

ABSTRACT
Polyamines are essential metabolites that play an important role in cell growth, stress adaptation, and microbial virulence1–3. In order to survive and multiply within a human host, pathogenic bacteria adjust the expression and activity of polyamine biosynthetic enzymes in response to different environmental stresses and metabolic cues2. Here, we show that ornithine capture by the ribosome and the nascent peptide SpeFL controls bacterial polyamine synthesis by inducing the expression of the ornithine decarboxylase SpeF4, via a mechanism involving ribosome stalling and transcription antitermination. In addition, we present the cryo-EM structure of an Escherichia coli (E. coli) ribosome stalled during translation of speFL in the presence of ornithine. The structure shows how the ribosome and the SpeFL sensor domain form a highly selective binding pocket that accommodates a single ornithine molecule but excludes near-cognate ligands. Ornithine pre-associates with the ribosome and is then held in place by the sensor domain, leading to the compaction of the SpeFL effector domain and blocking the action of release factor RF1. Thus, our study not only reveals basic strategies by which nascent peptides assist the ribosome in detecting specific metabolites, but also provides a framework for assessing how ornithine promotes virulence in several human pathogens.
INTRODUCTION
Putrescine is a naturally abundant polyamine that is produced from ornithine by the enzyme ornithine decarboxylase, whose expression and activity are tightly regulated2. Two ornithine decarboxylase genes exist in E. coli, the constitutive speC and the inducible speF, which along with its operon partner potE, an ornithine-putrescine antiporter, is expressed under mild acidic stress and high ornithine levels4–6. Searching for regulatory elements upstream of speF, we found a short open reading frame (ORF) encoding a putative 34-amino acid peptide, which we named speFL (leader of speF) (Fig. 1a). This ORF is conserved in many pathogenic γ-proteobacteria (Extended Data Fig. 1), including Salmonella typhimurium, where it was recently reported as orf347. Translation of orf34 in the presence of ornithine activates speF expression by preventing premature Rho-dependent transcription termination7. However, the mechanism by which ornithine triggers speF expression is unknown.

a, Schematic layout of the speF operon showing the sequence of the SpeFL peptide and the reaction catalyzed by SpeF. b, Toeprinting assay8,9 to monitor the translation of speFL in the absence (-) or presence (+) of 10 mM ornithine, 10 mM putrescine, release factors (RF1,2,3) or 90 μM puromycin. Arrows indicate ribosomes stalled with the codon for the indicated amino acid in the P-site (Ala33 – open triangle; Arg34 – filled triangle). c, E. coli TB1 cells13 transformed with a plasmid carrying a speF1–3-lacZα translational fusion whose expression is placed under the control of speFL and the speFL-speF intergenic region. Cells were grown on rich medium supplemented with 50 μg/ml streptomycin, 100 μg/ml ampicillin, 1 mM IPTG and 0.5 mM X-Gal in the absence (−) or presence (+) of 3 μmol ornithine or 20 μg bicyclomycin. Blue cells express the speF1–3-lacZα translational fusion. d, Model of speF induction following the ornithine-dependent stalling of ribosomes translating speFL. The speFL open reading frame is boxed and shown partly in turquoise, with the overlapping rut site in yellow. Consecutive rare arginine codons R12 and R13 are shown in red letters. The leading ribosome on speFL is outlined in black while the second and third ribosomes are outlined in gray. The SpeFL peptide is in turquoise. e, Codon frequency at each position of speFL in E. coli (red line) and in the Enterobacteriales order (nspecies=10 (see Extended Data Fig. 1); mean – blue lines; ± standard deviation (SD) – light blue boxes). Codon usage values were obtained from the Codon Usage Database (NCBI-Genbank Flat File Release 160.0 [June 15 2007])14. f, The same assay as in c, showing the induction of a speF1–3-lacZα translational fusion by wild-type (WT – R12R13) or synonymous speFL variants with different combinations of rare (r) or common (c) arginine codons at positions 12 and 13, in the absence (−) or presence (+) of different amounts of ornithine. Mutated codons are shown in red.
