

ABSTRACT
We describe the genome contents of six Protomyces species that are pathogenic within the typical host range of the genus and a novel Protomyces strain (SC29) that was previously isolated from the phylloplane of wild Arabidopsis thaliana (Arabidopsis), an atypical host. Genome-wide phylogenetic analysis defined SC29 as a distinct Protomyces species. Analysis of gene family expansions, gene retention, and gene loss patterns among the Protomyces suggests that SC29 has recently undergone a host jump and change in lifestyle. Genomic changes in SC29 were consistent with a phylloplane lifestyle. SC29 did not cause disease on Arabidopsis, but could persist in its phylloplane, while the closely related P. inouyei does not. SC29 treated Arabidopsis exhibited enhanced immunity against Botrytis cinerea infection, associated with activation of MAPK3/6, camalexin, and SA-signalling pathways. We conclude that SC29 is a novel Protomyces species adapted to life in the Arabidopsis phylloplane.
INTRODUCTION
The Protomyces are early diverging ascomycete yeasts and known phytopathogens, causing disease on host plants within the Compositae (Asteraceae) and Umbelliferae (Apiaceae) families (1, 2). The Protomyces and their sister clade Taphrina (order Taphrinales) (3) share similar dimorphic lifestyles, spreading as budding yeasts through the air to the plant phylloplane (as haploid ascospores or basidospores) and invading their hosts in their dikaryotic filamentous form (3). They also share a common virulence strategy; both typically cause plant tumours or gall-symptoms and are well known to produce plant hormones (2, 3). Plants and their pathogens are engaged in an evolutionary arms race where the pathogen develops new effectors to overcome the plant defences, which the host in turn tries to intercept (4). These organisms are assumed to co-evolve with their hosts based on congruent host and microbe phylogenetic trees (5, 6). As early diverging ascomycetes the Protomyces and Taphrina are of considerable phylogenetic and evolutionary interest. However, these fungi cause diseases primarily on wild and woody species and remain understudied (6). Recent reference genome assemblies have opened these taxa to molecular evolutionary studies, increasing interest and proving the value of these, and more widely, other Taphrinomycotina species (7-10).
The Protomyces also have scientific value as plant associated yeasts and phylloplane residents. Plant associated yeasts have a spectrum of relationships with their hosts ranging from the pathogenic, as mentioned above, to growth-promoting symbionts. Several yeasts have been identified as hub taxa, which restructure the microbiome community composition when they are present in the Arabidopsis phyllosphere (11, 12). The phyllosphere is gaining attention as a microbial habitat, but primarily prokaryotic residents have been under study. Eukaryotic resident species are now being isolated, but their genomic adaptation to the phyllosphere remains underexplored. Our understanding of phytopathogenic yeasts lags behind that of other pathogen classes. This is the case also for Arabidopsis thaliana (Arabidopsis), a model plant widely used when studying the molecular basis of plant immunity (13). Currently, no well-characterized systems for the study of Arabidopsis-yeast interactions exist.
We previously isolated a novel strain of Protomyces that is associated with Arabidopsis (11, 14). Here we characterize its interactions with Arabidopsis and compare the genome to six reference species of Protomyces. We present evidence of a new species that is adapted the phylloplane environment of Arabidopsis and positively affects plant immunity.
MATERIALS AND METHODS
Protomyces strains and culture
Protomyces reference species (Table 1) were obtained from the ARS culture collection. SC29 was isolated from wild Arabidopsis (14). All species were purified twice from single colonies and cultured on glucose, yeast-extract, peptone (GYP) agar at 21°C, unless otherwise indicated.
Genome sequencing, assemblies and annotation statistics of seven Protomyces species in this study. * Gene set completeness was estimated using BUSCO. ‡ Reference strains were obtained from the ARS culture collection (https://nrrl.ncaur.usda.gov/).
SC29 pre-treatment, Botrytis infection, and qPCR
Live and autoclaved SC29 were harvested, washed, and suspended at 1 OD600 (7 × 106 cfu/ml) in sterile water. Three-week-old soil-grown Arabidopsis were sprayed with one of the suspensions (0.2 ml/plant), or water as a control, then infected with a Botrytis spore suspension (15) three days after SC29 treatments. Plants were grown in a chamber with 12/12h (light/dark), 23/18°C, and 65/75% relative humidity. Pre-treated and infected plants were collected at 30 and 60 min for western blot, and at 24 and 72 h for qPCR. Four of five plants were pooled, frozen in liquid nitrogen, and stored in −80°C. Leaf lesions were photographed at 72 h and diameters measured in ImageJ. Lesion size data were statistically analysed with scripts in R (version 3.5.1) using the nlme package, a linear mixed model with fixed effect for treatment was fitted to the data with a random effect for biological repeat. The model contrasts were estimated with multcomp package, and the estimated p-values were subjected to single-step correction. RNA isolation and qPCR were performed as previously described (15). Primer sequences, amplification efficiencies, and reference genes are listed in File S1. Raw cycle threshold values were analysed with Qbase+ 2.1 (ref. 16). Significance was estimated by glht package in R (3.5.1) using false discovery rate adjustment of p-values.
Protomyces growth assay
Three-day-old P. inouyei (OD600 = 0.14, 5 µl/leaf) and SC29 (OD600 = 0.10, 5 µl/leaf) were drop inoculated on the adaxial side of 24-day-old Arabidopsis leaves, aseptically grown on 0.5xMS agar plates. Sterile foil on an agar plate was used as a control for non-specific surface growth. Growth in the phylloplane was assayed by re-isolating Protomyces from inoculated leaves by placing leaves in 2 ml Eppendorf tubes with 1 ml 0.025% Silwet-L77 in water and shaking (800 rpm; VWR microplate shaker) for 1 hour, after which 5 µl wash solution was serial diluted and pated on GYP agar for colony counting. Pooled data from five biological repeats were analysed using a linear mixed model in R, as above.
Genomic DNA extraction
Genomic DNA was isolated from cultures of the haploid yeast form for each species using a chromosomal DNA extraction method (17). Bead beating was done using a vortex (Vortex genie 2, Scientific Industries, Inc., United States) with multiple 2 ml tube adapter (2 min, full speed). Beads (0.2-0.3 g/tube) with 0.75-1.00 mm dimeter were used (Retsch GmbH, Germany). The quantity of isolated DNA was estimated using Nanodrop ND-1000 (Thermo Scientific, USA) and qubit fluorometer (Thermo Fisher Scientific, USA). The DNA yield was in average 23.9 µg/strain with standard deviation 12.6 µg assayed with qubit fluorometer. ITS fragments were amplified and sequenced as previously described (14) to confirm the species identity prior to sequencing.
Genome assemblies and annotations
Genome sequencing was done at the DNA Sequencing and Genomics Laboratory, Institute of Biotechnology, University of Helsinki. MiSeq Sequencing System (Illumina, California USA) was used to sequence pair end reads with average read length 231 bp with standard deviation 4.8 bp. Paired end sequencing libraries were prepared using Paired-End Sample Preparation Guide (Illumina). The four partial Illumina sequencing runs produced 758,373 read pairs in average (Standard deviation 17,341 read pairs). These reads were assembled using SPAdes v. 3.1.1 (18).
