

Abstract
The laminin N terminus (LaNt) proteins are a family of laminin and netrin related proteins derived by alternative splicing from laminin encoding genes. There are few reports to date on LaNt protein function. However, one family member, LaNt α31, has been shown to regulate epidermal keratinocyte migration and adhesion, although the mechanism for these effects is unknown. Based on its protein architecture, we predicted that LaNt α31 would influence laminin organisation. To test this prediction, we induced expression of GFP tagged LaNt α31 via adenoviral delivery into human epidermal and corneal keratinocytes. Consistent with the LaNt/laminin interaction hypothesis, LaNt α31 GFP codistributed with laminin β3 in the extracellular matrix and formed a complex together as indicated by co-immunoprecipitation, while live cell assays revealed the proteins to be deposited together during new matrix synthesis. Moreover, the induced expression of LaNt α31 led to changes to laminin α3 organisation into tight clusters compared with wide arcs in control cells. These changes were associated with early maturation of hemidesmosomes and mislocalization of focal adhesion complexes. At the cellular level, epithelial cells expressing LaNt α31 GFP displayed decreased scratch closure and single cell motility rates and increased cell spread area. These differences were rescued through the provision of a pre-formed cell-derived matrix. Together these data identify a new mechanism that influences the early stages of laminin matrix assembly and adds the LaNt proteins as new players in defining cell-to-matrix adhesion and migration characteristics.
Grant support This work was supported by funding from the Biotechnology and Biological Sciences Research Council (BB/L020513/1, KH), Fight For Sight (New lecturers’ award, KH), British Skin Foundation (PhD studentship, KH) and National Institute of Arthritis Musculoskeletal and Skin (K99/R00 1K99AR060242, KH).
Highlights
LaNt α31 is a new mediator of laminin-dependent processes.
LaNt α31 codistributes and is deposited with laminin 332 in the extracellular matrix of epithelial cells.
Increased LaNt α31 expression leads to changes in laminin 332 organisation into tight clusters compared with broad tracks.
Epithelial cells expressing increased LaNt α31 exhibit early hemidesmosome maturation and mislocalization of focal adhesion complexes.
LaNt α31 overexpression decreases scratch wound closure rates and single cell migration rates of epithelial keratinocytes which is rescues through the provision of a preformed matrix.
HighlightsThis manuscript identifies a new way in which laminin deposition and organisation is controlled, via LaNt α31. Specifically, these data demonstrate that this relatively unstudied laminin-gene splice isoform, induces the formation of laminin 332 clusters during times of new matrix synthesis, leading to changes in cell-matrix adhesion and behaviour.
- Laminin
- basement membrane
- migration
- keratinocyte
- hemidesmosome
Introduction
The Laminin N-terminus (LaNt) proteins are a relatively unstudied family of proteins generated by alternative splicing from the 5’ end of laminin (LM) encoding genes (Hamill et al., 2009b). To date, four members of the LaNt family have been identified, two each from the LMα3 (LAMA3) and LMα5 (LAMA5) encoding genes (Hamill et al., 2009b). LMs are critical structural proteins essential for the assembly and function of specialised regions of the extracellular matrix (ECM), termed basement membranes (BMs). BMs provide cell to matrix interaction sites for epithelial, endothelial, nerve, and muscle cells in intact tissue and are the substrate for cell interaction during tissue remodelling (for extensive reviews see (Hamill et al., 2009a; Aumailley, 2013; Hohenester and Yurchenco, 2013)).
Each LM is a heterotrimer consisting of three distinct subunits termed α, β and γ, which assemble via a laminin coiled-coil domain located toward the carboxyl terminus of each subunit (Hamill et al., 2009a; Aumailley, 2013; Hohenester and Yurchenco, 2013). In contrast to the LMs, LaNts are smaller, and do not contain the trimerisation domain, and are therefore unlikely to play the equivalent structural roles as their larger relatives (Hamill et al., 2009b). However, knockdown studies in epidermal keratinocytes have demonstrated a role for at least one family member, LaNt α31, in the regulation of keratinocyte adhesion and migration, although the mechanisms behind these effects are yet to be identified (Hamill et al.,HYPERLINK \l “bookmark13” 2009b).
LaNt α31 is generated by splice site read-through of the LAMA3 exon 9 donor splice site. This transcript, therefore, shares a promoter with that of LAMA3B and at the protein level consists of domains located toward the amino terminus of this LMα3b subunit. Working from the amino terminus, LaNt α31 therefore consists of a secretion signal sequence, a globular laminin N-terminal domain (LN domain), a short stretch of laminin-type epidermal growth factor-like repeats (LE domains) and a 54 amino acid unique carboxyl terminus with no known conserved domain structure (Fig 1A)(Hamill et al., 2009b). LE repeats adjacent to LN domains are required to maintain the appropriate folding of the LN domain and, in recombinant protein production, LE repeats are required for protein stability (Garbe et al., 2002; Kalkhof et al., 2008; Hussain et al., 2011). Therefore, in the case of LaNt α31, the LN domain is likely to be the key functional domain with the remainder of the protein required for protein folding and stability.

(A) Diagrammatic representation of LaNt α31 protein. LN – laminin N-terminal domain, LE – laminin-type epidermal growth factor-like repeats. Magenta box; unique C-terminus derived from exon 9e. (B) Western immunoblot of total cell lysates generated from cells transduced with either GFP or LaNt α31 GFP then probed with antibodies against LaNt α31 (upper panels) or GFP (middle panels). Numbers beneath lanes indicate expression level of endogenous LaNt α31 in each lane as determined by densitometry, normalised to total protein content. Numbers to left of blots in B and E indicate location molecular weight marker bands in kDa. In (C) HaCaT cells were either left untreated or transduced with GFP (+GFP), or LaNt α31 (+LaNt α31) then plated overnight at confluence and a scratch wound introduced 16 h later. (C) Representative images from immediately after scratching (T0 upper panels) and after 16 h of recovery (T16 lower panels), yellow lines delineate wound margins. (D) Scratch area closure measured 16 h after wounding plotted as percentage of the initial wound area. In (E) total cell lysates from HaCaT or hTCEpi cells were processed for fluorescent western blotting with mouse monoclonal and with rabbit polyclonal antibodies against LaNt α31 each detected in a different channel. Ponceau S stain refers to both halves of the blot. (F) and (G) as for (C) and (D) but using hTCEpi cells. (H), (I) and (J) Non-transduced hTCEpi or +GFP, +LaNt α31, transduced with LMβ3 mCherry (+LMβ3) were plated overnight onto tissue culture plastic at low density and migration paths of individual cells tracked over a 2 h period. (H) Vector diagrams showing representative paths of 10 individual cells with each colour representing a single cell. (I) Migration speed measured as total distance migrated over time. (J) Migration processivity; measured as maximum linear distance migrated over total distance migrated. Measurements in (D, G, I and J) are means from 3-6 independent biological replicates; 2-3 technical replicates per scratch or 20-40 cells per low density assay. * denotes significant differences compared to all controls with p<0.05 as determined by one way ANOVA followed by Tukey’s HSD (D, G) or Bonferroni post hoc analyses (I). Scale bar in C and F 100μm.