RESULTS
To investigate how speF is activated, we performed toeprinting assays8,9 to monitor the position of ribosomes on a transcript encoding SpeFL (Fig. 1b and Supplementary Data 1). A faint toeprint corresponding to ribosomes that reached the UAG stop codon was visible in the absence of exogenous ligand. Addition of ornithine resulted in two strong toeprint signals for ribosomes stalled with codons 33 or 34 of speFL in the ribosomal P-site. Ribosome stalling occurred in a dose-dependent manner with respect to ornithine concentration (Extended Data Fig. 2), but no toeprints were observed in the presence of putrescine, highlighting the strict dependence of the stalling process on ornithine availability. Translating a double-frameshifted speFLFS template that encodes a different amino acid sequence did not yield ornithine-dependent toeprints (Extended Data Fig. 3), indicating that ribosome stalling depends on the nascent peptide rather than on the mRNA structure. In the absence of release factors, the toeprint at position 34 intensified, reflecting impaired translation termination. Treatment with puromycin led to the disappearance of this toeprint, while ornithine-dependent toeprints remained visible. Puromycin causes premature peptide release and insensitivity to this antibiotic is characteristic of arrest peptides, a class of nascent regulatory peptides that stall the ribosomes that are translating them, often in a ligand-dependent manner10,11. Finally, we showed that an RNA element including speFL and the 257-nucleotide speFL-speF intergenic region induces the expression of a speF1–3-lacZα translational fusion in vivo in response to ornithine (Fig. 1c). Treatment with bicyclomycin, which specifically blocks the ATPase activity of Rho12, resulted in constitutive speF1–3-lacZα expression, confirming the previously reported7 involvement of Rho in the regulation of speF. Thus, ribosomes translating speFL stall in an ornithine-dependent manner, inducing speF through a Rho-dependent mechanism.
In S. typhimurium, Rho-dependent transcription termination occurs immediately downstream of an mRNA hairpin that includes the 3’ end of speFL7. This hairpin is conserved in E. coli and would cause an RNA polymerase that has just finished transcribing speFL to pause (Fig. 1d, Extended Data Fig. 4). A ribosome translating speFL can unwind the pause hairpin upon reaching the stop codon, freeing the RNA polymerase and allowing transcription to resume. When ornithine levels are low, the leading ribosome on speFL terminates and dissociates from the mRNA, exposing part of a predicted rut site15. For Rho to bind to the mRNA and cause premature transcription termination, however, a full rut site must be available. Since polysome accumulation on speFL would interfere with rut availability, we hypothesized that consecutive rare arginine codons at positions 12 and 13 of speFL may slow translation enough to fully expose the rut site and give Rho a chance to bind. As reported previously7, this region of speFL contains rare codons in many γ-proteobacteria, especially at position 12 (Fig. 1e). While