC and 3 parts media) was added and cells were incubated for 5 days. At end point, overlay was removed and cells were washed 2X with PBS. Infected monolayers were fixed in 100% methanol for 7 min, washed once with PBS and stained with 1% crystal violet (Catalog No. C581-25, Fisher Chemicals, Fair Lawn, NJ) for 15 min. Plates were finally washed with tap water, air dried and plaques with clear zone were quantified.
Glycoarrays
A dilution series of purified HCMV virions were incubated on two different custom glycoarrays (Table 1 and Table 2, Z-Biotech) using established protocols 40 and the arrays were analyzed to assess specific virus binding. Briefly, 105 to 108 pfu/ml of purified virions were incubated for an hour on glycoarrays containing six replicates of each glycosaminoglycan. After incubation, staining with primary antibody (mouse anti gB (clone 2F12, Virusys Inc, Taneytown, MD) was done at 100 µg/ml and secondary antibody (Goat anti mouse IgG AlexaFlour555) was done at 1µg/ml. Maximum strength fluorescent signal was obtained for 108 pfu/ml concentration of the virus, therefore, only this concentration is represented in the final data obtained for plotting the graphs.
A custom designed glycoarray containing hyaluronic acid, heparin, chondroitin sulfate and dermatan sulfate species. The structure, molecular weight, number of sugar residues and sulfate groups per disaccharide for each glycosaminoglycan are listed.
A custom designed glycoarray containing different heparan sulfate species. The structure, molecular weight, number of sugar residues and sulfate groups per disaccharide for each glycosaminoglycan are listed.
Effect of anti-HS and anti-3OS HS peptide on CMV entry
HFF cells were pre-treated with the phage display derived peptides (1 mg/ml) generated against wild-type HS (LRSRTKIIRIRH), and 3-O-S HS (MPRRRRIRRRQK) 41 or left mock treated for 4 hours before the cells were infected with β-galactosidase expressing CMV (ATCC) for 9 days. β-Galactosidase assay were performed using X-gal (Sigma). The effect of entry- blocking activity of peptide was examined by counting number of virus foci. Results are representative of three independent experiments.
Results
Purified HCMV extracellular virions preferentially bind to sulfated glycosaminoglycans with increased degree of polymerization
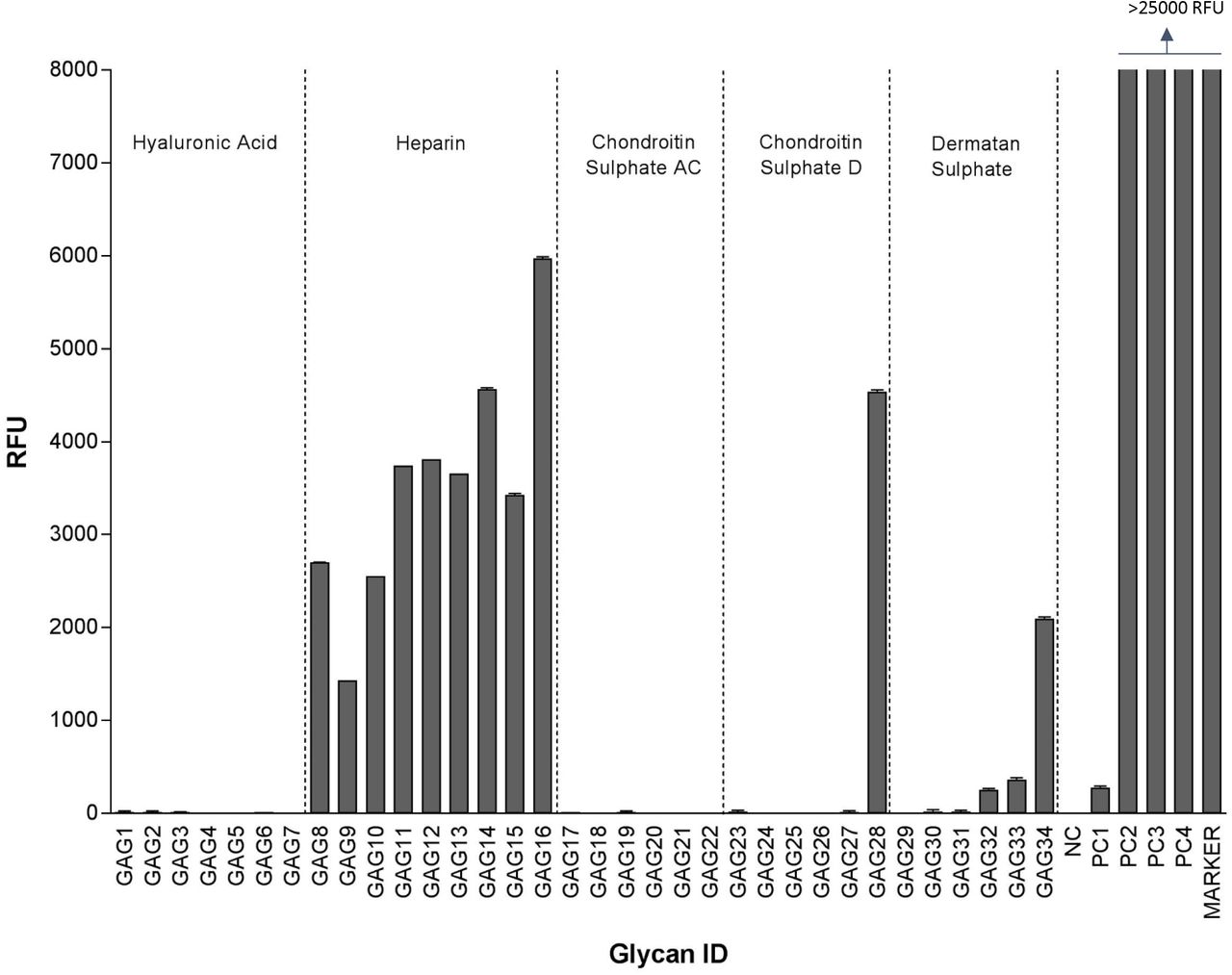