LN domains have been well established as the sites through which LM to LM interactions occur, and are therefore integral to the assembly of LM polymers or higher-order networks in BMs (Yurchenco et al., 1985; Hussain et al., 2011; Purvis and Hohenester, 2012; Hohenester and Yurchenco, 2013). The importance of LN domain interactions is exemplified by human genetic disorders such as Pierson syndrome and merosin-deficient congenital muscular atrophy (MDC1A), where missense mutations within the LN domain have been demonstrated to lead to loss of tissue integrity (Yurchenco et al., 2004; Patton et al., 2008; Matejas et al., 2010; Carafoli et al., 2012). Interestingly, the domain architecture of the LaNts are shared with a distinct family of ECM proteins; the netrins (Livesey, 1999; Koch et al., 2000). Recently one member of the netrin family, netrin-4, has been shown to disrupt laminin network assembly in vitro and in vivo with implications for axon outgrowth and angiogenesis (Oliver et al., 2016; Reuten et al., 2016). These findings raise the possibility that LaNt proteins may play a similar role to netrin-4, albeit being produced by alternative splicing from LM genes rather than from distinct genetic origins and with the LaNts providing an α LN domain compared with a β LN domain of netrin-4 (Livesey, 1999; Koch et al., 2000).
In this study, we have tested this hypothesis through inducing expression of fluorescently tagged LaNt α31 in epithelial cells and studying its dynamics relative to LMs as well as its effects on LM organisation, upon hemidesmosome and focal contact cell-to-LM adhesive devices, and the downstream cell behavioural outcomes in terms of cell adhesion and migration characteristics.
Results
Increased LaNt α31 expression leads to decreased scratch closure and single cell migration rates
In previous studies, LaNt α31 expression level was demonstrated to increase following the introduction of a scratch wound into human epidermal keratinocyte cultures (HaCaT), and scratch closure rates decreased when the expression level of the protein was reduced via siRNA-mediated knockdown (Hamill et al., 2009b). Here, we wanted to investigate LaNt α31 protein function, location and dynamics in live cells and, therefore, generated an adenovirally-delivered expression construct consisting of full-length LaNt α31 with C-terminal eGFP tag driven by the CMV promoter (cells expressing this construct are hereafter referred to as, +LaNt α31) (Fig 1A, B). Based on previous data (Hamill et al., 2009b), we predicted that increased expression of LaNt α31 driven by this construct would lead to more rapid scratch wound closure in HaCaT cells. However, surprisingly, +LaNt α31 HaCaTs displayed significantly reduced closure rates compared with non-transduced cells or HaCaTs induced to express eGFP only (+GFP) (Fig 1C, D) (non-transduced HaCaT 98.9% closure after 16hrs SD= +/-1.7%, +GFP 97.3 +/- 5.5%, +LaNt α31 52.1 +/- 12.3%, p<0.05). Equivalent findings were obtained in the limbal-derived corneal epithelial cell line hTCEpi (Robertson et al., 2005), which also express LaNt α31 as determined using previously published rabbit polyclonal antibodies and newly produced mouse monoclonal antibodies (Fig 1E). Specifically, hTCEpi displayed dramatically reduced ability to close the scratch when LaNt α31 expression was induced (Fig 1F, G) (non-transduced hTCEPi 95.2% closure after 16hrs SD= +/- 8.31%, +GFP 77.5 +/- 39.0 %, +LaNt α31 40.6 +/- 15.5%, p<0.05).
Unlike HaCaT cells, hTCEpi move rapidly as single cells and therefore are a useful platform to investigate the mechanisms underlying the differences in scratch closure rates. In low density migration assays; the +LaNt α31 hTCEpi displayed large reductions in total cell migration speed compared with controls (Fig. 1H) (non-transduced 1.15 μm/min SD= ± 0.26, +GFP 1.05 ± 0.11 μm/min, +LaNt α31 0.46 ± 0.17 μm/min, p<0.05). However, there were no differences in processivity, the ratio of total distance migrated to linear distance, (Fig. 1J) indicating that the ability of hTCEpi cells to establish and maintain polarity is not specifically affected by increased LaNt α31 levels (non-transduced 0.41 SD= +/- 0.11, +GFP 0.38 +/- 0.08, +LaNt α31 0.40 +/-0.11). As a control for later experiments, we also analysed hTCEpi expressing LMβ3 with an N-terminal mCherry tag (+LMβ3)(Hopkinson et al., 2008), hTCEpi cells expressing this construct did not display any differences in migration speed nor processivity compared with non-transduced or +GFP hTCEpi (Fig 1H, I) (speed= 1.06 +/- 0.32 μm/min, processivity= 0.28 +/- 0.10, p>0.05).
LaNt α31 interacts with LM332 and modifies its organisation
Imaging of the LaNt α31 GFP fluorescence in live hTCEpi cells indicated a diffuse distribution of intracellular LaNt α31 GFP protein throughout the cell (Fig. 2A). However, total internal reflection microscopy (TIRF) revealed organised clusters of LaNt α31 GFP along the matrix apposed surface of the cells (Fig 2A). This LaNt α31 GFP signal remained present in ECM preparations after hyperosmotic removal of cellular material using ammonium hydroxide (Fig. 2B). The ECM LaNt α31 GFP signal appears very similar to that observed with antibody staining of HaCaT ECM, though interestingly very little endogenous LaNt α31 was observed by antibody staining of hTCEpi ECM (Fig 2C).

(A) hTCEpi cells transduced with LaNt α31 GFP adenovirus were plated for 16 h on glass-bottomed dishes then imaged using epifluorescence (left) and total internal reflection fluorescence (TIRF) microscopy (right). (B) LaNt α31 GFP expressing hTCEpi were plated onto glass coverslips and 16h later cellular material removed through ammonium hydroxide treatment then remaining ECM material imaged. (C) Non-transduced HaCaT (top panels) or hTCEpi (bottom panels) were plated overnight onto glass coverslips then cellular material removed through ammonium hydroxide treatment and remaining ECM processed for indirect immunofluorescence microscopy with antibodies against LaNt α31 (left panel, pseudocoloured green in merge image in right hand panels) and laminin β3 (middle panel, pseudocoloured magenta in merged image). Phase contrast images in the inset in B and C show the removal of cells. Scale bars 10μm.