replacing codon 13 with a common synonymous codon caused a mild decrease in speF1–3-lacZα induction, the same mutation at position 12 or mutation of both codons gave rise to a basal level of speF expression that was not observed with wild-type speFL (Fig. 1f), consistent with a model whereby efficient Rho binding is dependent on polysomes not accumulating on rut. When ornithine levels are high, the leading ribosome on speFL undergoes nascent peptide-mediated translational arrest. Ribosome stalling masks the rut site and polysomes accumulate (Extended Data Fig. 5). This prevents Rho from binding, allowing transcription to proceed and speF to be expressed.
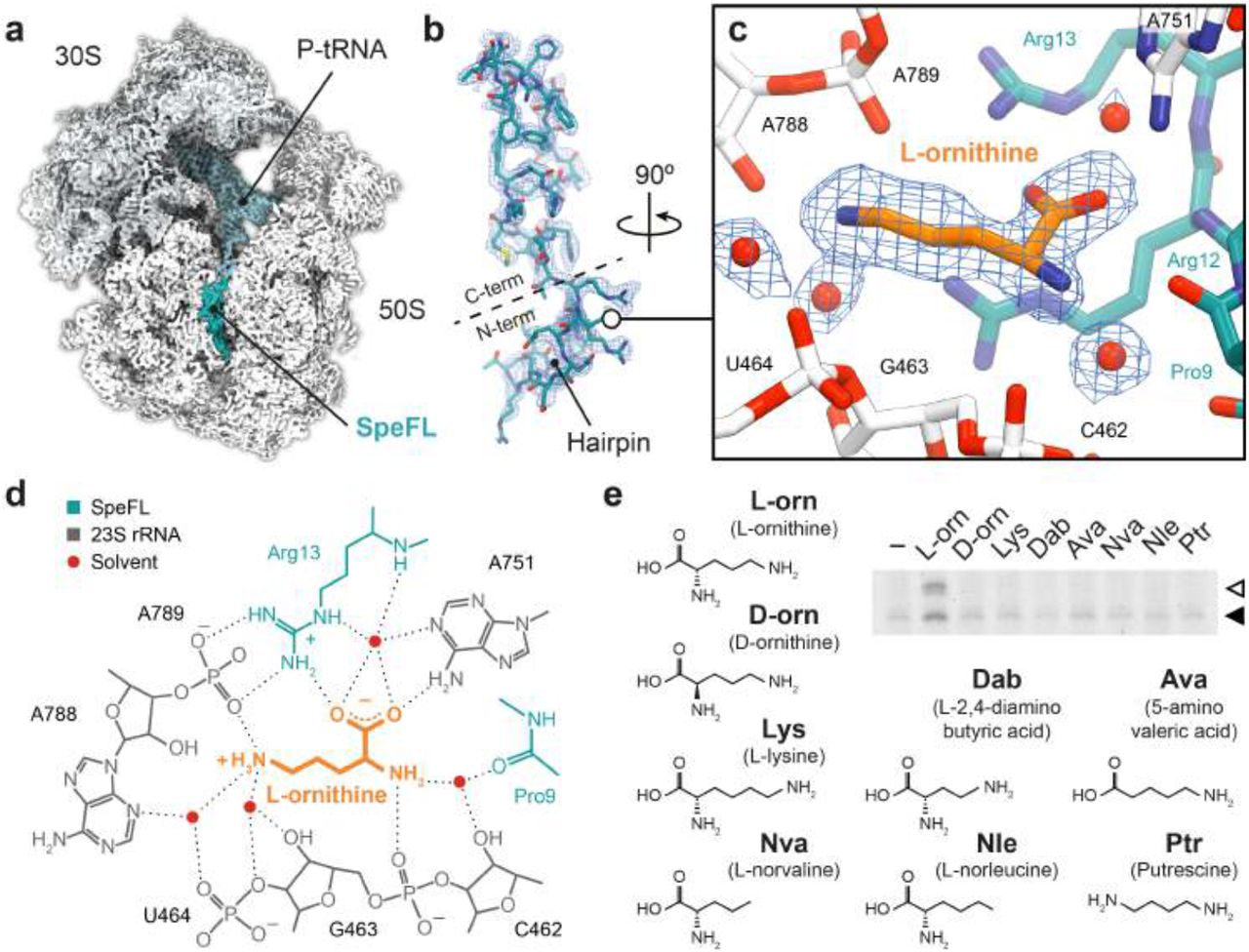
To determine how nascent SpeFL functions as an ornithine sensor, we used cryo-EM to obtain two structures of a SpeFL–70S ribosome complex stalled in the presence of ornithine at an overall resolution of 2.7 Å (Fig. 2a, Extended Data Fig. 5, 6 and 7, and Extended Data Table 2). We observed a major subpopulation corresponding to ribosomes with well-resolved density for a 34-residue peptidyl-tRNAArg bound to the P-site (Fig. 2b). SpeFL adopts a compact fold that completely obstructs the upper two-thirds of the exit tunnel and can be subdivided into N- and C-terminal domains, corresponding to residues 1–13 and 14–34 of SpeFL, respectively. The N-terminal domain forms a hairpin, while secondary structure elements stabilize the C-terminal domain, most notably two type I β-turns between residues 19–22 and 23–26, and one 310-helix between residues 27–32. In addition, SpeFL interacts extensively with the 23S ribosomal RNA (23S rRNA) and with ribosomal proteins uL4 and uL22 through a combination of π-stacking and hydrogen bonding (Extended Data Fig. 8). All of these structural elements contribute to stabilizing the complex fold adopted by SpeFL inside the exit tunnel.

a, Transverse section of a cryo-EM density map of the SpeFL–70S complex, showing the small (30S, light gray) and large (50S, white) ribosomal subunits, the P-site tRNA (pale blue) and the SpeFL peptide (turquoise). b, Cryo-EM density displayed as a mesh, fitted with a molecular model of SpeFL, with the N– and C–terminal domains highlighted. The N-terminal hairpin is also indicated. c, Binding pocket formed by the 23S rRNA (white) and SpeFL (turquoise), with a single L-ornithine molecule (orange) surrounded by 4 solvent molecules (red) fitted into the cryo-EM density of the SpeFL-ESRF complex. The existence of these solvent molecules was validated using two independently determined structures of the SpeFL-70S complex (see Extended Data Fig. 9). d, Chemical diagram showing interactions between the 23S rRNA (dark gray), SpeFL (turquoise), L-ornithine (orange) and solvent molecules (red) inside the ligand binding pocket. Possible hydrogen bonds are shown as dotted lines. e, Toeprinting assay8,9 to monitor the translation of speFL in the absence (−) or presence of 10 mM (+) of various small molecules. Arrows indicate ribosomes stalled with the indicated amino acid in the P-site (Ala33 – open triangle; Arg34 – filled triangle).