The Protomyces genome assemblies were annotated using the Hidden Markov Model based gene predictor Augustus version 2.5.5 (19). The gene predictor was trained using RNASeq data obtained from Taphrina betulina (Bioproject: PRJNA188318) (20). Paired RNA raw reads (Run 1) were aligned against the Taphrina betulina genome to generate the BAM alignment file using TopHat version 2.0.11 which internally used Bowtie version 2.1.0.0 (21). The unpaired RNA raw reads (Run 2) (22) were also aligned against the genome using TopHat using junction information from the paired read alignment. The BAM alignment files coming from Run 1 and Run 2 were merged using Samtools version 0.1.19-44428cd (23). Transcripts were then generated from the merged BAM alignment file using Cufflinks version 2.2.1 (24). These transcripts were then splice-aligned against the Taphrina betulina genome using PASA version r20130605p1 (25) to generate complete ORFs. These ORFs combined with 950 manually annotated genes and CEGMA (Core Eukaryotic Genes Mapping Approach) genes (26) were used to train Augustus. The gene set completeness per genome was estimated using BUSCO (Benchmarking Universal Single-Copy Orthologs) version 3.0.2 (27) internally using the Ascomycota odb9 fungal dataset. The predicted gene models were assigned human-readable functional descriptions using the tool AHRD (Automated Assignment of Human Readable Descriptions) version 3.3.3 (https://github.com/groupschoof/AHRD). Conserved protein domains were defined using the Pfam database (Supplemental file S4) (28). The gene models were initially aligned against two custom BLAST databases containing fungal proteins from SWISSPROT and TREMBL respectively. The BLAST results along with GO annotation information of the fungal proteins in UniProt format were then passed to the AHRD tool which parses the results and assigns the best description and GO information to the gene models. Gene family evolutionary rate and ancestral size estimation of the orthogroups were carried out using badirate v.1.35 (29). A species-level rooted phylogenetic tree and orthogroup size information were provided as input for the badirate software. The BDI turnover rate model (Birth, Death and Innovation) was selected. The FR branch model (Free Rates) was used which assumes that every branch in the phylogenetic tree has its own turnover rate. Maximum Likelihood statistical method was used to estimate the turnover rates. The outlier branches reported per orthogroup, which did not evolve under the estimated turnover rates, if any, were identified as significant branches for further analysis.
Genome-wide phylogeny analysis
Orthologous protein families from 15 yeast species were inferred with OrthoFinder (default) (30) utilizing the Guidance2 algorithm (31) multiple sequence alignment. Totally, 1035 single-copy proteins with confidence scores >= 0.8 were selected and concatenated with the FASconCAT_v1.0 script (32). A Maximum likelihood phylogeny tree was built by RAxML v8 (33) with the rapid bootstrap algorithm (100 bootstraps). The tree was viewed and edited in iTOL (34). ANI and AAI values were estimated by genome-based distance matrix calculator (35).
Selection and analysis of CSSPs
Automated annotation did not capture candidate effector-like proteins (CELPs); thus a manual screen was used. We identified CELPs by defining small secreted proteins encoded in fungal genomes as follows. Open reading frames (ORFs) between 80 to 333 amino acids long were chosen and filtered for a secretion signal with SignalP 4.1 (36) to defines short secreted proteins (SSPs). After removing signal peptides, cysteines were counted; SSPs with ≥ 4 cysteines were regarded as cysteine-rich SSPs (CSSPs). OrthoVenn (37), orthofinder (30), and Badirate (29) were used to analyse gene family evolution in CELPs. Known virulence factors were searched from SSPs and CSSPs using PHI base (38). Pfam domains of genomic proteins and SSPs were searched with HMMER v3.2.1 (http://hmmer.org/) using hmmscan.
Genomic mining for multiple enzymes of important traits
Genome assemblies and annotations of all seven Protomyces species were applied as local database. Fungal HHK (hybrid histidine kinases) protein sequences were collected from prominent plant-pathogenic fungi (39) for blastp (E value < 1e-30) (40). Proteins involved in carotenoid biosynthesis (Supplementary file S9) were collected from NCBI and UniProt for blastp (E value <1e-30) (40). Sequences of enzymes involved in IAA synthesis of T. deformans (Supplementary file S10) were selected as reference for tblastn (E value <1e-5, score >100 and identity >50%) (40). Duplicate hits were manually removed for all blast. Carbohydrate-active enzymes (CAZymes) in Protomyces genomes were searched with dbCAN (41).
Carbon assimilation, indolic-compound production, and auxin-activity assays
Carbon assimilation patterns among seven species were tested with API 50 CH strips (bioMerieux SA) cultured at 21°C for seven days, according to the manufacturer’s instructions. Sterile filtrates (0.20 µm filter) from cultures on, GYP, GYP + 0.1% tryptophan, or nitrogen base with glucose, were tested for indolic compound production, as previously described (14), (42). Statistics were performed with t-test in R 3.5.1. Sterile seeds of Arabidopsis thaliana (arabidopsis) bearing the artificial auxin-responsive promoter:reporter (DR5:GUS) fusion, were grown in 0.5xMS agar (1% sucrose, pH 5.7) for seven days under long-day conditions at 23°C. Staining followed previous protocol (14). Root hair phenotypes were captured using a LEICA MZ10F microscope with camera LEICA DFC490 24h after treatment of five-day-old arabidopsis seedlings with culture supernatant, as above.
Protein extraction, SDS-PAGE, and Western blotting
Total protein was isolated from frozen rosettes using 100 mg plant material in 100 µl Lacus buffer (43). Protein concentration determined by Bradford assay (www.bio-rad.com/) with BSA as standard. Total protein (100 µg) was loaded in SDS-PAGE, transferred to a PVDF (immobilon–FL) membrane, and scanned with LI-COR Odyssey scanner. Equal loading was examined by amido black staining. Primary anti-phosphorylated-MAPK (rabbit monoclonal) antibody was used at a 1:2000 dilution and the secondary anti-rabbit IgG antibody (IRDye 800CW Goat anti-Rabbit) at 1:10,000.