TIRF images of hTCEpi doubly transduced with the LaNt α31 GFP and LMβ3 mCherry constructs revealed regions where co-distribution between the GFP and mCherry signals were present, however, not all the LaNt α31 clusters were at sites of the LMβ3 mCherry signal (Fig 3A). In ECM preparations, in contrast, LaNt α31 GFP signal was only observed codistributed with LMβ3 (Fig. 3B). These data suggest a subset of the LaNt α31 protein associates with LMβ3. To determine whether this co-distribution of signals indicates an ability to associate together, we performed an immunoprecipitation experiment using covalently conjugated anti-GFP beads, and lysates derived from hTCEpi transduced with either GFP or LaNt α31 GFP then probed with antibodies against LMβ3 (Fig. 3C). Consistent with complex formation, these experiments revealed LMβ3 to be precipitated along with LaNt α31 GFP but not when GFP alone is pulled-down (Fig. 3C).

(A) TIRF images of hTCEpi cells doubly transduced with LaNt α31 GFP (left panel, pseudocoloured green in merge) and laminin β3 mCherry (second panel, pseudocoloured magenta in merge) imaged 16 h after plating on glass-bottomed dishes. Yellow boxed region is shown at higher magnification in panels to the right. (B) ECM preparation of doubly transduced cells generated by ammonium hydroxide mediated removal of cells 16 h after plating, phase contrast image in inset shows removal of cellular material. (C) Total cell lysates from hTCEpi transduced with GFP (+GFP) or LaNt α31 GFP (+LaNt α31) were processed with anti-GFP antibody-conjugated beads. Input lysates equivalent to 8% of the total immunoprecipitation volume (left 2 lanes) and anti-GFP pull-down lanes (IP-anti-GFP, right two lanes) were immunoblotted (IB) with antibodies against LMβ3 (upper panel) or GFP (lower panel). (D) hTCEpi, +GFP or +LaNt α31 hTCEpi were seeded at low (left panels) or high (right panels) density on uncoated glass coverslips then processed for immunofluorescence microscopy with antibodies against laminin α3 after 5 h (left panels) or 24 h (right panels). (E and F) LMα3 cluster area was measured in untreated hTCEpi (WT), +GFP or +LaNt α31 cells plated at low (E) or high (F) density. N=2 independent biological replicates; 25 measurements per cell type per assay. * In (E) denotes significant differences between +LaNt α31 and both controls, p<0.05 determined by one way ANOVA followed by Tukey’s post-hoc test. Scale bars in A, B and D; 20 μm.