A clear peak of density that could be unambiguously attributed to a single L-ornithine molecule was visible inside a cavity formed by 23S rRNA residues C462, G463, U464, A751, A788 and A789 and by Pro9, Arg12 and Arg13 of the N-terminal domain of SpeFL, referred to here as the sensor domain (Fig. 2c, d and Extended Data Fig. 9). To our knowledge, this cavity represents a novel binding site for small molecules on the ribosome, which could be targeted for future antibiotic development. The ornithine recognition loop of SpeFL consists of a HIRRXXH motif spanning residues 10–16, among which His10, Arg13 and His16 help form the ligand binding pocket by interacting with 23S rRNA and ribosomal protein uL22 residues (Fig 2c, d and Extended Data Fig. 8f). Deletion of residues 1–7 of SpeFL, which disrupts the hairpin but retains the HIRRXXH motif (Extended Data Fig. 10a), or mutation of the strictly conserved Arg12 and Arg13 to alanine or lysine (Extended Data Fig. 10b) abolished ornithine-dependent translational arrest in vitro, highlighting the importance of the hairpin and of these residues for the stalling process. The side chain and α-amino groups of ornithine interact with the backbone phosphates of 23S rRNA residues A789 and G463, respectively (Fig. 2d). Ornithine is further stabilized via hydrogen bonding between its α-carboxyl group and both the guanidino group of SpeFL residue Arg13 and the N6-amino group of 23S rRNA residue A751. Four ordered solvent molecules fill the cavity and make additional bridging interactions between ornithine and the 23S rRNA or SpeFL (Fig. 2c, d and Extended Data Fig. 9). Thus, small molecules differing from ornithine by only a single methylene group are either too short (L-2,4–diaminobutyric acid) or too long (L-lysine) for the binding pocket, while the deletion of ligand functional groups (putrescine, L-norvaline, L-norleucine, 5-aminovaleric acid) or the use of a D-enantiomer (D-ornithine) abolish stalling by preventing the formation of certain hydrogen bonds (Fig. 2e and Supplementary Data 2). The tight coordination of L-ornithine via each of its potential hydrogen bond donors and acceptors therefore explains the high selectivity of SpeFL and the ribosome for their cognate ligand.
To understand how ornithine capture by the sensor domain stalls the ribosome, we must focus on the C-terminal effector domain of SpeFL, which consists of a hydrophobic core composed of four phenylalanine residues (Phe20, Phe28, Phe30 and Phe31) nucleated around the strictly conserved Phe26 (Fig. 3a and Extended Data Fig. 8c). Residues Phe28, Phe30 and Phe31 establish π-stacking interactions with the bases of 23S rRNA residues U2586, G2505 and A2062, respectively, which help to position the effector domain in the upper part of the ribosomal exit tunnel (Fig. 3a, Extended Data Fig. 8). Mutation of Phe26 or any of these three aromatic residues to alanine abolishes ribosome stalling in vitro (Fig. 3b and Supplementary Data 3), highlighting their importance for translational arrest. Since the SpeFL-70S structure corresponds to stalled ribosomes with a UAG stop codon in the A-site, it is clear that SpeFL must inhibit the action of RF1, the release factor responsible for recognizing this stop codon. Comparing our structure with that of a Thermus thermophilus 70S ribosome in complex with RF1 and a P-site tRNA16 reveals that the binding of RF1 to the SpeFL-70S complex is prevented by 23S rRNA residue U2585, which adopts a rotated conformation that would sterically clash with the GGQ loop of RF1. Rotation of U2585 is caused by residue Asn32 of SpeFL, which takes the place of its base in the 70S–RF1–P-tRNA complex (Fig. 3c, d). Thus, the continued synthesis of SpeFL after the recognition of ornithine by the sensor domain leads to the compaction of the effector domain and forces U2585 into a conformation that prevents peptide release by RF1, causing the ribosome to stall.

a, Close-up of the ribosomal exit tunnel showing the sensor and effector domains of SpeFL (turquoise) interacting with residues of the 23S rRNA (white). Ornithine (orange) is trapped between the tunnel wall and the sensor domain, while synthesis of all 34 amino acids of SpeFL leads to compaction of the effector domain and blockage of the peptidyl transferase center (PTC). b, Toeprinting assay8,9 to monitor the translation of wild-type (WT) and mutant speFL in the absence (−) or presence (+) of 10 mM ornithine. All samples were treated with 90 μM puromycin. Arrows indicate ribosomes stalled with the indicated amino acid in the P-site (Ala33 – open triangle; Arg34 – filled triangle). c, Structure of an E. coli 70S–RF1–P-tRNA complex (PDB 5J3C)16, showing the GGQ loop of RF1 (peach, with residue Gln-235 labeled) and 23S rRNA residue U2585 (white). d, Close-up of the SpeFL–70S structure showing the same view as in c. The side chain of residue Asn32 of SpeFL (turquoise) forces U2585 to adopt a rotated conformation that prevents RF1 binding.