RESULTS
Novel Protomyces species from Arabidopsis
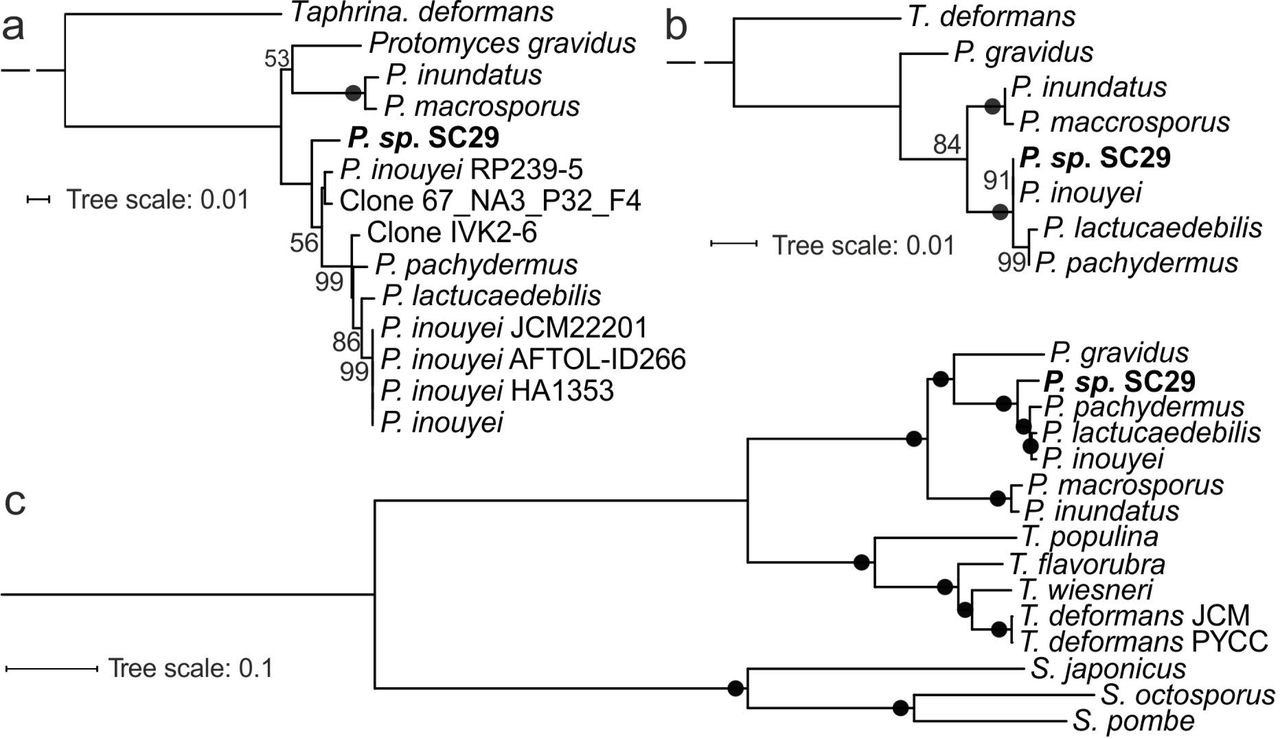
We previously isolated and characterized phylloplane yeasts from wild Arabidopsis (11). This included OTU1 comprising three strains from two distinct locations, which was subsequently identified as a putative new Protomyces species. Strain C29 (SC29) was selected for characterization of morphological and carbon utilization traits, in comparison to six species representing the diversity of the genus (Table 1). Reference species used and their abbreviations were; P. gravidus (Pgra), P. inouyei (Pino), P. inundatus (Pinu), P. lactucaedebilis (Plac), P. macrosporus (Pmac) and P. pachydermus (Ppac). Differences in cell size (Table S1), cell morphology, and colony morphology (Fig. S1) support SC29 as a novel species (Fig. S1). Our results for reference species agree with previously published data (1). SC29 exhibited a distinct profile of carbon utilization traits, especially for D-cellobiose, amygdalin, L-arabinose and D-arabinose (Table S2). Phylogenetic analysis was conducted with ITS and D1/D2 sequences from the seven Protomyces. Also, publicly available ITS sequences from several Protomyces strains sequenced from environmental samples that were related to SC29 were included. These trees resolved all reference Protomyces species with varied levels of confidence (Fig. 1a,b). However, only the ITS analysis differentiated between SC29 and Pino (Fig. 1a). The ITS phylogeny also revealed uncultured strains similar to SC29 that have been found on broad bean (Vicia faba) and in arctic soil (clone 67_NA3_P32_F4 and RP239-5, respectively) (44). To study the association of Protomyces with Arabidopsis, we queried Illumina MIseq amplicon sequencing data targeting ITS1 from a three-year Arabidopsis garden time-course experiment in Cologne, Germany. This revealed the persistent presence of Arabidopsis-associated Protomyces OTUs on multiple Arabidopsis accessions, in varied abundance across the growth season (Fig. S2). These data suggest SC29 is a distinct species in the genus Protomyces and yeasts of the genus Protomyces are reproducibly associated with Arabidopsis.

ITS and D1D2 regions for all species were sequenced in this study and confirmed against sequences mined from NCBI for all species, except SC29. Species and abbreviations used are: unnamed Protomyces strain C29 isolated from wild Arabidopsis (SC29), P. gravidus (Pgra), P. inouyei (Pino), P. inundatus (Pinu), P. lactucaedebilis (Plac), P. macrosporus (Pmac) and P. pachydermus (Ppac). a) Internal transcribed spacer (ITS) based tree that utilizes ITS1-5.6S rDNA-ITS2 sequences. Additionally, publicly available ITS sequences from species closely related to SC29 were included; including P. inouyei RP239-5 isolated from bean. b) D1D2 based tree that utilizes sequences from the large rDNA subunit. Trees in (a and b) were constructed by the maximum likelihood method with ClustalX2 using Taphrina deformans as an outgroup. c) Whole genome phylogenetic tree of Protomyces and Taphrina spp. with three Schizosaccharomyces spp. used as an outgroup. RAxML and rapid bootstrapping (100x) were chosen for constructing the tree utilizing 1035 concatenated single-copy conserved protein sequences. Alignment quality control was achieved by applying sequence scores >= 0.8 in MAFFT analysis using Guidance2. Multiple aligned sequences of each species/strain were concatenated using FASconCAT_V1.0 and bootstrap values are indicated at the nodes, black circles denote 100% support. For whole genome alignment (synteny) data see Figure S3.
To define relationships within the Protomyces, we sequenced SC29 and six reference genomes. For genome statistics, conserved protein domains, and conserved synteny see (Table 1, File S2, Fig. S3). Phylogenetic analysis with 1035 single-copy proteins (Fig. 1c) places SC29 as a distinct species within the Protomyces, as an outgroup to the clade composed of Ppac, Plac, and Pino, and as sister clade to Pgra, which are all pathogenic on Compositae family hosts. Pmac and Pinu form another distinct clade and infect Umbelliferae family hosts (Table 1).
SC29 persists on Arabidopsis phylloplane
Since all known Protomyces are phytopathogens, we tested many Arabidopsis-infection protocols, including chamber experiments at low temperatures and long-term field infections, mimicing the natural growth conditions of wild Arabidopsis (Table S3). No visible disease symptoms were observed. However, in field infections, SC29 survived overwinter on Arabidopsis and was re-isolated from infected plants the following spring. The growth of SC29 and Pino was assayed on the leaf surface of soil grown (not shown) and in vitro aseptic Arabidopsis cultures (Fig. 2a). In both experiments, SC29 persisted on the Arabidopsis leaf surface. In contrast, its closest relative, Pino, was unable to survive, demonstrating that adaptation to the Arabidopsis phyllosphere was specific to SC29. Further, SC29 was unable to survive on sterile foil placed on top of agar media plates, indicating it was not generally adapted to survive on surfaces without specificity.