Due to the observed potential interaction and co-distribution of LaNt α31 with LMβ3 and the known roles of LN domains in LM network assembly, we next hypothesised that overexpression of LaNt α31 could influence the organisational status of LM332 deposited by hTCEpi cells. To assess this, we performed indirect immunofluorescence staining for the LMα3 subunit. In cells fixed 5 h after seeding, control hTCEpi had deposited arcs and contiguous lines of LMα3, however in comparison, the LMα3 deposited by+LaNt α31 cells were in tight, small clusters (Fig. 3D, quantified in Fig. 3E; average LMα3 cluster area hTCEpi 236 ± 5 pixel2, +GFP 234 ± 16 pixel2, +LaNt α31 80 ± 4 pixel2, p<0.05). However, when cells were plated at higher density and allowed 24 h to form their matrix there were no significant differences in LMα3 organisational status (Fig. 3D, E; hTCEpi 274 ± 5 pixel2, +GFP 280 ± 8 pixel2, +LaNt α31 254 ± 3 pixel2).
LaNt α31 co-distributes with laminin β3 during new matrix deposition
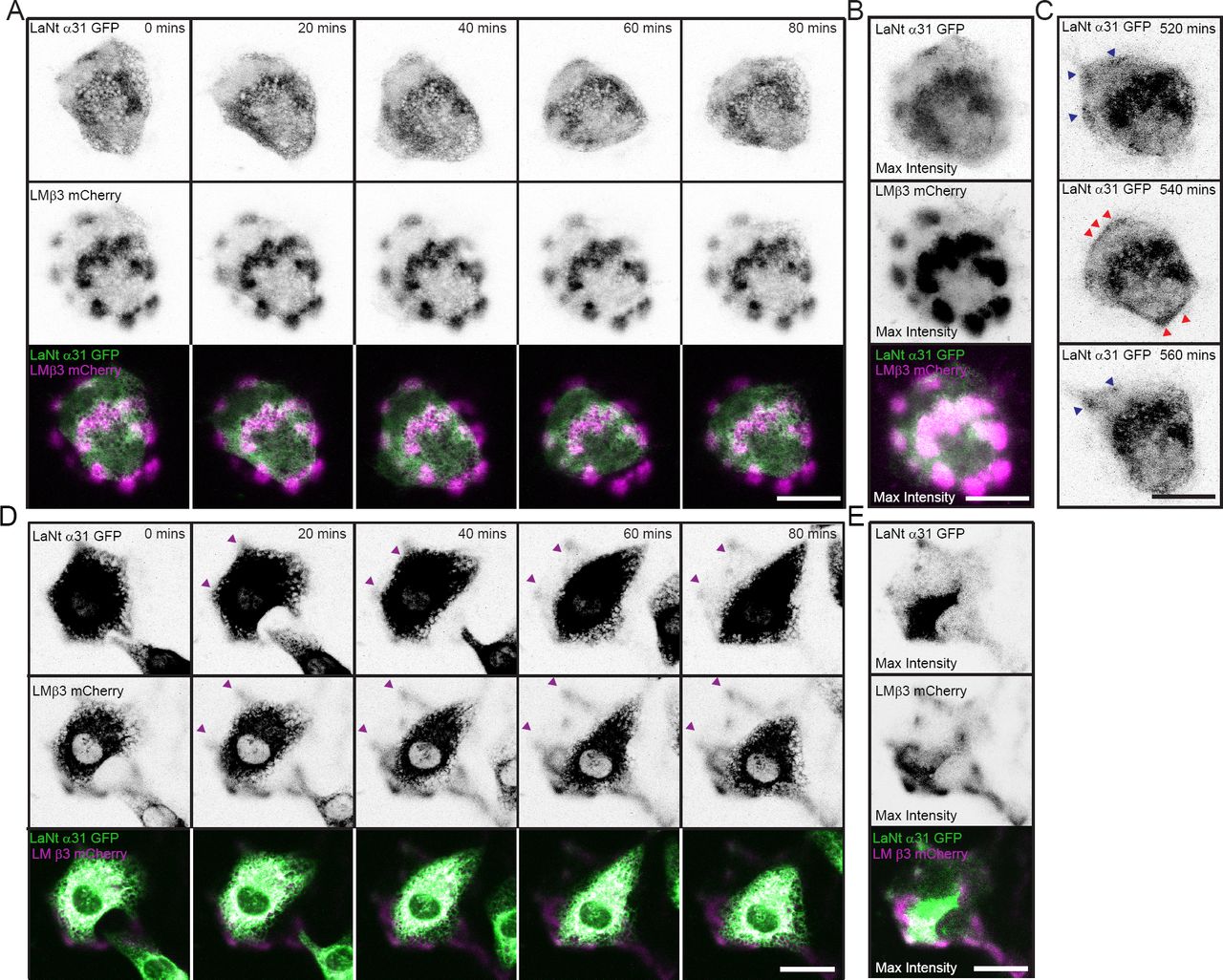
As the preceding data suggested an interaction between LaNt α31 and LMs, we next investigated the protein dynamics of LaNt α31 relative to LMβ3 in live cells. hTCEpi double transduced with LaNt α31 GFP and LMβ3 mCherry were plated for 16 h at low density on uncoated glass-bottomed dishes then imaged using confocal microscopy every 20 min over the next 12 h (Fig. 4A, supplemental movie 1). Throughout the time course of these experiments, rather than stable association with LMβ3, the LaNt α31 GFP rapidly formed and dissociated clusters at sites of LMβ3 deposits (Fig. 4A). Although individually transient, the LaNt α31 temporal enrichment at LMβ3 clusters is strongly apparent when a maximum intensity projection of each frame of the movie is generated (Fig. 4B). Closer analysis of these time-lapse movies also revealed frames where LaNt α31 GFP rather than being assembled into discrete clusters, formed an almost contiguous line of signal along cell peripheries (Fig. 4C). These were relatively short-lived structures and were followed by a return to clustered distribution (Fig. 4C).
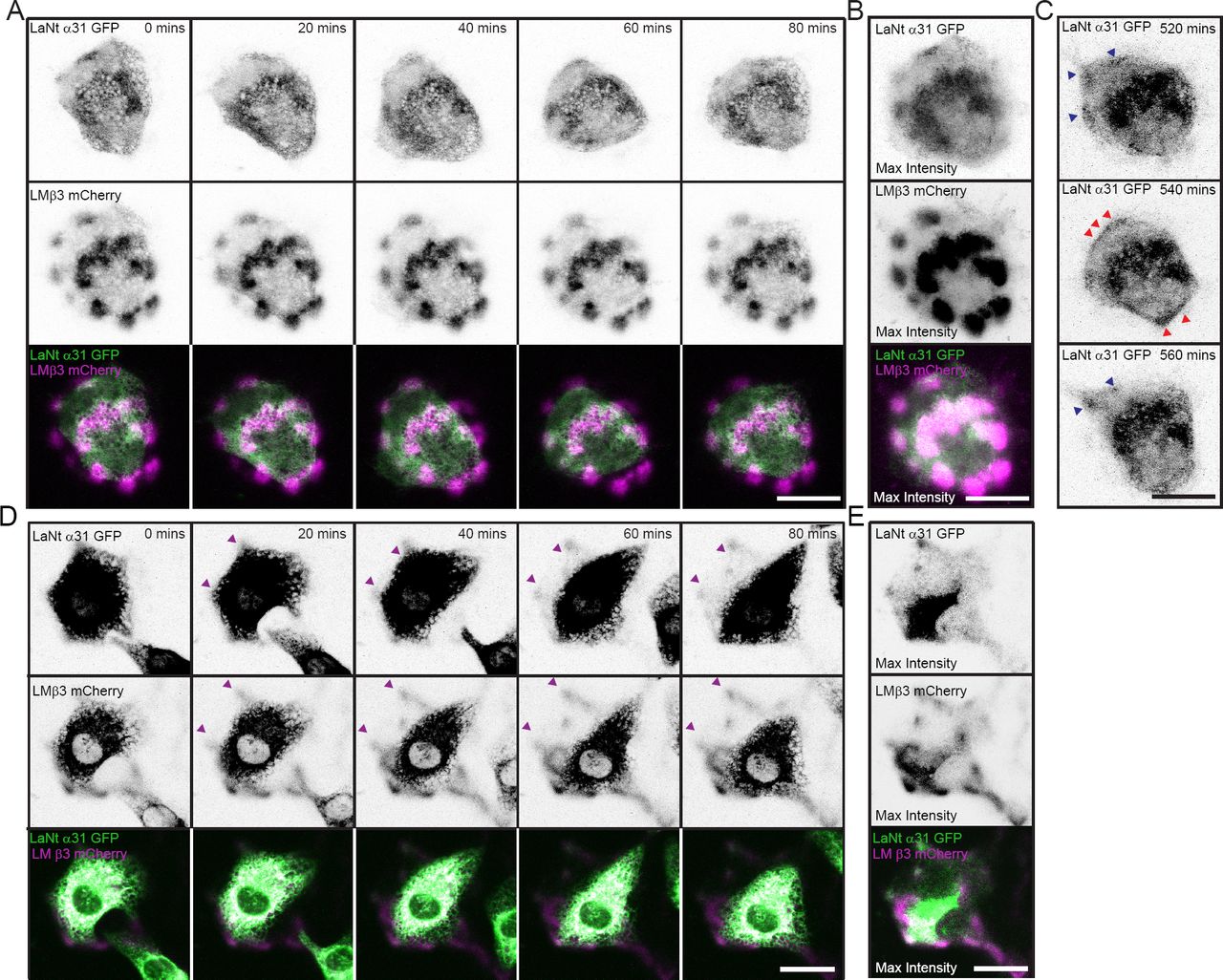
hTCEpi doubly transduced with LaNt α31 GFP and LMβ3 mCherry were plated overnight (A, B and C) or for 2h (D and E) on uncoated glass-bottomed dishes then imaged by confocal microscopy every 20 min over 16 h. (A and D) Representative stills from 0 to 80 min of LaNt α31 GFP (upper panels) and LMβ3 mCherry (middle panels), with signals inverted to aid visualisation. Bottom panel, merged images of LaNt α31 (pseudocoloured green) and LMβ3 (pseudocoloured magenta). (B and E) Maximum intensity projection of the entire 16 h time course images. (C) Three consecutive images where LaNt α31 localisation changes from clusters (blue arrowheads in 520 min and 560 min) to linear at cell margins (red arrowheads in 540 min). Scale bars 20 μm.