Metabolite sensing by a translating ribosome is a complex and dynamic process whose understanding has been hampered by the lack of high-resolution structural data11,17–19. With the exception of antibiotic-dependent translational arrest20–22, in which the drug binds directly to the empty ribosome, it is not known if the metabolite helps to create a binding surface for the nascent peptide or vice versa11. In the SpeFL–70S structure, ornithine interacts primarily with the ribosome, either directly or via bridging solvent molecules, whereas SpeFL provides only a few stabilizing interactions that help capture the cognate ligand (Fig. 2d). This implies that ornithine is already loosely associated with the 23S rRNA prior to the arrival of the SpeFL sensor domain (Fig. 4), as suggested by molecular dynamics simulations pointing to the existence of binding crevices for different amino acid side chains within the ribosomal exit tunnel23. Additionally, the close proximity of the sensor domain to the tunnel constriction formed by ribosomal proteins uL4 and uL22 raises the possibility that the sensor domain begins to fold in the upper part of the exit tunnel, consistent with the decreased speF expression seen for the R12rR13c mutant (Fig. 1f). Indeed, a partially folded sensor hairpin emerging from the tunnel constriction would rapidly contact and fix an ornithine molecule present within its adjacent binding crevice on the 23S rRNA. Once the interaction between the sensor domain and the tunnel wall has been stabilized through the binding of ornithine, the effector domain can be synthesized and compacted, resulting in inhibition of peptide release by the ribosome. These basic strategies for ligand recognition are likely to be used by many other metabolite-sensing nascent peptides.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model for the binding of ornithine (orange) by the ribosome and SpeFL (turquoise), leading to inactivation of the peptide release activity of the ribosome (red cross). The tunnel constriction is shown as a blue dotted line.
In conclusion, our data reveal how the ribosome aided by SpeFL functions as a highly selective ornithine sensor to regulate polyamine biosynthesis in pathogenic bacteria like E. coli or S. typhimurium. Both speF and potE, whose expression is regulated by SpeFL in response to fluctuating ornithine levels, have been linked to biofilm formation and microbial virulence24–26. SpeFL is therefore likely to play a role in polymicrobial and biofilm-associated infections by opportunistic pathogens, which often rely on metabolic signaling and cross-feeding to overcome host defenses27–29. For example, ornithine secreted by Enterococcus faecalis helps uropathogenic E. coli grow in the iron-restricted environment of a human host by redirecting its metabolism towards siderophore production30. Our findings strongly suggest that ribosomes translating speFL could be responsible for this behavior and that ornithine signaling may be an important feature of bacterial virulence.
Author Contributions
C.A.I. and B.S. designed the study. I.C-M. identified speFL. A.H.V., B.S., G.S. and A.C.S. performed biochemical experiments. A.H.V. performed bacterial assays. A.H.V. prepared the cryo-EM sample. A.H.V. and C.A.I. processed the cryo-EM data. A.H.V., B.S., G.S., A.C.S. and C.A.I. interpreted the results. A.H.V., B.S. and C.A.I. wrote the paper.
Author Information
The authors declare no competing financial interests.
METHODS
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
Bioinformatic identification of speFL
Homologs of E. coli speF were identified using tblastn31 (E-value lower than 10−4, >70% coverage), redundancy was minimized using CD-Hit32 (95% sequence identity cutoff) and regions between speF and the nearest upstream annotated genes were compiled (unknown, hypothetical, uncharacterized or leader genes were considered not to be annotated). All possible forward ORFs within the last 500 nucleotides of these upstream regions were extracted, considering ATG and alternative start codons defined for bacterial, archaeal and plant plastid genetic codes (NCBI Genetic codes Table 11). Possible Shine-Dalgarno sequences were not taken into account. In cases where more than one ORF was possible, the longest ORF was kept. Redundancy within the ORFs thus obtained was minimized with CD-Hit (95% sequence identity cutoff). A pairwise comparison of the resulting ORFs was then carried out as follows. First, nucleotide sequences were translated into amino acid sequences. A sliding window of 10 amino acids was applied to each sequence and an alignment score based on a BLOSUM6233 substitution matrix was computed for all possible combinations of 10-amino acid fragments from each pair of ORFs (no gaps allowed). A graph in which each node represents a 10-amino acid fragment and each edge represents an alignment score greater than 10 between two fragments (with the alignment score as a weight) was constructed. Finally, we used MCL34,35 to identify clusters within this graph and found a major cluster of conserved upstream ORFs corresponding to speFL.