a) Persistence assay of SC29 and P. inouyei (Pino) on the surface of Arabidopsis leaves aseptically grown on 0.5xMS agar. Cells were re-isolated from drop inoculated plants at the indicated times and plated to determine cell numbers, which are presented as the number of colony forming units (CFUx1000 cm-2). Persistence on sterile foil was used as a control for the ability to survive non-specifically on surfaces. Pooled data from five independent biological repeats (n=25 total) were analysed by computing a linear mixed model in R (3.5.1). b) Lesion diameters of Botrytis drop infections on Arabidopsis leaves pre-treated with water (mock), autoclave killed SC29 (A-SC29), or live SC29 cells. The bars represent the mean lesion size ±SD (n=24 total). Statistics performed with pooled data from six independent biological repeats by computing a linear mixed model in R. c) Arabidopsis MAPK3/6 activation was monitored by western blot with anti-phospho MAPK antibodies following pre-treatment with A-SC29 or live SC29 at 30 and 60 min. Equal loading was confirmed by amido black staining. Three independent experiments were repeated with the same result, representative results are shown. d) Relative gene expression of plant defence signalling genes 24 (white bars) and 72 (grey bars) hours post infection (hpi) following SC29 treatments only (top row) and after Botrytis infections with and without SC29 pre-treatments (bottom row). Gene expression was normalized to water control 24 hpi for each gene. Data are presented as mean relative expression levels ±SD of three pooled biological repeats. Statistics was performed with glht package in R (3.5.1) with fdr method for adjusted p-values. Key to abbreviations: A-SC29, autoclaved SC29; B.c, Botrytis cinerea. * p < 0.05; ** p < 0.01; *** p < 0.001.
SC29 activates MAPK and hormone immune signalling pathways
To define the SC29-Arabidopsis interaction we tested the ability of SC29 to enhance resistance against a broad-host-range fungal pathogen. Plants were pre-inoculated with water, SC29, or SC29 killed by autoclaving (A-SC29), prior to infection with the necrotrophic pathogen Botrytis cinerea and disease progression was monitored as Botrytis-induced lesion size. Significantly smaller lesion diameters in Arabidopsis pre-treated with both live and killed SC29 (Fig. 2b) was observed, suggesting increased resistance. Additionally, live SC29 pre-treated plants had significantly smaller lesions than those treated with A-SC29. This suggests that yeast MAMP molecules liberated from dead SC29 could trigger plant immunity, but the full effect required the live SC29. Co-cultivation on artificial media demonstrated that SC29 was not able to inhibit B. cinerea growth (Fig. S4), ruling out possible direct interactions. These results suggest SC29 interacts with Arabidopsis to activate immune signalling.
To test early immune signalling, we assayed MAPK activation in response to SC29 pre-treatment, both alone and with subsequent Botrytis infection. Western blots probed with an anti-TEpY antibody specific for the active phosphorylation site of MAPKs revealed activation of Arabidopsis MAPK3 and MAPK6 (MAPK3/6) in response to treatment by both live and dead SC29 at 60 min post infection (Fig. 2c). Botrytis infection also resulted in MAPK3/6 activation.
To further explore activation of defence signalling, we employed qPCR to monitor expression of defence signalling marker genes (for gene names and AGI codes, see File S1). Strikingly, we observed evidence of immune priming, seen as enhanced Botrytis-induced transcriptional responses, in samples pre-treated with both live and dead SC29 (Fig. 2d), for PDF1.1, a jasmonic acid (JA) marker; CYP71a13 and PAD3, markers of camalexin - the primary antimicrobial compound in Arabidopsis; and PR1, a marker of salicylic acid (SA). Live and dead SC29 alone had limited effect, showing induction at 72 hpi of PAD3 (Fig. 2d). These results indicate SC29 was able to activate some of the known canonical defense pathways and implicates JA, SA, and camalexin in the Arabidopsis response to this yeast. Markers of other signaling pathways such as ethylene, ABA, auxin, and cytokinin exhibited no significant changes (Fig. S5), suggesting the SC29 response is independent of these pathways.
Gene expansion and loss
We identified candidate effector-like proteins (CELPs) in the Protomyces and categorized them as small secreted proteins (SSPs), some of which were cysteine-rich (CSSPs; Table S4). All Protomyces had a higher number of SSPs and CSSPs compared to Taphrina spp. and the non-pathogenic Schizosaccharomyces spp., exceptionally, T. flavorubra had more SSPs than Pgra (Table S4). All Protomyces SSPs had similar features and were slightly smaller and more cysteine-rich compared to T. deformans (Fig. S6). Positive hits in the PHIbase indicated the presence of conserved fungal virulence proteins or effector-like proteins in Protomyces genomes (File S3). Identification of conserved protein domains in Protomyces CELPs (File S2) revealed the remarkable lack of LysM domains. However, the carbohydrate-binding, legume-like lectin (PF03388.13) domain was present in Protomyces, except Ppac. Topology of the phylogenetic tree constructed with SSPs (File S4), suggests that Pmac, Pinu, and Pgra CELP evolution diverged early from the other Protomyces species.
BadiRate analysis for estimation of gene family turnover rates (29) with SSP gene families (orthogroups, OGs) identified eight significant expansions (File S4), including one species-specific expansion in SC29 in SSP OG12. Public database queries with genes in the expanded OGs revealed that they are all Protomyces-specific genes with no conserved domains. These results suggest that although Pgra, Pino, Plac, and Ppac are all adapted to Compositae family hosts, CELPs in Pgra have a distinct evolutionary history. The expansion in SC29 suggests potential adaption to a different lifestyle or host, prompting us to seek additional evidence of this.