We also wanted to investigate the protein dynamics during new matrix synthesis. Therefore LaNt α31 GFP and LMβ3 mCherry co-expressing hTCEpi cells were imaged shortly after seeding onto uncoated glass-bottomed dishes (Fig. 4D, E supplemental movie 2). Over the first 3 h of this experiment, LMβ3 and LaNt α31 GFP were deposited in clusters that assembled toward cell peripheries with the two proteins displaying near identical patterns (Fig. 4D). These data are consistent with a role for LaNt α31 in early matrix assembly and the establishment of the LM clusters we observed in the fixed cell analyses.
LaNt α31 overexpressing motility phenotype is rescued through provision of a cell-derived matrix
The live deposition data and immunofluorescence analyses indicate a role for LaNt α31 in new matrix deposition. We, therefore, predicted that the cell migration defects might not occur on preformed matrixes. To test this prediction, cell-derived ECMs from untreated hTCEpi (hTCEpi ECM) and +LaNt α31 (+LaNt α31 ECM) were generated from cells plated for 24 h at high density. Onto these prepared matrices fresh hTCEpi or +LaNt α31 cells were plated, allowed to attach for 2 h, then single cell motility behaviour analysed (Fig. 5A, B and C). These analyses revealed that the migration speed defects of the +LaNt α31 cells’ phenotype are rescued by provision of an extensive cell-derived matrix, suggesting that +LaNt α31 cells migration defects are not an inherent defect to cell migration machinery but rather their ability to establish an ECM (Fig. 5A, B and H hTCEpi on hTCEpi ECM 1.16 ± 0.08 μm/min, +LaNt α31 on hTCEpi ECM 1.20 ± 0.05 μm/min,). Consistent with our LMα3 organisational analyses, the matrix deposited by high density +LaNt α31 cells was indistinguishable from non-transduced hTCEpi ECM in terms of supporting cell movement (Fig 5A, B and C hTCEpi on +LaNt α31 ECM 1.26 ± 0.05 μm/min, +LaNt α31 on +LaNt α31 ECM 1.14 ± 0.12 μm/min). Together with the scratch wound and low-density migration data, these findings indicate that the +LaNt α31 migration defects appear to be restricted to times when new matrix synthesis is required.

In (A), (B) and (C) hTCEpi or +LaNt α31 hTCEpi were plated overnight at high density and cell-derived matrixes prepared by ammonium hydroxide removal of cellular material. Onto prepared matrixes, fresh hTCEpi or +LaNt α31 hTCEpi were seeded at low density, allowed to adhere for 2 h then migration paths of individual cells tracked over a 2 h period. (A) Vector diagrams of 10 randomly selected cells (B) migration speed (C) migration processivity. In panels (D-I) 2D cell area and aspect ratio measured as width at widest point / length at longest point of untreated, +GFP, +LaNt α31 or +LMβ3 hTCEpi were seeded for 2 h onto uncoated tissue culture plastic (D-F), or untreated or +LaNt α31 hTCEpi were plated onto ECM derived from untreated of +LaNt α31 hTCEpi (G-I). Representative phase contrast images (D and G), with area plotted in (E and H), and aspect ratio in (F and I). Data are plotted as mean values from three independent biological replicates with 40-60 cells per type measured per assay. * In (B) denotes significant differences between +LaNt α31 and all controls, p<0.05, determined by one way ANOVA followed by Bonferroni post-hoc analysis.
Also consistent with a LaNt α31 role in cell-matrix interaction, during routine handling of +LaNt α31 cells, we noticed an apparent increase in cell spread area. To quantify this observation, the 2D area of live individual cells, plated at low density on uncoated tissue culture plastic were measured. +LaNt α31 cells displayed significantly increased 2D area compared with controls (Fig. 5D, E)(non-transduced 839 µm2 SD= +/- 41 µm2, GFP 943 +/- 33.09 µm2, LaNt 2249 +/- 107 µm2, LM 868 +/- 48 µm2, p<0.05). No significant differences were observed in length/width aspect ratio, again suggesting no polarity defects (Fig. 5F) (non-transduced 0.65 +/- 0.05, GFP 0.59 +/- 0.11, LaNt 0.67 +/- 0.08, LM 0.65 +/- 0.09). As above with the motility assays, provision of a preformed matrix rescued this increased cell spread area, further supporting a role for LaNt α31 that is specific to new matrix or new cell-matrix adhesions (Fig. 5G, H and I, +LaNt α31 on hTCEpi ECM 1303 ± 282 µm2 vs 1180 ± 278 µm2 hTCEpi on hTCEpi matrix; +LaNt α31 on +LaNt α31 matrix 1194 ± 298 µm2 vs 1266 ± 230 µm2 hTCEpi on +LaNt α31 ECM).
Increasing LaNt α31 expression leads to mislocalized focal contacts and mature hemidesmosomes
A likely explanation for the observed increased cell area phenotype and migration defects is differences in the cell-to-matrix attachment complexes assembled by the +LaNt α31 cells compared with controls. Epithelial cells assemble two major integrin-based cell to laminin adhesive complexes; focal adhesions (FA) and hemidesmosomes (HD) both of which contribute to migratory and adhesion characteristics (Carter et al., 1990; DiPersio et al., 1995; Baker et al., 1996; Tsuruta et al., 2011). To analyse the effect of induced LaNt α31 expression on FAs and HDs, we performed indirect immunofluorescence for the FA protein paxillin and the core HD protein β4 integrin 5 h after seeding (Turner et al., 1990; Litjens et al., 2006). These analyses revealed that while paxillin formed discrete puncta in control hTCEpi, in the +LaNt α31 cells paxillin was assembled into linear regions around cell margins (Fig. 6A). As expected, the distribution of β4 integrin mirrored that observed for LMα3, with swirls and arcs in control cells compared with tight clusters in the +LaNt α31 cells (Fig. 6A). These tight clusters are reminiscent of the “cat paw” staining pattern obtained from tightly adhered cell lines such as the 804G rat bladder epithelial line (Langhofer et al., 1993).