HCMV extracellular virions were purified as described above and incubated with custom glycoarrays containing increasing molecular weight species of hyaluronic acid, heparin, chondroitin sulfate, and dermatan sulfate (Table 1). As indicated in Fig. 3 HCMV binding to non-sulfated hyaluronic acid (HA10 to HA20 and HA93 polymer) was negligent but significant binding to all heparin species was detected with a trend of increased binding to heparins as their dp increased. HCMV also showed binding to large size chondroitin sulfate D (CS-D 20), and dermatan sulfate oligosaccharides (DS16-20) but not to Chondroitin Sulphate AC (CS-AC). It is important to note that while the CS-A is sulfated at C4 of the GalNAc, and the CS-C is sulfated at the C6 of the GalNAc only, the CS-D is sulfated at C2 of the glucuronic acid as well as the C6 of the GalNAc sugar and hence has double the amount of sulfation compared to CS-A and CS-C. Dermatan sulfate, formerly referred to as CS-B, is formed from the polymer backbone of chondroitin sulfate by the action of chondroitin-glucuronate C5 epimerase, which epimerizes individual d-glucuronic acid residues to l-iduronic acid. The binding affinity to DS was also size-dependent increasing from DS16 to DS20. Heparin (dp30) was the best binder in this assay.

Relative fluorescence units (RFU), which are directly proportional to the amount of virus binding, are plotted on the Y-axis in the graph. Ligand descriptions and chain structures are provided in Table 1. Six replicates for each GAG were used in the assay. NC: Negative control (print buffer), PC1: positive control (Biotinylated Glycan), PC2: human IgG (0.1 mg/ml), PC3: mouse IgG (0.1 mg/ml), PC4: rabbit IgG (0.1 mg/ml).
On a second HS specific array (Table 2), HCMV showed decent specific binding to sulfated HS with stronger binding to the HS with longer disaccharide chains (HS007 to HS024) (Fig 4). HCMV showed minimal binding to unsulfated glycans (HS001-HS006). The maximum binding was observed for HS014, HS015 and HS016, which are all 6-O-S 9-mers with moderate amount of sulfation (1.3-1.8 sulfate group per disaccharide). Also, significant amount of binding was observed for 2-O-S (HS17-HS19), 6-O-S/2-O-S (HS20-22) and 2-O-S/6-O-S/3-O-S (HS23-24) HS that had high amount of sulfation (1.3-2.7 sulfate group per disaccharide) and 6-8 disaccharide per chain. Overall the data from these experiments indicate that the dp of HS as well as sulfation is important for HCMV binding.

Relative fluorescence units (RFU), which are directly proportional to the amount of virus binding, are plotted on the Y-axis in the graph. Ligand descriptions and chain structures are provided in table 2. Twelve replicates for each ligand were used. NC: negative control (print buffer) PC1: positive control (biotinylated glycan), PC2: human IgG (0.1 mg/ml), PC3: mouse IgG (0.1 mg/ml), PC4: rabbit IgG (0.1 mg/ml).
The degree of polymerization of GAG chains impacts CMV infectivity
Glycosaminoglycans of different dp were fractionated from enoxaparin (a low molecular weight heparin). All of these GAGs are based on a HS backbone and differ in either dp or degree/place of sulfation or both (Fig 5, S1-S3). These GAGs, along with heparin and Arixtra (fondaparinux sodium), were first screened in a GFP-based virus focus reduction assay using GFP tagged HCMV (Towne strain). The viral GFP expression was most efficiently reduced by heparin salt (PIHSS; Heparin sodium salt from porcine intestinal mucosa) whereas arixtra, 6-O-desulfated arixtra and Enoxaparin had little to no impact on GFP expression (Fig 5). In general, enoxaparin derived GAGs with higher dp were more efficient in reducing viral GFP compared to low dp derivatives. This screening assay when performed at a range of GAG concentrations (10nM to 100µM) determined 10µM as the most effective concentration at reducing viral titers with no additional reduction seen at >10µM concentrations (data not shown). To follow up on this primary GFP based screening, we performed viral titer assay using HCMV (Towne strain) that measures total virus yields at 5 days post infection. Most reduction in viral titers was observed for heparin (PIHSS) followed by enoxaparin derivative with >20 dp (Fig 6A). Plotting of viral titer reduction as a function of dp revealed a general trend where higher dp derivatives lead to higher reduction in viral titers (Fig 6B). Thus, this experiment indicated that longer HS chains are more efficient at reducing HCMV titers in cells. To investigate whether this inhibitory effect was due to an increase in avidity of longer chain GAGs towards virus particles, the experiments were repeated at 0.05 g/L concentrations of GAGs (Fig 7A). Similar trend of inhibitory results leaning towards efficacy of higher dp against HCMV infection were obtained at 0.05 g/L indicating that this effect is dictated by the molecular composition of GAG and is not a mere effect of increased avidity. A line graph for each concentration of GAGs was generated that demonstrates the relationship of viral titer and degree of polymerization (Fig 7B). To investigate whether some of these effects on virus titers could be attributed to cell death, we performed cell viability assays in both uninfected and infected settings. Cell viability was not affected at the treated concentrations of any of our test GAGs (Fig 8A). Moreover, heparin (PIHSS) and enoxaparin derivatives (dp 12 or greater) efficiently protected cells from virus induced lytic death (Fig 8B). These results corroborate the results of our glycoarray experiments that showed that GAG with higher dp have higher CMV binding compared to GAG with lower dp (Fig 4).

Primary human foreskin fibroblasts (HFF) grown in 96 well plate were pretreated for one hour with 10 µM of 1) 6-O-desulfated Arixtra, 2) Regular Arixtra, 3) Heparin sodium salt from porcine intestinal mucosa (PIHSS), 4) Enoxaparin, or series of heparin oligosaccharide from enoxaparin: 5) dp2, 6) dp4, 7) dp6, 8) dp8, 9) dp10, 10) dp12, 11) dp14, 12) dp16, 13) dp18, 14) dp20 15) > dp20 or control (dH2O). Cells were infected with GFP tagged HCMV (Towne strain) virus at an MOI of 3.0. At 5 days post infection, cells were fixed and number of foci (GFP) was counted under an epifluorescent microscope. Percent of viral GFP was calculated compared to virus only infected control (100% GFP expression).

Primary human foreskin fibroblasts (HFF) were pretreated for one hour with 10 µM of 1) 6-O-desulfated Arixtra, 2) Regular Arixtra, 3) Heparin sodium salt from porcine intestinal mucosa (PIHSS), 4) Enoxaparin, or series of heparin oligosaccharide from enoxaparin: 5) dp2, 6) dp4, 7) dp6, 8) dp8, 9) dp10, 10) dp12, 11) dp14, 12) dp16, 13) dp18, 14) dp20 15) > dp20 or control (dH2O). Cells were infected with HCMV (Towne strain) virus at an MOI of 3.0. Cells and media were harvested at 5 days post infection and titered for HCMV plaque forming units (pfu) on fresh fibroblasts in tissue culture dishes. Individual samples (3 replicates each) were quantified and displayed as total pfu/ml on Y-axis. (B) Virus titer is plotted (Y-axis) against degree of polymerization (X-axis). Data points ahead of the broken line is for a mixture of GAGs (dp>20).

Primary human foreskin fibroblasts (HFF) were pretreated for one hour with 0.05 g/L (B) of 1) 6-O-desulfated Arixtra, 2) Regular Arixtra, 3) Heparin sodium salt from porcine intestinal mucosa (PIHSS), 4) Enoxaparin, or series of heparin oligosaccharide from enoxaparin: 5) dp2, 6) dp4, 7) dp6, 8) dp8, 9) dp10, 10) dp12, 11) dp14, 12) dp16, 13) dp18, 14) dp20 15) > dp20 or control (dH2O). Cells were infected with HCMV (Towne strain) virus at an MOI of 3.0. Cells and media were harvested at 5 days post infection and titered for HCMV plaque forming units (pfu) on fresh fibroblasts in tissue culture dishes. Individual samples (3 replicates each) were quantified and displayed as total pfu/ml on Y-axis. At the bottom of each panel, titer is plotted (Y-axis) against degree of polymerization (X-axis). Data points after the broken line is for a mixture of GAGs (dp>20).

Primary HFF were pretreated for one hour with 10 µM of 1) 6-O-desulfated Arixtra, 2) Regular Arixtra, 3) Heparin sodium salt from porcine intestinal mucosa (PIHSS), 4) Enoxaparin, or series of heparin oligosaccharide from enoxaparin: 5) dp2, 6) dp4, 7) dp6, 8) dp8, 9) dp10, 10) dp12, 11) dp14, 12) dp16, 13) dp18, 14) dp20, 15) > dp20 or control (dH2O). Cells were either mock infected (A) or infected with HCMV (Towne strain) virus at an MOI of 3.0 (B). Cells were harvested at 5 days post infection and cell viability was assessed using Trypan Blue exclusion assay.
Cell lines defective in expression of specific sulfation enzymes have reduced CMV titers
Due to species specificity of HCMV, animal models are frequently used to study CMV pathogenesis 42,43. Studies of murine CMV (MCMV) infections of mice have served a major role as a model of CMV biology and pathogenesis 44. Mutant mouse lung endothelial cell lines were from adult mice were mutated for specific sulfotransferase enzymes by a CRISPR-Cas9 based gene editing system 38,45,46. Since previous studies showed that 3-O-S HS is important for HCMV entry in human iris stromal cells 28, we analyzed virus replication in Hs3st1 and Hs3st4 (Glucosaminyl 3-O-sulfotransferase 1 and 4, respectively) knockout cell lines as well as the Hs3st1/4 double knockout cell line. At high (5.0) as well as low (0.01) multiplicity of infection (MOI), MCMV growth was significantly reduced in the single Hs3st1 and Hs3st4 knockouts as well as in the double Hs3st1/4 knockouts, indicating that 3-O-sulfation of HS is important for HCMV infection (Fig 9).

Cells were grown to 90% confluency and infected with wild-type MCMV (strain K181) at low (0.01, 6A, B) and high (5.0, 6C, D) MOI. Cells and the medium were harvested at 3- and 5-days post infection, sonicated to release the virus and diluted for plating on to wild-type MEF in tissue culture dishes in order to enumerate total MCMV pfu/ml. Hs3st1 and Hs3st4: Glucosaminyl 3-O-sulfotransferase 1 and 4, respectively. WT: wild-type; KO: knockout. P values of <0.05 were considered significant (*) compared to WT cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HFF cells were pre-treated with wild-type HS peptide, anti 3-OS HS peptide for 4 hrs. The mock treated cells were used as a positive control. The cells were infected with β-galactosidase expressing CMV for 9 days. β-Galactosidase assay were performed using X-gal (Sigma). The effect of entry-blocking activity of peptide was examined by counting number of foci. Results are representative of three independent experiments.
Peptide generated against 3-O-S HS blocks HCMV infection
In order to examine the effect of sulfated HS on HCMV infection, we utilized phage display derived anti-HS and anti-3-O-S HS peptide 41. The HFF cells were pre-treated either with anti-HS peptide or anti-3-O-S HS peptide. The mock treated cells were considered as a positive control. As indicated in Fig. 9, the anti-3-O-S HS peptide treatment resulted in a significant reduction of HCMV titers in HFF cells compared to an anti-HS peptide or the mock-treated cells.
Discussion
In this study, we utilized multiple tools such as glycoarray binding analysis, HS mimics, HS mutant cell lines, and anti-HS/3-OS HS peptides to provide convincing evidence that specifically sulfated HS with higher degree of polymerization determine CMV infection and binding. We first screened several GAGs that are sulfated or unsulfated and have complex sugar structure to investigate which GAGs are more efficient at binding to HCMV virions. This glycoarray analysis indicated that HCMV bound with heparins with strong affinity and showed increased binding for longer chain length (Fig. 3). To further investigate this binding, we utilized another glycoarray consisting of HS of varied polymerization and sulphation levels. The results from this glycoarray indicated that HCMV binds strongly with HS having both longer sugar residues and a moderate level of sulphation. Thus, sulfated HS with more complex branches and sulfation patterns preferentially bind to HCMV. Next, we fractioned HS by length (2-20) from enoxaparin and tested their ability to inhibit HCMV growth in cell culture by competing with HCMV binding. In the preliminary experiment, GFP tagged HCMV was used and the number of GFP+ foci was quantified. The data from this experiment indicated that viral GFP was more effectively reduced when cells were pretreated with GAGs having a higher dp (Fig 5). For a deeper understanding of this reduction, we performed a similar experiment where HCMV towne strain was used and viral load was quantified at 5 days post infection. Significant reduction in virus titers was observed in samples treated with higher dp of GAG but not with lower dp corroborating the results from glycoarray experiments that chain length of GAG is an important factor in determining HCMV binding. Also, this effect was not due to combined affinities of multiple binding sites on GAGs as evidenced by similar trend of inhibition obtained when treating cells with equivalent µM or µg/ml concentrations of GAGs. Treatment of cells with these GAGs did not affect their viability for the duration of treatment (Fig 8A) confirming that the observed reduction in virus titer was not due to the cell death. Moreover, cells pretreated with GAGs associated with longer dp resisted infection induced cell death at late time post infection (Fig 8B). We also tested the impact of specific HS sulfation mutants on MCMV infection. As 3-O sulfation has been reported to be critical for herpesvirus entry 25,28, we tested MCMV growth in Hs3st1, Hs3st4 and dual Hs3st1/4 knockout cells. For both high and low MOI, virus titer was significantly reduced in Hs3st1, Hs3st4 and dual Hs3st1/4 knockout cells (Fig 9). Additional data from anti-3-O- S HS peptide confirmed the significance of sulfation in HCMV infectivity.
Overall, the data from these studies indicate that dp of GAGs as well as specific sulfation patterns govern HCMV infection of cells. These studies show the promise that highly polymerized sulfated-HS targeted to develop effective anti-CMV agents. Future studies would be aimed at confirming the CMV glycoproteins that specifically bind to HS on cell surface and their possible structural illustrations. It would also be interesting to pursue specifically designed glycomimetics that inhibit these specific virus-host interactions for their effectiveness in a mouse model of CMV infection as well as in future clinical trials.
Author Contributions
RT, JSS, and LW designed the experiments; MHH, RBP, QL, JSS, VT and RT performed the experiments and analyzed the data. RT and MHH wrote and edited the manuscript.
Acknowledgments
The research was supported by American Heart Association (Award 14SDG20390009, PI: Tandon) and NIH (Award R21HL131553, PI: L.W.). QL, JSS and LW acknowledge funding from the National Institute of General Medical Sciences through the Research Resource for Integrated Glycotechnology (P41GM103390).
References