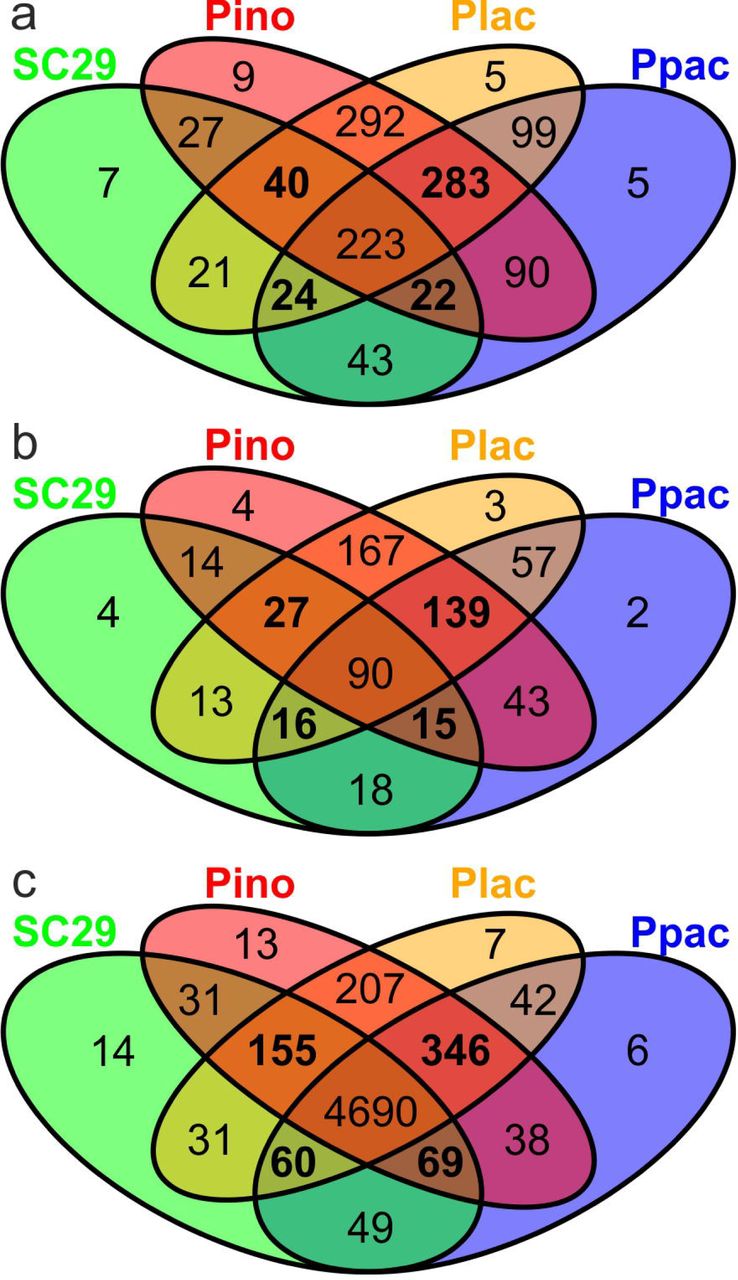
Gene loss is a feature in fungal genomes that have undergone a host jump (45). To test for this, gene content was compared between the closely related SC29, Pino, Plac, and Ppac (Fig. 3). Altogether 283 SSP OGs were absent from SC29 but present in the other three species, while 24, 40, and 22 were missing in Pino, Ppac, and Plac, respectively, but present in all others (Fig. 3a). Many, but not all, of these SSPs were cysteine rich (Fig. 3b). Although loss of a small number of genes from each OG were not detected by BadiRate as significant, cumulatively these losses are likely biologically relevant.

Species used form a closely related clade and include; Protomyces strain C29 (SC29), P. inouyei (Pino), P. lactucaedebilis (Plac), and P. pachydermus (Ppac). a) Orthologous clusters of candidate effector-like proteins (CELPs) of the small secreted protein (SSP) class. b) Orthologous clusters of CELPs of the cysteine-rich SSPs (CSSPs) class. This is a subset of the genes in (a). c) Orthologous clusters found in all annotated genes in the genomes of these species. Clusters common to three of the four species are indicated in bold.
Gene loss in SC29 was also evident at the whole genome level. SC29 only encoded 5514 annotated genes, compared to 5956, 5612, and 5952 for Pino, Ppac, and Plac, respectively (Table 1). Comparisons of the whole genome data revealed 346 OGs missing from SC29, while 60,155, and 69 were absent from for Pino, Ppac, and Plac, respectively (Fig. 3c). Gene ontology (GO) analysis of genes absent from SC29 indicated enrichment in GO:0036267 (invasive filamentous growth; File S5). These four genomes show high levels of similarity as evaluated by ANI, AAI (Table 1), and synteny (Fig. S3) and their genome completeness levels estimated with BUSCO were similar (86.0-86.3%), indicating similar amounts of unannotated genes.
Genes found only in one of these four species were also identified; 14 OGs (32 genes), 13 OGs (28 genes), six OGs (16 genes), and seven OGs (19 genes), were found to be species specific for SC29, Pino, Ppac, and Plac, respectively (Fig. 3c). SC29 specific genes were enriched for GO:0036349 (galactose-specific flocculation) and GO:1900233 (regulation of biofilm formation on inanimate substrates) (File S5).
Taken together, we conclude that SC29 exhibits genomic changes consistent with an adaptation to a new host and new lifestyle.
Carotenoids and auxin-like compounds in Protomyces
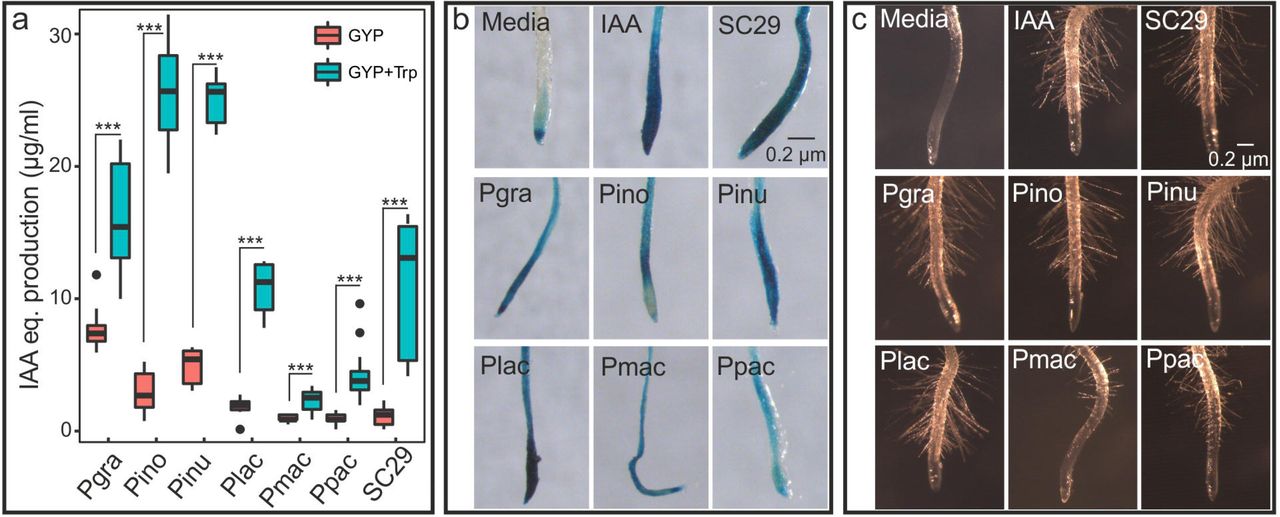
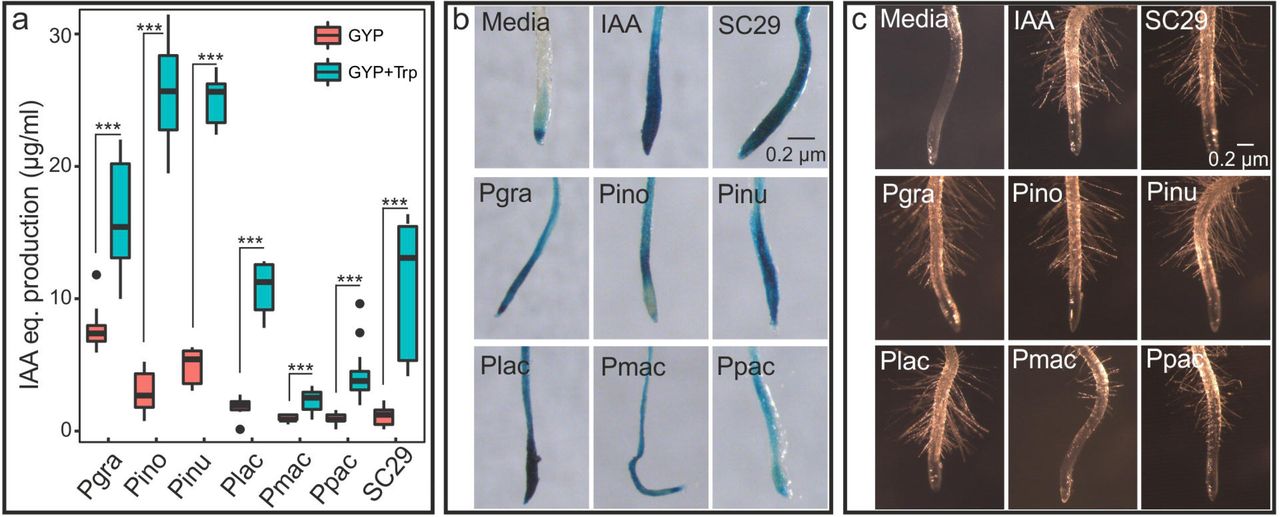
Assays of Protomyces indolic-compound production showed variation among species, ranging from 0.9 to 7.7 µg/ml when cultured in GYP medium and from 2.3 to 25.6 µg/ml with added tryptophan (Fig. 4a). Indolic-compound production was elevated in all cultures with additional tryptophan. Protomyces-derived indolic compounds in culture supernatants activated the artificial auxin-responsive promoter (DR5) in transgenic Arabidopsis bearing a DR5::GUS fusion, resulting in deposition of blue GUS stain (Fig. 4b). Additionally, we assayed auxin activity phenotypically, as the induction of root hair growth by Protomyces culture filtrates (Fig. 4c). Taken together, these results indicate that Protomyces produce indolic compounds with auxin-like activity, prompting us to identify the possible indole acetic acid (IAA) biosynthesis pathways encoded in these genomes. Of the five queried pathways, genes encoding all required enzymes were present only for the indole-3-pyruvic acid (IPyA) pathway (Table S5). Remarkably, all genes for this pathway were found in a single contig in the genome of SC29 (Fig. S3).