hTCEpi, +GFP or +LaNt α31 hTCEpi were seeded on uncoated glass coverslips and fixed after 5 h then processed for indirect immunofluorescence microscopy with antibodies against paxillin and β4 integrin (A) or Col XVII and β4 integrin (B). Numbers in (B) represent Pearson’s Correlation Coefficient (PCC) of signals obtained in Col XVII and β4 integrin channels. Scale bars 10 μm.
The HD-like structures assembled by epithelial cells in culture change over time from an initial α6β4 integrin/plectin containing complex to more mature complexes via the recruitment of collagen type XVII (Col XVII) and bullous pemphigoid antigen 1e (Hopkinson et al., 1998; Koster et al., 2003). We therefore next investigated if there were differences in HD complexes assembled between control and +LaNt α31 hTCEpi in short term, low-density cultures. Immunofluorescence analysis of the distribution of Col XVII in comparison to β4 integrin revealed higher levels of colocalisation in +LaNt α31 cells compared with a general lack of co-distribution in non-transduced hTCEpi or +GFP hTCEpi in cultures at this time point (Fig. 6B, Pearson’s correlation coefficient hTCEpi 0.13, +GFP 0.12, +LaNt α31 0.57), suggesting that the HD-like complexes are more mature at this time point in +LaNt α31 cells compared with controls.
Discussion
Taken together, the data presented here demonstrate that LaNt α31 influences LM organisation during times of new matrix deposition. Specifically, we have shown that LaNt α31 is deposited alongside LMβ3 and leads to clustering of the LM in the matrix. These LM organisational changes are associated with FA and HD changes including more rapid maturation of HD-like complexes, increased cell spreading and decreased corneal and epidermal epithelial cell migration. Together these findings implicate the LaNt protein family as new regulators of LM-dependent cellular processes.
The finding that increased expression of LaNt α31 leads to reduced scratch closure rates is somewhat surprising in the light of previous reports where knockdown cells also presented with the same phenotype (Hamill et al., 2009b). In both cases, however, the defect relates specifically to times of new matrix deposition and taken together, these data indicate that control of LaNt α31 levels could provide a new mechanism to regulate the switch from stably attached to a migratory, matrix depositing phenotype or vice versa. In other words LaNt α31, and by extension, the other LaNt family members may help to define the “Goldilocks” level of cell-matrix adhesion for the required cell behaviour.
The question remains open as to what is driving the differences in matrix organisation in LaNt α31 overexpressing cells. As has been elegantly demonstrated, LN-LN interactions, and therefore higher order network assembly, requires formation of a ternary node consisting of an α, a β and γ LN domain, where the α LN domain stabilises transient βγ interactions (Cheng et al., 1997; Hussain et al., 2011; Carafoli et al., 2012). Here, by introducing an extra α domain we would predict competition with the native LM α chains which would lead to destabilisation of the overall LM network, in a similar way to that reported for netrin-4, which contains a β LN domain, and introduction of which disrupts BMs (Staquicini et al., 2009; Reuten et al., 2016). However, our findings of a tighter, more restricted LM deposition pattern that supports maturation of HD complexes suggest the possibility that, at early time points, when LM concentration is low, the extra α chain LN domain in the matrix provides partial stabilisation of weak interactions that would not usually occur until LM concentrations increased (Fig 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In interpreting these data, it is also important to consider that in epithelial cells, which we selected due to their high levels of LAMA3 promoter activity and therefore LaNt α31 production, produce predominantly LM332 as their major LM heterotrimers, along with smaller amounts of LM511, 521 and 111 (Ferrigno et al., 1997; Qin and Kurpakus, 1998; Hasenson et al., 2005; Bystrom et al., 2006; Lu et al., 2006). LM332 contains only a single LN domain on the LMβ3 subunit, and LM332, therefore, cannot self-polymerise; therefore any LaNt α31 induced or stabilised LM interaction would still not lead to true polymer formation (Ljubimov et al., 1995; Cheng et al., 1997; Bystrom et al., 2006; Kabosova et al., 2007; Schlotzer-Schrehardt et al., 2007; Torricelli et al., 2013). The finding that the LM matrix from high-density +LaNt α31 cells is indistinguishable from control cells both in terms of LM organisation and cell behaviour suggest that the effects observed are restricted to when LM abundance is relatively low. Previous studies on LaNt transcripts indicate wide distribution of LaNt family members (Hamill et al., 2009b) suggesting that these relatively unstudied proteins may have widespread roles in multiple tissues and different developmental situations; pointing toward a role for the LaNt proteins in regulating any of the processes where BM remodelling is required. It will be interesting to determine LaNt effects in cell types which produce LMs with 2 or 3 LN domains where differences to LM networks assembled are likely to be long-lasting.
In addition to LN-LN domain interactions, cell-surface receptor-binding capabilities have been described for some LN domains (Garbe et al., 2002; Odenthal et al., 2004). In LMs these interactions are believed to combine with stronger LG domain interactions to ensure that the deposited LMs are locally enriched, thereby allowing initiation of the LM network assembly (Yurchenco et al., 1985; Hohenester and Yurchenco, 2013). Here, LaNt α31 to receptor binding could be playing a role; by locally enriching the LaNt concentration the LaNt α31 LN domain may help to nucleate or stabilise early or weak LM-LM interactions. Consistent with this, our live experiments demonstrate that LaNt α31 and LMβ3, as far as we can temporally resolve, are deposited or, at least assembled into visible deposits, together suggesting their initial assembly points are in close proximity rather than the two proteins being independently secreted and subsequently organised. The two mechanisms are not mutually exclusive. Determining whether LaNt α31 is exerting its effect through LN network nucleation or via a more general stabilisation of LN interactions, or a combination of both mechanisms, is a complicated question which will form an exciting future research direction.
Together, our data have identified a new player in cell-LM interaction and therefore in BM assembly. As the LaNt proteins are produced by alternative splicing from LM genes, control of the LaNt to LM ratio is represents a new layer of self-regulation of LM function that occurs at the post-transcriptional level. As BM remodelling is integral to numerous physiological and pathological situations such as wound repair, tissue morphogenesis, differentiation, angiogenesis, and in tumour cell invasion and metastasis, it will be interesting to determine how LaNts and this new LM self-regulation mechanism contributes to and is controlled within these diverse processes.
Materials and Methods
Antibodies and other reagents
Rabbit polyclonal antibodies against paxillin and mouse monoclonal antibodies against β4 integrin (clone M126) were from Abcam, (Cambridge, UK). Rabbit polyclonal antibodies against LMβ3 and mouse monoclonal antibodies against GFP (a mixture of clones 7.1 and 13.1) were from Thermo Scientific (Loughborough, UK). RG13, mouse monoclonal antibodies against laminin α3 (Gonzales et al., 1999) and J17, rabbit polyclonal antibodies against type XVII collagen (Hopkinson et al., 1992) were generous gifts from Jonathan Jones, Washington State University, WA. Fluorescein isothiocyanate, rhodamine and Cy5 conjugated goat anti-mouse and goat anti-rabbit secondary antibodies were obtained from Jackson Immunoresearch (West Grove, PA). Goat anti-mouse IRDye 800CW and goat anti-rabbit IRDye 680CW were obtained from LiCor BioSciences (Rugby, UK).
Mouse monoclonal antibody clone 3E11 was raised against the synthetic peptide VLPQRSHQANFGSV (GenWay Biotech, San Diego, CA) corresponding to human LaNt α31 residues 437-451, conjugated to keyhole limpet haemocyanin following the procedure described in (Langhofer et al., 1993) with help from the recombinant protein production core, Northwestern University, Chicago IL. Five days following the final boost, spleens were removed, and isolated splenocytes were fused with the myeloma cell line Sp2 for the production of hybridomas using standard techniques (Galfre and Milstein, 1981). Hybridoma cells producing antibodies against the LaNt α31-derived peptide were selected based on their ELISA reactivity to the unconjugated peptide and immunoblotting reactivity against a human epidermal keratinocyte (HaCaT) protein extract. Selected hybridoma cells were cloned twice by limited cell dilution.
Cell Culture
Telomerase-immortalized human corneal epithelial cells, hTCEpi cells (Robertson et al., 2005), were cultured at 37 °C with 5% CO2 in Keratinocyte-Serum Free Medium supplemented with bovine pituitary extract (0.05 mg/ml), human recombinant Epidermal Growth Factor (5 ng/ml)(Life Technologies, Carlsbad, CA) and 0.15 mM CaCl2 (Sigma-Aldrich, St Louis, MO). HaCaT spontaneously transformed human epidermal keratinocytes (Boukamp et al., 1988) were cultured using Dulbecco’s Modified Eagle Medium (DMEM, Sigma Aldrich) supplemented with 10% Fetal Calf Serum (LabTech, East Sussex, UK) and 4mM L-glutamine (Sigma-Aldrich).
SDS PAGE and immunoblotting
Cells were plated at 1 × 106 in 100 mm dishes (Greiner-BioOne, Stonehouse, Gloucestershire, UK) and lysed after 24 h using RIPA buffer (50 mM Tris-HCl pH=7.5, 1 mM EDTA, 150 mM, NaCl, 0.1% (w/v) Sodium deoxycholate) containing 50 μM phenylmethylsulfonyl fluoride (PMSF) and 50 μM N-ethylmaleimide, or Urea/SDS buffer (10 mM Tris-HCl pH=6.8, 6.7 M Urea, 35 mM SDS, 10% Glycerol and bromophenol blue) containing 50 μM phenylmethylsulfonyl fluoride (PMSF) and 50 μM N-ethylmaleimide (all Sigma). Before loading, lysates were sonicated, and 15% β-mercaptoethanol (final volume) added. Proteins were separated by SDS PAGE using 10% polyacrylamide gels (Biorad, Hercules, CA), transferred to a nitrocellulose membrane using a Biorad TurboBlot system (Biorad) and blocked at room temperature in Odyssey®TBS-Blocking Buffer (Li-Cor BioSciences, Nebraska, USA) for 1 h. The blocked membranes were probed overnight at 4 °C with primary antibodies diluted in blocking buffer, washed 3 × 5 min in TBS/0.1% Tween and probed for 1 h at room temperature with IRDye® 800CW and/or IRDye® 680CW conjugated secondary antibodies (Li-Cor) diluted in blocking buffer (1:20,000). Membranes were then washed for 3 × 5 min in TBS/0.1% Tween and imaged using an Odyssey® CLx 9120 Infrared Imaging System.
Immunoprecipitation
hTCEpi cells were plated at 1 × 106/plate in 100 mm plates and 24 h later transduced with GFP and LaNt α31 GFP adenovirus. 48 h later cells were extracted in 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40, 150 mM NaCl, 1 mM CaCl2 in 50 mM Tris-HCl, pH 7.5 containing a mixture of protease and phosphatase inhibitors (Sigma-Aldrich). Cell extracts were clarified by centrifugation and a 50% slurry of sepharose beads covalently conjugated with rabbit anti-GFP polyclonal antibodies (Abcam) added to the supernatant. The bead/supernatant mixture was incubated overnight at 4 °C, the beads were washed in lysis buffer, collected by centrifugation and boiled in SDS-PAGE sample buffer before immunoblotting as above.
Adenovirus production and cell transduction
Full-length LAMA3LN1 was PCR amplified from cDNA generated from cultured human keratinocytes and cloned into pCR2.1 (Life Technologies) and sequence verified by DNA sequencing (DNA Sequencing and Services, University of Dundee). The native translational stop codon was converted to an AgeI restriction enzyme site by site-directed mutagenesis following the manufacturer’s directions of QuikChange II XL mutagenesis kit (Agilent, Santa Clara, CA). The mutated LAMA3LN1 was then subcloned using KpnI and AgeI (New England Biolabs, Hitchin, UK) into pENTR4 (Life Technologies) with eGFP inserted into the BglII and KpnI sites of the multiple cloning site (a kind gift from Jonathan Jones, Washington State University, WA). LR recombination was used to transfer the LAMA3LN1-eGFP construct from pENTR to pAD-CMV/V5-DEST (Life Technologies) and adenoviral particles produced following the standard Gateway-adapted ViralPower adenoviral expression protocol (Life Technologies). Adenoviruses inducing CMV driven expression of LMβ3-mCherry (Hopkinson et al., 2008) and eGFP were kind gifts from Jonathan Jones, Washington State University, WA.
3 × 105 hTCEpi cells were seeded in 60 mm dishes (Greiner-Bio One) and transduced the day after seeding, with eGFP, LaNt α31-GFP and/or LMβ3-mCherry. All analyses of LaNt α31-GFP (+LaNt α31), LMβ3-mCherry (+LMβ3), GFP (+GFP) or untreated cells were conducted 48-72 h following transduction.
Scratch closure, cell migration assays, and cell morphology analyses
For scratch closure assays, hTCEpi or HaCaT cells were seeded at 2 × 105 cells/well in a 24 well-plate (Greiner-Bio One) and 16 h later were adenovirally transduced. 24 h after adenoviral transduction a scratch was introduced into the confluent monolayers using a 200 μl tip. Cell debris was removed with several PBS washes. Scratch closure was imaged using a Nikon TiE epifluorescence microscope with a 10X objective for 16 h. Scratch closure rates were determined by measuring the scratch area after 16 h using the freehand image tool in FIJI (National Institutes of Health, Bethesda, MA)
For cell morphology analyses and low-density migration assays, cells were seeded at 2.5 × 104 cells/well onto uncoated 24 well plates (Greiner-Bio One). Where cell-derived preformed matrixes were required, non-transduced hTCEpi or +LaNt α31 hTCEpi were plated at 1.8 × 105 cells/well overnight on a 24 well plate, after which cellular material was removed through exposure to 0.18% NH4 OH for 5 min followed by extensive PBS washes (Langhofer et al., 1993). Fresh untreated or +LaNt α31 hTCEpi were then seeded on top of the prepared matrixes for 2 h before analysis. For morphology, 20X phase contrast images were acquired on a Nikon TiE microscope (Nikon, Surrey, UK). Images of individual cells were analysed using FIJI software (NIH). Cell perimeters were manually traced to define cell area; cell length was measured as the longest linear axis, with width measured at widest point at right angles to the length measurement. The aspect ratio was defined as the ratio of width to length. For low-density migration assays, cells were imaged every 2 min over a 2 h period, using a 20X objective on a Nikon TiE fluorescent microscope, then individual cells tracked using the MTrackJ plugin of the Fiji32-ImageJ (NIH). Speed (total distance travelled/time) and processivity (total distance/linear distance) were calculated for each cell.
Immunofluorescence microscopy
1 × 105 cells were seeded either for 5 h or overnight on uncoated glass coverslips then fixed and extracted in ice-cold methanol or ethanol for 4 min then air dried, or fixed in 3.7% formaldehyde (Sigma) for 5 min and extracted in 0.05% Triton X-100 in PBS for 7 min (Sigma Aldrich). For ECM analysis, cellular material was removed by ammonium hydroxide treatment as described above prior to fixation. Primary antibodies were diluted in PBS with 20% normal goat serum (Jackson labs) and incubated at 37 °C for 2 h; coverslips were then washed extensively with PBS before probing for 1 h at 37 °C with FITC, Rhodamine or Cy5 conjugated secondary antibodies diluted in PBS. Coverslips were washed in PBS then mounted with polyvinyl alcohol mounting medium with DABCO (Sigma Aldrich). Images were obtained using a Zeiss LSM510 confocal microscope or Zeiss ApoTome.2 fluorescence microscope (Zeiss, Cambridge, UK).
LMα3 staining area was measured for 50 regions of staining per cell treatment using the freehand selection tool on FIJI (NIH). Pearson’s correlation coefficients were calculated using the Coloc 2 plugin on FIJI (NIH) for all pixels above channel thresholds in both acquired channels.
Live protein imaging
For TIRF and new matrix deposition assays, 1 × 105 hTCEpi cells, doubly transduced with LaNt α31 GFP and LMβ3 mCherry adenoviruses were seeded onto uncoated 35 mm glass-bottomed dishes (MatTek Corporation, Ashland, MA) then imaged either 16 h, or 2 h after seeding for new matrix deposition experiments using a Zeiss 510 Multiphoton 2 confocal microscope or Zeiss LSM880 confocal laser scanning microscope with TIRF capability. For preformed matrix studies, 5 × 105 singly transduced LMβ3 mCherry hTCEpi were seeded for 24 h, then ECM extracts prepared as above, fresh LaNt α31 GFP expressing cells were then plated onto the preformed matrix for 2 h prior to imaging. In live experiments, images were acquired using a 63X objective every 20 min over 16 h. Images were processed using FIJI.
Statistical analyses
Quantification of experiments was derived from a minimum of 3 independent biological replicates. One-way ANOVA and Tukey’s HSD or Bonferroni post hoc analysis were used to analyse and interpret the data using SPSS software (IBM, Hampshire, UK). Differences were deemed significant where p<0.05.
Author contributions
VI, LT, VB, and KH designed, performed and analysed experiments, prepared figures, and wrote the manuscript.
Additional information
The authors declare no competing financial interest.
Supplemental Movie 1
hTCEpi double transduced with LaNt α31 GFP and LMβ3 mCherry were plated overnight on uncoated glass-bottomed dishes and imaged by confocal microscopy every 20 min. Left panel LaNt α31, middle panel LMβ3 with signals inverted, right panel, merged LaNt α31 GFP (pseudocoloured green) and LMβ3 mCherry (pseudocoloured magenta).
Supplemental Movie 2
hTCEpi doubly transduced with LaNt α31 GFP and LMβ3 mCherry were plated overnight in a plastic cloning ring on an uncoated glass-bottomed dish and imaged by confocal microscopy every 10 min for 16 h. Left panel LaNt α31, middle panel LMβ3 with signals inverted, right panel, merged LaNt α31 GFP (pseudocoloured green) and LMβ3 mCherry (pseudocoloured magenta).
Acknowledgements
This work was supported by funding from the Biotechnology and Biological Sciences Research Council (BB/L020513/1), Fight For Sight (New lecturers’ award), British Skin Foundation (PhD studentship) and National Institute of Arthritis Musculoskeletal and Skin (K99/R00 1K99AR060242, KH). The authors would like to acknowledge Jonathan Jones and Susan Hopkinson (Washington State University) and Robert Lavker (Northwestern University) for generous gifts of reagents and cells. In addition technical help was greatly appreciated from Izolda Popova of the monoclonal antibody production facility and Alex Yemelyanov at the DNA/RNA delivery core at Northwestern University, and David Mason, Marco Marcello, Violaine See and Joanna Majchrowska at the Centre for Cell Imaging at the University of Liverpool with the TIRF microscope purchase supported the BBSRC (BB/M012441/1).
Abbreviations
- LM
- laminin
- hTCEpi
- human telomerase reverse transcriptase immortalised corneal epithelial cells
- LN
- laminin N-terminal domain
- LaNt
- laminin N-terminus protein
- HD
- hemidesmosome
- FA
- focal adhesion
- HaCaT
- human adult low calcium high temperature epidermal keratinocytes
- ECM
- extracellular matrix
- BM
- basement membrane
- eGFP
- enhanced green fluorescent protein
- IP
- immunoprecipitation
- IB
- immunoblot
- LE
- laminin-type epidermal growth factor-like domain
- LG
- laminin globular domain
- TIRF
- total internal reflection microscopy
References