Phylogeny of speFL
Homologs of E. coli speFL were identified using tblastn31 and a phylogenetic analysis of speFL from 15 representative species of γ–proteobacteria was carried out using the EMBL-EBI Simple Phylogeny server36. The resulting tree was displayed with Dendroscope37 (Extended Data Fig. 1).
Toeprinting assays
Toeprinting was performed as described previously9. Briefly, DNA templates containing a T7 promoter, a ribosome binding site, wild-type or mutant speFL, the first 75 nucleotides of the speFL-speF intergenic region and the NV1 sequence38 were generated by polymerase chain reaction (PCR). The wild-type template was generated using oligonucleotides 1–9 (see Extended Data Table 1 for the sequences of all oligonucleotides used). Point mutations were introduced by substituting oligonucleotide 5 for the relevant oligonucleotide (10-18). The double frame-shifted template speFLFS was generated using oligonucleotides 1, 2, 19–21 and 24. The speFL-Δ1–7 template was amplified by PCR from E. coli DH5α genomic DNA using oligonucleotides 1, 24 and 23 followed by PCR amplification of the product with oligonucleotides 2 and 9. DNA templates were transcribed and translated in vitro using the PURExpress Δ RF123 system (New England Biolabs). Ligands were dissolved in water and added as needed at the beginning of the reaction. A Yakima Yellow-labeled probe (2 μM) complementary to the NV1 sequence38 was added to the 5 μL reaction after incubating for 60 minutes at 30°C, and the sample was incubated for another 5 minutes at the same temperature. When needed, samples were treated with 90 μM puromycin at 30°C for 3 minutes, immediately followed by reverse transcription with 50 U of AMV reverse transcriptase (Promega) for 20 minutes at 30°C. RNA was degraded by adding 0.5 μL of a 10 M NaOH stock at 30°C for 15 minutes. Samples were neutralized with 0.7 μL of a 7.5 M HCl stock and the remaining cDNA was purified using a nucleotide removal kit (Qiagen). Sequencing reactions were performed according to the method of Sanger. Briefly, 1 pmol of DNA template was mixed with 10 pmol of oligonucleotide 9 labeled with Yakima Yellow and 1μL of HemoKlen Taq DNA Polymerase (New England Biolabs) in a 6μL reaction mixture containing 50 mM Tris-HCl pH 9.0, 2 mM MgCl2, 6.6 μM dNTPs, 10 μM ddGTP, 117 μM ddATP, 200 μM ddTTP or 66 μM ddCTP. Primers were extended with 30 cycles of 30 seconds of annealing at 42 °C and 1 minute of elongation at 70 °C. The purified cDNA and sequencing reactions were dried using a SpeedVac and resuspended in 6 μl or 3.5 μl gel-loading buffer (95 % formamide, 0.25 % (w/v) xylene cyanol, 0.25 % (w/v) SDS), respectively. Samples were denatured at 95°C for 5 minutes, and 2μL of the sequencing reactions and 3 μL of the toeprinting were separated by 7.5 % sequencing PAGE (2000 V, 40 W for 2-2.5h) followed by detection on a Typhoon imager (GE).
β-galactosidase assay
To test for in vivo activity, a translational reporter plasmid was obtained by fusing a region containing speFL, the speFL–speF intergenic region and the first three codons of speF to lacZα. The insert was prepared by PCR amplification from the E. coli K12 genome using oligonucleotides 25 and 26. Oligonucleotides 27 and 28 were used to linearize the pErmZα plasmid39. The insert and linearized plasmid were mixed and transformed following the AQUA cloning protocol40. Plasmids containing point mutations in the R12–R13 region were generated by site-directed mutagenesis as follows. The wild-type plasmid was linearized by PCR amplification with oligonucleotides 32 and 29, 30 or 31 (the latter included the mutations). The PCR product was purified from a 2% TAE-agarose gel with a Gel Extraction Kit (Qiagen) and phosphorylated for 30 minutes at 37 °C with 4U of T4 Polynucleotide Kinase (New England Biolabs) in a total volume of 20 μl according to the manufacturer’s instructions. The plasmid was circularized again by incubating the phosphorylated product with 400 U of T4 DNA ligase (New England Biolabs) for 2 hours at 16°C. The plasmids were transformed into E. coli TB139 and the cells were grown in lysogeny broth (LB) at 37°C (200 rpm) with streptomycin (50 μg/ml) and ampicillin (100 μg/ml) until they reached an optical density of 0.6 at 600 nm. 5 μL of the cell culture were plated onto LB-agar plates supplemented with streptomycin, ampicillin, 1 mM Isopropyl-β-D-1-thiogalactopyranoside (IPTG) and 0.5 mM 5-bromo-4-chloro-3-indolyl-beta-D-galactopyranoside (X-gal). 20 μg of bicyclomycin (Santa Cruz Biotechnology) or 0–3 μmol of L-ornithine (Sigma) were added after a 6 hour incubation at 37°C. The plates were then incubated at 37°C overnight and pictures were taken the next day.
Preparation of an E. coli SpeFL–70S complex for cryo-EM
The SpeFL–70S complex was prepared using a modified disome purification strategy21. Briefly, SpeFL was expressed in an RTS 100 E. coli HY Kit (Biotechrabbit) for 1 hour at 30°C in the presence of 10 mM L-ornithine if indicated, using a pEX-K4-SpeFL_2x plasmid (Eurofins) that carries two copies of speFL arranged as a bicistronic mRNA (Extended Data Fig. 5; insert sequence: 5’-CGA-TCG-AAT-TCT-AAT-ACG-ACT-CAC-TAT-AGG-GCT-TAA-GTA-TAA-GGA-GGA-AAA-AAT-ATG-GAA-AAT-AAC-AGC-CGC-ACT-ATG-CCC-CAT-ATA-AGG-CGG-ACA-ACT-CAT-ATT-ATG-AAG-TTT-GCT-CAT-CGC-AAT-AGC-TTC-GAC-TTT-CAC-TTC-TTC-AAT-GCC-CGT-TAG-TCT-ACC-GAC-TAA-GGG-CAC-TTC-AGC-TAA-AGT-TTT-ATA-AGG-AGG-AAA-AAA-TAT-GGA-AAA-TAA-CAG-CCG-CAC-TAT-GCC-CCA-TAT-AAG-GCG-GAC-AAC-TCA-TAT-TAT-GAA-GTT-TGC-TCA-TCG-CAA-TAG-CTT-CGA-CTT-TCA-CTT-CTT-CAA-TGC-CCG-TTA-GTC-TAC-CGA-CTA-AGG-GCA-CTT-CAG-CTA-GAT-ATC-TAG-CAT-AAC-CCC-TTG-GGG-CCT-CTA-AAC-GGG-TCT-TGA-GGG-GTT-TTT-TG-3’). Reaction volumes of 50 μL and 750 μL were used for analytical and preparative purposes, respectively. When indicated, the reaction was treated with 100 μM puromycin for 3 minutes at 30°C before being layered over 10-40% (w/v) sucrose gradients containing Buffer A (50 mM HEPES-KOH pH 7.5, 100 mM K-acetate and 25 mM Mg-acetate), prepared using a Gradient Master 108 (Biocomp). Sucrose gradient ultracentrifugation was performed for 2 hours and 45 minutes at 35,000 rpm in a SW 41 Ti rotor (Beckman-Coulter) at 4°C. Polysome fractions were detected and collected using a UV detection system (UA-6, Teledyne ISCO) coupled to a gradient fractionator (Foxy R1, Teledyne ISCO). Polysomes were washed in 100 kDa molecular weight cutoff (MWCO) spin concentrators to remove sucrose, concentrate ribosomes and replace the solution with Storage Buffer (Buffer A supplemented with 10 mM L-ornithine). The concentration of ribosomes was inferred by measuring the absorbance of the sample at 260 nm (1 A260 = 60 μg/ml or 24 nM) with a NanoDrop One (ThermoFisher). For analytical purposes, 13.2 pmol of ribosomes with an excess of 10 nmol of rnaseH oligonucleotide were incubated for 1 hour at 25°C with 7.5 U of RNase H or without it (RNase H–control). The sample for cryo-EM grid preparation was treated with 75 U of RNase H (New England Biolabs) per 250 pmol of ribosomes for one hour at 25°C in the presence of 5 nmol of oligonucleotide 33. The monosomes obtained after RNAse H treatment were isolated by sucrose gradient ultracentrifugation as described above. The sample was flash frozen in liquid nitrogen and stored at −80 °C.
Cryo-EM grid preparation
Frozen SpeFL–70S complex was thawed and diluted in Storage Buffer to yield a final concentration of 120 nM. Quantifoil carbon grids (QF-R2/2-Cu) were coated with a thin carbon layer prepared using an Edwards Vacuum Carbon Coater E306. Grids were glow discharged for 30 seconds at 2 mA before application of 4 μL of the SpeFL–70S complex. After blotting for 2 seconds and waiting for 30 seconds, grids were plunge-frozen in liquid ethane using a Vitrobot (FEI) set to 4°C and 100% humidity.
Cryo-EM data acquisition and processing
Grids were imaged using two 300-keV Titan Krios (FEI) equipped with a K2 Summit direct electron detector (Gatan) at ESRF (France) and at the Diamond Light Source (eBIC, UK) producing the SpeFL-ESRF and SpeFL-DLS datasets, respectively. Images were recorded with EPU in counting mode with a magnified pixel size of 1.067 Å (Extended Data Table 2). 30 frames were collected to have a total accumulated dose of 30 electrons per Å2. Data were processed in Relion 2.141, Relion 3.042 and Cryosparc 0.643 according to the scheme presented in Extended Data Figure 6. Briefly, MotionCor244 was used for movie alignment, Gctf45 for CTF estimation and either Relion 2.1 or Cryosparc for 2D classification of the particles obtained by automated picking in Relion 2.1. The csparc2star.py and star.py scripts46 were used for exporting particles selected in Cryosparc back into Relion. 3D classification was performed in Relion 2.1 in three steps: (i) unsupervised classification with 4 times downsized particles, (ii) focused classification on all 3 tRNA sites with background subtraction and 3 times downsized particles, and (iii) focused classification on the P-site tRNA with background subtraction and 2 times downsized particles. Classes containing a single P-tRNA or both P- and E-site tRNAs were combined after ensuring that each class contained a peptide with the same conformation in the ribosomal exit tunnel when refined individually. Movie refinement and particle polishing were first performed with Relion 2.1. Refined particle coordinates were then used to re-extract particles in Relion 3.0 in order to perform per particle CTF and beam tilt refinement, followed by Bayesian polishing.
Model building and refinement
An initial model of the SpeFL–70S complex was obtained by placing the coordinates for an E. coli 70S ribosome (PDB 4U 27)47 into the cryo-EM density map with Situs48, using the colores routine for the initial fit at 15 Å and the collage routine for fitting subdomains of the ribosome (30S body, 30S head, 30S spur, 50S body and L1 stalk) as independent rigid bodies at progressively higher resolutions until reaching the map resolution. The pixel size was optimized by generating post-processed maps with different pixel sizes in Relion 2.1 and assessing the map-to-model correlation after real space refinement in Phenix49 with the initial model. A model for the SpeFL peptide was built manually with Coot50 and refined through multiple rounds of real-space refinement in Phenix and manual rebuilding in Coot. The model was validated with MolProbity51. Automatic map sharpening was performed in Phenix using a refined model from which L-ornithine and surrounding solvent molecules had been removed (Extended Data Fig. 7 and 9). The resulting map was used to prepare all figures except Fig. 2a, for which a post-processed map from Relion was used (sharpening B-factor of – 10).
Figure preparation
Figures showing cryo-EM density or atomic models were prepared using Chimera52, Chimera X53 or Pymol Molecular Graphics Systems (version 1.7.4 Schrödinger)54.
Data availability
The speFL sequence was annotated in GenBank with the primary accession code [To be provided]. The SpeFL-DLS and SpeFL-ESRF structures were deposited in the RCSB PDB with accession codes [To be provided] and [To be provided], and cryo-EM maps were deposited in the EMDB with accession codes [To be provided] and [To be provided].
Acknowledgements
A.H.V. is funded by a doctoral grant from the French Ministère de l’Enseignement Supérieur et de la Recherche. C.A.I., B.S. and G.S. have received funding for this project from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant Agreement No. 724040). C.A.I. is an EMBO YIP and has received funding from the Fondation Bettencourt-Schueller. I.C-M. is funded by Inserm and A.C.S. is funded by a joint doctoral grant from Inserm and the Aquitaine Regional Council. We acknowledge Diamond for access and support of the Cryo-EM facilities at the UK national electron bio-imaging centre (eBIC), (proposal EM 19716-1), funded by the Wellcome Trust, MRC and BBSRC, and Sarah Neumann, Andrew Howe and James Gilchrist for assistance. We also acknowledge the European Synchrotron Radiation Facility for provision of microscope time on CM01 and we thank Michael Hons for his assistance. This work has been supported by iNEXT, grant number 3901, funded by the Horizon 2020 program of the European Union. We thank Isabelle Iost for help with gradient fractionation, Yaser Hashem, Armel Bezault and Marion Decossas for help with grid preparation and screening, Rémi Fronzes for access to the GPU cluster, Chiara Rapisarda for help with cryo-EM data processing and Nora Vazquez-Laslop for providing the E. coli TB1 strain and pErmZα plasmid for the in vivo lacZ assay.
REFERENCES