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Species used and their abbreviations are: Protomyces strain C29 isolated from wild arabidopsis (SC29), P. gravidus (Pgra), P. inouyei (Pino), P. inundatus (Pinu), P. lactucaedebilis (Plac), P. macrosporus (Pmac) and P. pachydermus (Ppac). a) Quantification of indolic compounds produced by each Protomyces species using the Salkowski reagent assay on supernatants from five-day-old cultures in the specified liquid media [glucose yeast extract peptone (GYP) and GYP with additional tryptophan (GYP+Trp)]. Results are presented as equivalents (IAA eq.) from a standard curve with the auxin, indole acetic acid (IAA) and are means ±SD. Statistics were performed with t-test in R (3.5.1), *** p < 0.001. b) In vivo activation of the auxin transcriptional response is shown in roots of Arabidopsis bearing an artificial auxin-inducible promoter-reporter fusion (DR5:GUS) treated with sterile filtered culture supernatants. Auxin-induced promoter activity is visualized by deposition of blue stain indicating the presence of β-glucuronidase (GUS) reporter activity. c) Root hair formation as a biological assay for auxin activity. Experimental details are as in b. Uncultured media was used as a negative control (media) and IAA (5µM in b and 1µM in c) was used as a positive control (IAA). Three independent biological repeats were conducted for all experiments.
We further queried for possible carotenoid biosynthesis pathways within these genomes (Fig. S7). These were found in all seven Protomyces spp. with homologs of key enzymes phytoene desaturase (albino-1) and lycopene beta-cyclase (albino-2), but not lycopene epsilon-cyclase (CrtL-e).
Other features of Protomyces genomes
To probe for genomic features associated with the dual phylloplane resident and pathogenic lifestyle of the Protomyces, BadiRate was utilized in comparisons within and between these Protomyces species and non-pathogenic ascomycete yeasts using proteome data (File S4). Selected gene families with statistically significant lineage-specific expansions related to Protomyces lifestyle are highlighted below.
Orthogroup (OG)184 are a group I sensory histidine kinases. OG184 is absent from Schizosaccharomyces spp., present in variable copy number in all Taphrina and Protomyces; in SC29 it shows a significant expansion to five copies (Table S6). We manually curated all hybrid histidine kinases (HHKs) present in these Protomyces genomes (Table S6; File S6). Remarkably, in Pmac a dual HHK (g3206) was found with another type XI HHK (g3205) immediately adjacent. Several adjacent HHK gene pairs were found in other Protomyces; in Pinu two type XI HHKs (g5765-g5766) and two pairs in SC29 involving the type I HHK gene family (the expanded OG184) adjacent to a type IX and XI HHK (g3356-g3357 and g3889-g3890, respectively).
OG3409 and OG226 encode two related pectin degradation activities. OG102 encodes a family of secreted subtilisin-like serine proteases. These OGs both are absent from Taphrina and Schizosaccharomyces species, present in all Protomyces species, and show significant expansions in some Protomyces lineages (File S4). OG46 encodes the serine protease component of the SPS sensor. Members of this gene family are present in a single copy in two Taphrina species, and show a significant expansion in all but one Protomyces where they are present in high (three to nine) copy number. OG53 encodes cutinase gene palindrome-binding proteins, which were present with one or two copies in all species examined and significantly expanded to seven copies in SC29.
Finally, we investigated carbohydrate-active enzymes (CAZymes) in Protomyces genomes using dbCAN (41). All Protomyces genomes harboured a similar and relatively low number of CAZymes (241-261 total; File S7).
DISCUSSION
Phylogenetic implications
Our results have several implications for species delineations within the Protomyces and the definition of species that should be included in this genus. These issues are beyond the scope of this paper; thus, the naming of SC29 to P. arabidopsidis and other phylogenetic issues will be handled elsewhere (Wang, Sipilä, and Overmyer, manuscript in preparation).
An Arabidopsis associated Protomyces
We previously presented Protomyces strains isolated from an Arabidopsis population in Helsinki, Finland (14). Here we expand those results with Arabidopsis-associated Protomyces evidence from ITS sequencing in Germany (Fig. S2). The occurrence of Protomyces on Arabidopsis, together with the ability of SC29 to both persist in the Arabidopsis phylloplane and interact with the Arabidopsis immune system, give evidence that this is a true interaction. The genomic signatures of a host jump observed in SC29 genome further supports this. This is the first instance of a Protomyces species associated with a host plant outside of the Umbelliferae or Compositae families and suggests this novel Protomyces sp. has become adapted to a new host.
In defining this interaction, we have established a model experimental system with a phytopathogenic yeast and the genetic model-plant Arabidopsis, which will facilitate future genomic studies into the biology of early diverging ascomycetes, the evolution of fungal virulence, and plant immunity against yeasts. The activation of Arabidopsis defence signalling pathways by SC29 provides evidence of yeast-specific MAMPs that can trigger plant immunity. The human innate immune system deploys a distinct set of pattern recognition receptors (PRRs) activated by mostly mannose containing linkages that are highly abundant in the outer layers of cell walls of pathogenic yeasts (46, 47). Engagement of Arabidopsis immune signalling has been previously observed with autoclaved cell suspensions of S. cerevisiae resulting in the activation of SA signalling, camalexin biosynthesis, and enhanced resistance against other pathogens (48). Protein-depleted (protease-treated) yeast extract also triggered immune signalling, suggesting a carbohydrate MAMP was responsible (49). Accordingly, with SC29 we observed activation of SA and camalexin pathways and enhanced Botrytis resistance.
Protomyces reside in the phyllosphere as yeasts, but invade their hosts in the hyphal form. The genomes of filamentous (hyphal) fungal pathogens have chitin containing cell walls and frequently contain lysine motif (LysM) domain effectors involved in sequestering chitin or otherwise blocking immune responses elicited by this MAMP (50). The absence of LysM domain containing CELPs in the Protomyces suggests that chitin is of lesser importance for their host interactions. However, CELPs bearing another conserved carbohydrate-binding domain, the legume (L)-type lectin domain were present in all but one Protomyces genome. This lectin domain mediates the binding of mannose linkages (51-53). Pinu has been shown to have a cell wall composed of glucan and mannose, similar to other yeasts (54). Remarkably, human pathogenic yeasts also lack LysM effectors (50). Taken together, these results further support the importance of mannose-linkages, or other yeast MAMPs, in the interactions of Protomyces with their plant hosts. Forward and reverse genetic screens aimed at discovering PRRs involved in detecting yeast MAMPs (yeast cell wall components) are underway in our laboratory.
Comparative genomics within the Protomyces
The phyllosphere is estimated to be one of the largest microbial habitats on earth with an estimated size of 109 km2 (55). However, this habitat presents abiotic hazards such as lack of nutrients, exposure to full solar irradiation, extreme temperature fluctuations, and long periods of water shortage punctuated by periodic deluge of raindrops, which threaten to dislodge microbes (55, 56). Biotic threats are also present; for instance, microbe-microbe interactions such as competition for resource acquisition (57) and growth inhibition by antibiotics (58). Although not yet documented in the phylloplane, effector proteins can be deployed in direct inter-microbial competition (59-61). The host is also a threat; host derived antimicrobial secondary metabolites are present in the phyllosphere (62) and increasingly a picture is emerging where changes in the cuticle can trigger plant immune signalling, extending plant immune surveillance out into the phyllosphere (63-65). Thus, the phyllosphere is a remarkably hostile environment that presents a significant barrier to be overcome by resident microorganisms and pathogens utilizing this space for host access. Accordingly, phyllosphere microbiome communities comprise microbes specifically adapted to this environment (11, 55).
Genome sequencing enables the elucidation of genomic features of plant-associated microbes. Significant advances have been made, especially in root associated microbial communities (66, 67). Phyllosphere microbes are now gaining considerable interest, but our understanding still lags behind. The Protomyces lifecycle is highly dependent on survival of the yeast phase on host leaf surfaces, presenting an opportunity to explore genomic adaptations to the phyllosphere in this eukaryote. This is especially the case for SC29, which shows genomic adaptations consistent with our observations of a change toward a more phyllosphere-based lifestyle.
Several features related to nutrient acquisition were found in the Protomyces. Gene families for serine proteases belonging to the SPS sensor were expanded in the Protomyces. The SPC system is involved in sensing extracellular amino acids. Intriguingly, in Candida albicans the SPS system has also been implicated in immune evasion, in addition to nutrient acquisition (68). Other expanded gene families include: pectinase gene families that may be involved in utilization of pectin as a nutrient in the plant phylloplane and a family of secreted subtilisin-like serine proteases previously linked with nutrient acquisition during soil-residency (69). Finally, carbohydrate-active enzyme (CAZyme) gene content was defined; all Protomyces species had similar and low number of CAZymes relative to other fungal pathogens, consistent with their small genomes and facultative-biotrophic pathogen lifestyles (70).
Auxin of microbial origin is multifunctional and has been implicated in growth promotion by beneficial microbes (71, 72) and immune suppression by pathogens (73-75). Auxins function in the phyllosphere to differentially inhibit bacteria, but not yeasts (76), and to enhance nutrient leaching by loosening host cell walls and releasing sugars (55, 77, 78). The production of indolic compounds with the ability to activate plant auxin-induced transcriptional response suggests that Protomyces produce IAA. The pathways responsible for fungal IAA biosynthesis are not well defined; several pathways are suggested (79, 80). Feeding exogenous tryptophan in growth media increased the production of indolic compounds by Protomyces species suggesting a tryptophan-dependent IAA biosynthesis pathway. All seven Protomyces genomes had a complete indole-3-pyruvic acid (IPyA) pathway and orthologs of efflux carriers responsible for IAA export (Table S5), suggesting this as a candidate IAA biosynthesis pathway in Protomyces. Remarkably, all the genes for this pathway are located on a single contig in the SC29 genome. The IPyA has been suggested for other Taphrinomycotina species, Taphrina deformans (7, 10) and three other Taphrina species (10), based on the presence of orthologs of TAM and IAD genes, similar to the pathways previously found in the Basidiomycete, Ustilago maydis (81). Tsai et al also found orthologs of the YUC Flavin monoxygenase in four Taphrina species, suggesting the presence of an additional plant-like IAA biosynthesis pathway (10); however, YUC orthologs were absent from the Protomyces (Table S5).
Carotenoids are important stress protectants (82) whose high level accumulation is common in phylloplane yeasts (83). Carotenoid pigments are responsible for coloured colonies observed in Protomyces cultures (1) and three Protomyces species (Pino, Ppac, and Plac) previously assayed produce high levels (65-99 ug/g dry weight) of carotenoids (84). Carotenoid biosynthesis machinery was present in all Protomyces, with the exception of the enzymes CrtL-e and CrtL-b, suggesting that these Protomyces all produce lycopene, γ-carotene, and β-carotene, but not δ-carotene or α-carotene.
The inability of SC29 to cause disease on Arabidopsis, but ability to persist on the leaf surface suggest a change to a lifestyle where phylloplane residency plays a larger role. Some genomic changes in SC29 were consistent with this. The family of genes encoding the DNA repair protein Mre11 were expanded. Mre11 is the nuclease subunit of the widely conserved double-strand break repair MRX complex with Rad50p and Xrs2p. These genes are only found in SC29 where they are significantly expanded to three copies. DNA repair mechanisms represent a mechanism by which phyllosphere microbe protect themselves from direct solar irradiation and reactive oxygen species produced by plant defence responses (30). The gene family for cutinase transcription factor 1 (CTF1) was specifically expanded in SC29. CTF1 is required for the induction of fungal cutinase by plant hydroxyl fatty acids from cutin (85, 86), a component of the plant cuticle and potential energy source for phyllosphere resident microbes.
Sensory histidine kinases
HHKs are among the most important sensory proteins that transmit both internal and environmental information in the cell (39, 87). Most groups of HHKs were found in Protomyces genomes, with some exception (Table S6). Several genomic features suggest that the HHKs are actively evolving in the Protomyces. Groups III and X are involved in regulation of pathogenicity and morphogenesis in pathogenic fungi (39). Group III HHKs are normally present as a single copy in pathogenic fungi (39), but are seen in two copies in several Protomyces. Group I HHK genes have undergone a significantly expansion in SC29. Group I HHK function is not well studied (39); in rice blast fungus mutants lacking a type I HHK (MoHik3) exhibited slight alterations in development of virulence (88). The expansion of this gene family may be indicative of the change of lifestyle observed in SC29. Two of these group I HHK genes are immediately adjacent to other HHKs in SC29 suggesting a mechanism for innovation of signalling components in these taxa. Tandemly duplicated type XI HKKs were also observed in Pinu. Remarkably, in Pmac a dual HKK was adjacent to a type XI HKK. Dual HHKs have only been found in the genomes of fungi in the Basidiomycota. Protomyces belong to an early diverging lineage of the Ascomycota; this first instance of a dual HHK in an ascomycete is consistent with the observation of basidiomycete-like and primitive features in other fungi in the order Taphrinales (6, 89).
The GO class for invasive filamentous growth was enriched in genes that were absent from SC29, but present all the three most closely related Protomyces. These genes encode ubiquitin-related modifier1 (Urm1) orthologs, responsible for urmylation, an ubiquitin-like protein modification, which does not lead to proteasomal degradation (90, 91). These genes are conserved in yeasts and are involved in invasive growth in response to glucose starvation. In S. cerevisiae urmylation was required for invasive and pseudohyphal growth (91). The absence of this gene in SC29 is consistent with lifestyle change.
Two GO categories were enriched in the SC29 specific genes, galactose-specific flocculation and positive regulation of single-species biofilm formation on inanimate substrate. These SC29 specific genes are related to the S. pombe galactose-specific cell agglutination protein (gsf2), an outer PM protein promoting cell-to-cell adhesion (92, 93) and the Candida albicans biofilm regulator 1 (BRG1) transcription factor (94, 95). Microbial aggregates and biofilms facilitate water retention and wetting in the phylloplane; in addition to their roles in attachment these are adaptations promoting drought tolerance and nutrient leaching (55).
Taken together, the above results support that SC29 is adapted to the Arabidopsis phyllosphere. The model best supported by our results is that SC29 is a phylloplane resident on one or more hosts and lacks the ability to cause disease. This would be analogous to several known Taphrina species originally assigned to the defunct genus Lalaria, which are Taphrina isolated in the yeast (anamorphic) state from asymptomatic hosts (96, 97). These Taphrina not associated with host disease are thought to have lost virulence and live only in the phylloplane (6, 97). Given the relatedness of Taphrina and Protomyces and their similarity in lifestyle and pathogenicity strategy, the discovery of phyllosphere resident Protomyces species is plausible. Although the anamorph genus Lalaria has been rendered invalid by abandonment of the dual name system, the questions raised by the non-pathogenic phylloplane-resident lifestyle that defined the Lalaria remain relevant to understanding the biology of yeasts of the order Taphrinales.
We cannot formally exclude that SC29 is pathogenic on Arabidopsis or another plant species. Conditions required for SC29 virulence may not have been met in controlled infections. Although, the inability of SC29 to infect Arabidopsis in the experimental field argues against this. Assuming this is the case, it does not change our basic findings; SC29 is the first Protomyces found to be adapted to persist in the phyllosphere of a Brassicaceae (Cruciferae) family host, well outside the host range of all other Protomyces. In this case, our work illustrates the importance to a pathogenic yeast of phylloplane survival while waiting for favourable infections conditions.
Future prospects
While our results with SC29 support that it has transitioned to a phylloplane resident lifestyle on a new host Arabidopsis, much of the core Protomyces genome and virulence machinery remain intact. This is supported by the core CELPs and annotated genes common to SC29 and the three other closely related Protomyces (Fig. 3). This suggests latent potential for returning to the pathogenic lifestyle, but on a new host. Thus moving as a phylloplane resident onto different plants may be a general strategy utilized to expand into new hosts by organisms like the dimorphic Protomyces and Taphrina that have a dual phyllosphere resident-pathogenic lifestyle. Living as a phyllosphere resident with intimate access to the host and its microbiome as a novel adaptive zone (98) may facilitate development of pathogenicity on the new host and drive new speciation. Finally, the congruence of host and pathogen phylogenetic trees is taken as evidence of co-speciation, however, similar patterns can emerge from speciation that is driven by a jump to a new host (pseudo-cospeciation) (99). Thus, our evidence of a host shift in the Protomyces indicates that assumptions concerning host pathogen co-speciation should be re-examined in these taxa.
ACKNOWLEDGMENTS
We thank Tuomas Puukko, Airi Lamminmäki, and Leena Grönholm, for excellent technical support and Katariina Vuorinen for assistance with infection assays, Mikeal Broché and Tiina Blomster for advice of qPCR, Julia Krasensky-Wrzaczek for instruction on MAPKs work, Ansa Palojärvi (Joint ‘SUCCESS’ project) for the control Paenibacillus strain with antifungal activity, and the personnel of the DNA sequencing and genomics laboratory performing NGS sequencing. This work was supported by the following grants: Academy of Finland Fellowship (decisions no. 251397, 256073 and 283254) to KO and the Academy of Finland Center of Excellence in Primary Producers 2014-2019 (decisions #271832 and 307335). KW, SR, and OM are members of the University of Helsinki Doctoral Programs in Plant Science (DPPS) and AA in Microbiology and Biotechnology (MBDP). Computing resources provided by the Finnish IT Center for Science (CSC; www.csc.fi) are gratefully acknowledged.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.
- 9.
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.
- 61.↵
- 62.↵
- 63.↵
- 64.
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵




