

Abstract
Alu repeats contribute to lineage specific novelties in conserved transcriptional regulatory networks. We report for the first time the origin of a multi-miRNA human specific sponge through exaptation of 23 Alu repeats that forms a novel principal isoform of CYP20A1 gene with a 9kb 3’UTR. This 3’UTR, confirmed by RACE, is an outlier in terms of its length, with expression in multiple cell lines including brain as evidenced from single nucleus RNA-seq data of ∼16000 human cortical neurons. It has diverged from its parent gene through exon skipping into a novel lncRNA. Its uniqueness in humans was validated by its presence in rosehip neurons and absence in closely related primate species through RNA-seq datasets. Strikingly, prediction by miRanda revealed ∼4700 MREs for ∼1000 different miRNAs, majorly in Alu repeats, in this 3’UTR. Permutations on 1000 random sets suggest their creation is non-random and post Alu exaptation. We hypothesise this lncRNA is an miRNA sponge as it has cytosolic localization and harbors ≥10 MREs for 140 miRNAs (threshold > −25kcal/mol). Under experimental conditions where CYP20A1 displays differential expression, we probed the expression of the miRNAs that map to this 3’UTR and their cognate targets through small RNA and mRNA-seq, respectively. We observed correlated expression of our lncRNA and a set of 380 genes with downregulation in heat shock and upregulation in HIV1-Tat treatment in primary neurons. GO analyses suggest the involvement of this sponge lncRNA in modulation of processes linked to neuronal development and hemostasis pathways.
Introduction
Transposable elements (TEs), comprising half of the human genome, fine-tune conserved gene regulatory networks in a lineage specific manner (Lander et al. 2001; Chen et al. 2017; Trizzino et al. 2017; Wang et al. 2017). Depending upon the genomic context, TEs contribute to gene expression divergence through large scale transcriptional rewiring (Lynch et al. 2011; Rebollo et al. 2012; Trizzino et al. 2017). These could be through providing non-canonical TFBS (Polak and Domany 2006), epigenetic modifications (Xie et al. 2009; Lynch et al. 2011; Bakshi et al. 2016; Tristán-Flores et al. 2018)), alternate splice isoforms (Sorek et al. 2002; Payer et al. 2019) and differential mRNA stability (An et al. 2004). Primate specific Alu retrotransposons, which occupy 11% of the human genome, are major players in this process (Lander et al. 2001; Grover et al. 2005). Alus can bring about structural variations (Payer et al. 2017) via indels and exhibit a non-random, gene-biased distribution (Lander et al. 2001; Grover et al. 2003; Versteeg et al. 2003; Grover et al. 2004; Xing et al. 2009). Alus harbour cryptic splice sites which potentiate their inclusion into splice isoforms, some of which are ubiquitously expressed. Alu derived exons exhibit lineage specificity, high transcript inclusion levels as well as have much higher rates of evolution (Lin et al. 2008; Sorek 2009; Shen et al. 2011). Exonization is more frequent in genes that have arisen de novo in primates, with a strong preference for 3’UTRs (Toll-Riera et al. 2009; Mandal et al. 2013). ∼14% of the human transcriptome contains exonised Alus, predominantly in the principal isoforms. These provide substrate for different regulatory events through gain of poly (A) sites (Roy-Engel et al. 2005; Lee et al. 2008), formation of specific secondary structures (Sobczak and Krzyzosiak 2002), AU-rich elements and miRNA recognition elements (MREs) leading to alternative polyadenylation, mRNA decay or interfering with translation (An et al. 2004; Häsler and Strub 2006; Häsler et al. 2007).
Among the TEs, Alu contains the maximum number of MREs (Spengler et al. 2014). A few cases of their involvement in modulating gene expression as well as modifying existing miRNA regulatory networks in a lineage specific manner have been reported. For example, miR-1285-1 is processed from an Alu and predominantly targets exonized Alus (Spengler et al. 2014). In another instance, 3’UTRs of MDM2 and MDM4 harbouring Alu-MREs for primate specific miR-661, have been shown to provide an additional layer of regulation in the conserved p53 pathway in certain primate species (Hoffman et al. 2014). We have also shown that Alu-MREs can alter transcript isoform dynamics during heat shock response and some of these sites are still evolving in humans (Pandey et al. 2016). Although these are majorly non-canonical, Alu-MREs can be responsive in conditions where miRNA levels or targeting efficiency is altered. For example, in patient derived cells from two different cancerous states, it has been shown that elevated levels of Pol III transcribed, free Alu RNA can sequester miR-566 which correlates with disease progression (Di Ruocco et al. 2018).
Exonized Alus can increase the regulatory possibilities for a transcript by engaging antisense as well as A-to-I RNA editing in a spatio-temporal manner. Since 3’UTR is the hotspot for Alu exonization, miRNAs would be the most common antisense encountered. The transcript isoform dynamics i.e., the relative proportion of exonized versus non-exonized isoforms, depends on the levels of editing as well as antisense RNA expression including the miRNAs and has been shown to be relevant in conditions such as viral and heat shock response (Mandal et al. 2013; Pandey et al. 2016). From our earlier study on 3177 Alu-exonized genes, we had observed the co-occurrence of cis Alu antisense and A-to-I editing at the level of single Alu exons in 319 genes. Enrichment analysis revealed that genes related to apoptosis and lysosomal processes were overrepresented in this set (Mandal et al. 2013). While mapping these 319 genes onto the apoptosis pathway, a serendipitous observation led us to an orphan gene CYP20A1 that has acquired an unusually long 3’UTR due to exaptation of 23 different Alus and contains target sites for ∼1000 different miRNAs. We propose it functions as an Alu-derived, endogenous multi-miRNA sponge to orchestrate systemic changes in associated miRNA regulatory networks.
Results
CYP20A1 - a unique 3’UTR with Alu-driven divergence
Apoptosis entails a cascade of sequential events at the molecular level. We performed an extensive, systematic manual curation and text mining to ascertain where in the apoptotic pathway the list of 319 genes mapped (supplementary information). 91 out of 319 genes mapped to apoptosis with 75% non-randomly clustered around three discrete hubs: cell cycle-DNA damage response (p53 hub; 31 genes), mitochondrial events (mito hub; 22 genes) and proteostasis (ubi hub; 15 genes) (Supplementary Table S1).
Since majority of the events occur in the 3’UTRs of the exonized transcripts which can also modulate miRNA regulatory networks in a lineage specific manner (Hoffman et al. 2014; Pandey et al. 2016), we further focussed on looking at phylogenetic novelties in the 3’UTRs of these genes. Interestingly, we identified a novel principal isoform of orphan CYP20A1 gene with a 8.93kb long 3’UTR, 65% of which is contributed by exonization of 23 Alus (Figure 1a). The orthologs of this 3’UTR in mouse, rat and zebrafish are extremely short, well within 1kb and there is only a sparse presence of two B1 SINEs, one simple repeat and one low complexity repeat in mouse whereas the zebrafish 3’UTR lacks repeats altogether. Amongst the eight transcripts of human CYP20A1 annotated in NCBI, experimental evidence is available only for the longest isoform (NM_177538). This 10.94kb RNA not only appears to be unique to humans, but also its 3’UTR is an outlier in terms of size and occupies the 85th position in the genome-wide length distribution (Figure 1b). Such extended stretches of UTR are extremely rare as less than 3% of Alu-exonized genes have 3’UTRs greater than 6kb (Supplementary Figure S1). 3’UTR length also does not seem to correlate with the density of exonized Alus (r= 0.25) and compared to the genomic average of exonization events of 5.42 per UTR, CYP20A1 3’UTR is clearly an outlier with 23 Alus. The longest annotated CYP20A1 transcripts for mouse (NM_030013.3), rat (NM_199401.1) and zebrafish (NM_213332.2) were 2.27, 2.03 and 1.79kb, respectively. Even among the non-human primates, we did not find any annotated transcripts beyond 3kb from this locus. Taken together, these suggest that the event of CYP20A1 3’UTR extension is lineage specific. Interestingly, these Alus belong to different subfamilies which differ in their evolutionary ages, suggesting that their exaptation had happened gradually over a period of time.

a. UCSC track representing the four transcript isoforms of CYP20A1 with varying 3’UTR length. Only isoform 1 (NM_177538) contains the full length 8932bp 3’UTR. The RepeatMasker track shows this 3’UTR to be populated with 23 Alu repeats belonging to different subfamilies.
b. Genome wide 3’UTR length distribution shows CYP20A1 to be an outlier. Mean and median 3’UTR lengths were 1553bp and 1007bp, respectively.
c. Cladogram representing the CYP20A1 protein sequence divergence among different classes of vertebrates suggest that it has diverged minimally.
d. DNA level conservation analysis of 5’UTR and 3’UTR among 20 mammals reveals that 5’UTR is well conserved among all primate lineages, suggesting that divergence is unique to 3’UTR (also see Supplementary Figure S3).
Unlike its 3’UTR, the coding region of CYP20A1 is remarkably well conserved among vertebrates, both at the sequence level as well as in the length of the mature protein (Table 1). The chimpanzee, macaque and mouse CYP20A1 all code for the same 462-470aa protein as humans although their transcript orthologs range between 1-3kb. The protein sequence identity is greater than 60% between zebrafish and human. Multiple sequence alignment across vertebrates reveals a strong conservation at both the N and the C terminals (Supplementary Figure S2); of the first 100aa, 62 are completely conserved while 18 contain lineage specific substitutions with residues that contain strongly similar functional groups. This is also corroborated by the minimal evolutionary divergence across vertebrate CYP20A1 proteins (Figure 1c) and a strong purifying selection operative on the CDS (Ka/Ks ∼0.2 in mammals and <0.1 in non-mammalian vertebrates) (Table 1). The 3’UTR, however, becomes increasingly divergent as we move from the great apes to rhesus macaque and is maximum for mouse (the non-primate evolutionary out-group) and is maximally contributed by the Alu repeats, as evidenced through Jukes Cantor divergence.
CYP20A1 protein sequences among different vertebrate classes were compared using NCBI pBLAST. Drosophila, sea urchin and Arabidopsis were used as the evolutionary outgroups. All the pairwise comparisons are done with respect to the 462aa human CYP20A1 protein (NP_803882) and a query cover of ∼95% was obtained in each case. CYP20A1 is well conserved among vertebrates. Except for the three great apes (chimpanzee, bonobo and gorilla), all the Ka/Ks comparisons are significant (Fisher’s exact test; p<0.01; marked in red). Lesser the value of Ka/Ks, the more stringent is the negative selection operative on the protein i.e., fewer non-synonymous substitutions are tolerated in it.
Next, we asked if the patterns of sequence conservation and divergence are also reflected at the DNA level. We observed that though the 5’UTR appears to be well conserved across the primate lineage (except lemur and proboscis monkey), the 3’UTR exhibits divergence right from bonobo, with the breakpoints mostly coinciding with an Alu insertion (Figure 1d). To control for the length difference between the 5’ and 3’UTRs, we also checked for conservation in the 10kb region upstream of the first TSS of CYP20A1 and 10kb downstream of transcription end site (TES) and found it to be almost perfectly conserved among the higher primates, except for some New world monkeys (Supplementary Figure S3). Hence, a human specific divergence in this 3’UTR seems to have occurred in a genomic context that is per se conserved, at least among the higher primates.
CYP20A1 full length transcript may be a lncRNA
Multiple lines of evidence suggesting human-specific divergence of this orphan 3’UTR prompted us to investigate if this is transcribed. As two thirds of the 3’UTR is primarily comprised of repetitive sequences, the full length transcript is not captured adequately in expression arrays nor does it map uniquely from sequencing reads. Moreover, there are disparities regarding the annotation of the full length 3’UTR-containing isoform in different genomic portals (supplementary information). Thus instead of using publically available expression data, we confirmed the expression on this long 3’UTR experimentally using eleven pairs of primers that span the entire length of the transcript and also validated three amplicons by sequencing (see methods for details). Although highly variable, we observed the transcript of our interest was expressed in all the six cell lines tested (Figure 2a). The cell lines tested are cancerous and aberrant transcriptional profiles are reported in cancer (Suzuki et al. 2014), so next we compared its expression in a neuroblastoma cell line (SKNSH) with those in primary neuron, glia (astrocyte) and progenitor cells (NPC) to check if it is expressed due to the cancerous state of the cell. Neuroblastoma shares features with both mature neurons and NPCs, but is distinct from glia. CYP20A1 expression differs significantly only between glia and SKNSH (Figure 2b) but not in neurons or NPCs, suggesting it exhibits tissue specificity and is unlikely to be an artifact of the cancerous state of the cell.

a. A schematic representation of the primers designed on the full length CYP20A1 long-3’UTR transcript. Representative gel images showing this isoform is expressed in multiple cell lines of different tissue origin using primer pairs 1 and 10 from 5’UTR and 3’ UTR, respectively. Amplicons (1, 5 and 10) were also confirmed by Sanger sequencing.
* genomic DNA was used as positive control for PCR
b. RT-qPCR for CYP20A1 expression in cancerous versus non cancerous cell types of neuronal origin. Fold change was calculated with respect to SKNSH, after normalization with the geometric mean of expression values from β-actin, GAPDH and 18S rRNA. The error bars represent the SD of three biological replicates and the average of three technical replicates were taken for each biological replicate (** → p<0.01, *** → p<0.005; Student’s t test).
c. 3’RACE confirms the expression of the full length transcript. The schematic depicts the oligo(dT) (attached to a tag sequence) primed reverse transcription, followed by nested PCR. The amplification products corresponding to the bands below 900bp and above 700bp mapped to CYP20A1 3’UTR, suggesting that the full length transcript is expressed in untreated MCF-7 cells (N=3).
d. Differentiating the long-3’UTR transcript from other isoforms: the schematic highlights the skipped exon 6 and the position of flanking primers on shared exons. The presence of at least two different types of transcripts was confirmed. 277bp amplicon corresponds to isoform(s) that contain exon 6, but have shorter 3’UTRs (isoforms 2 and 3 in figure 2e) and 196bp amplicon corresponds to the long-3’UTR isoform (isoform 1). None of the six translation frames of the long 3’UTR isoform match with the annotated protein. The amino acids marked in red are common to both isoform 2 and 3, blue exclusive to isoform 3 and green represents the sequence from isoform 1.
e. Schematic representation summarizing the differences between CYP20A1 isoform 1 and isoforms 2 and 3.
We selected MCF-7 for the subsequent experiments as this cell line has been extensively used for drug screening and studying the effect of xenobiotics on different CYP family genes (Coumoul et al. 2001; Ptak et al. 2010). The copy number of CYP20A1 is not altered in this line (2n=2) (human CYP20A1 lacks paralogs (Pan et al. 2016) and the long 3’UTR isoform is expressed at a low level although CYP20A1 protein is relatively abundant (Supplementary Figure S4). Further, 3’RACE followed by nested PCR and amplicon sequencing confirmed the presence of this long 3’UTR (Figure 2c, supplementary information).
The sixth exon is skipped in the transcript of our interest but retained in the other isoforms containing shorter 3’UTRs. Using primers encompassing the sixth exon, we could distinguish between the transcripts as this results in 196bp amplicon from the transcript of our interest compared to the 277bp from the shorter 3’UTR isoforms whose expression was also observed to be much higher (Figure 2d). Notably, the 277bp amplicon could be obtained using cDNA synthesized using regular RTs, unlike the 196bp amplicon which could only be obtained using cDNA prepared with a very efficient enzyme (such as SuperScript III). Although the long 3’UTR transcript is annotated as the principal isoform, its expression level did not correlate with CYP20A1 protein which is relatively abundant in MCF-7 cells (Supplementary Figure S4). To probe further, we performed six-frame in silico translation of all CYP20A1 isoforms and compared them to the annotated human CYP20A1 protein. The two short 3’UTR isoforms matched – one perfectly and another with an additional amino acid stretch (Figure 2d), but the long 3’UTR isoform goes out of frame in the sixth and seventh exon and the resulting truncated 24 amino acid peptide has no hits in the human proteome as evidenced by BLAST analysis. Taken together, these data suggest that the long-3’UTR isoform is unlikely to be protein coding and may represent a novel lncRNA transcribed from the same locus. This presents a classic case of evolutionary sub-functionalization of a gene into two different classes of transcripts that might have evolved for different functions (Force et al. 1999) (Figure 2e).
In order to assess the relative contribution of different isoforms to the overall expression of CYP20A1, we used RNA-seq data from 15928 single nuclei derived from the different layers of human cerebral cortex (https://doi.org/10.1101/384826). NM_177538 is expressed in 75% of the nuclei whereas all the other RefSeq isoforms are found in <1% (cut-off CPM≥50). There are 7038, 5134 and 1841 single nuclei in which NM_177538 (but no other isoform) is expressed with ≥10, 50 and 100 reads, respectively (Supplementary Table S2). We did not find any reads mapping to the extended 3’UTR in chimpanzee and macaque (prefrontal cortex, CD4+ T cells), suggesting its expression may be human specific. Interestingly, it is expressed in rosehip neurons - a highly specialized cell type in humans(Boldog et al. 2018), suggesting co-evolution (Supplementary Table S3).
9kb 3’UTR: an evolving regulatory hub
The 3’UTRs of transcripts can harbor many regulatory motifs that govern their stability (half life), cellular localization and translational fate (Matoulkova et al. 2012). Alu repeats have contributed to many such functional regulatory elements in the primate genomes(Pandey and Mukerji 2011; Elbarbary et al. 2013: 1; Chen and Yang 2017). Hence, we hypothesized that this recently diverged, Alu-rich 3’UTR could be a potential hotspot of many such motifs and act as an evolving regulatory hub. Our earlier study had revealed that 3’UTR-resident Alus contain functional miRNA sites, making exonized isoforms amenable to miRNA-mediated regulation post-transcriptionally(Pandey et al. 2016). Hence, we wanted to check if this long UTR is also targeted by miRNAs. Using miRTarBase (release 6.0) (Hsu et al. 2011), we identified a set of 169 miRNAs (supported by microarray/ sequencing data) with predicted target sites on CYP20A1 3’UTR, of which 46 were also listed as “functional miRNAs” in FuncMir (miRDB) (Supplementary Table S4). Surprisingly, approx. 50% of these are either primate-specific or human-specific miRNAs (microRNAviewer) (Kiezun et al. 2012). The occurrence of target sites for human-specific miRNAs in this recently evolved UTR prompted us to carry out further in-depth analysis of miRNA recognition elements (MREs) in this 3’UTR as databases only report a few top target sites for each miRNA and provide limited information from a few experimental datasets.
It is generally recommended to obtain a consensus of MREs predicted using two or more algorithms for reducing the false positives. Since our 3’UTR has diverged across the vertebrate phylogeny, we decided against using algorithms that employ evolutionary conservation as a prediction criteria. Many of them also predict target sites based on seed sequence matches, which is increasingly being considered as an error-prone approach as the length and position of the seed sequence is variable amongst miRNAs (Ellwanger et al. 2011; Mullany et al. 2016). To circumvent this issue of inaccurate MRE prediction in non-conserved regions, we used miRanda which employs a two-step strategy: sequence complementarity, followed by thermodynamic stability of the predicted miRNA-mRNA duplex (Enright et al. 2003). Using stringent cut-off criteria, we obtained a total of 4742 MREs for 994 miRNAs, 4500 of which overlap with Alus (4382 MREs, if a conservative estimate of <50% overlap is considered) (Supplementary Table S5). These MREs are spread across the length of the 3’UTR, although there are several high density pockets (Figure 3a). The top 3 miRNAs with maximum number of MREs were miR-6089 (43), miR-762 (36) and miR-4763-3p (33), respectively.

a. Circos plot representing the MREs for the 994 miRNAs on CYP20A1 3’UTR. miRNAs are grouped on the basis of number of MREs. 23 Alus in this 3’UTR contribute to 65% of its length and are distributed throughout the UTR. Only 14% of miRNAs have MREs >10 (117 and 23 in G3 and G4, respectively).
b. Distribution of MREs for these 994 miRNAs on 1000 random sets of 23 length and subfamily matched Alu repeats. Only 6 sets contain MREs in range of 4701-4800 suggesting this is a non random phenomenon and MREs are created post alu exaptations. Highlighted in green are sets with more than 4500 MREs.
c. Schematic representation illustrates CYP20A1 acting as an miRNA sponge. In the condition where CYP20A1 is highly expressed, it will recruit multiple miRISC complexes thereby relieving the repression of cognate targets and subsequently leading to their translation; whereas in case of its low expression, those miRISC complexes remain free to load on the cognate targets and affect translational repression or promote mRNA degradation. CYP20A1 has the potential to sponge multiple miRNAs at the same time thereby regulating a large repertoire of transcripts.
Since Alus harbor regulatory sites, it is possible that the accumulation of so many Alu-MREs in this 3’UTR was due to the retrotransposition or recombination of Alus with pre-existing target sites. However, analysis of a 1000 sets of 23 Alus with similar composition with respect to length and subfamily, taken randomly from the genome, did not show a similar distribution of Alu-MREs. Just 0.5 and 4.2% of these random sets had MREs ≥4742 and 4500, respectively (Figure 3b), suggesting that the chance of Alus having retrotransposed into this 3’UTR with pre-existing MREs is extremely low. Given these observations, it is clear that the presence of such large number of MREs is not artifactual and these have been created within Alus post exaptation into CYP20A1 3’UTR. This implies a directional evolution of this UTR for a novel function. One possibility is that accumulation of MREs for so many miRNAs could potentiate its function as an endogenous “miRNA sponge” for a regulatory network (Figure 3c).
Next, we inquired how many of these 994 miRNAs were expressed in a given cell type. We observed low concordance across replicates and high variation across experiments in publically available miRNA expression datasets. These experiments were mostly microarray based (supplementary information). We selected MCF-7 and primary neurons for further experiments. Since CYP20A1 has also been implicated in neurophysiological roles (Stark et al. 2008: 1) it is convincing that besides its function as protein, lncRNA could have assumed novel role. Also, primary neurons preferentially express longer 3’UTRs which can increase the chances of miRNA-mediated regulation (Miura et al. 2013; Tushev et al. 2018). Using a cut-off of at least 10 MREs on CYP20A1 3’UTR and TPM value of 50, we obtained a set of 21 and 9 miRNAs in MCF-7 and neurons, respectively, of which 7 were common to both the cell types (Table 2).
miRNAs were prioritized based on their expression level (≥50 TPM), number of MREs ≥10 with binding energy ≤ −25kcal/mol. * expression values <50 TPM have not been represented.
CYP20A1 long 3’UTR isoform functions as an endogenous miRNA sponge
To determine if CYP20A1 long 3’UTR isoform can be a potential miRNA sponge, we compared its features with previously characterized sponge RNAs. An effective sponge is predominantly cytosolic and typically contains 4 to 10 low binding energy MREs for a particular miRNA, each separated by a few nucleotides. It is devoid of destabilizing RNA elements, resistant to cellular nucleases and stably produced at high levels. It contains mispaired bases or bulge within the MREs that allows it to sequester miRNAs without triggering its own degradation and exhibits RNA induced silencing complex (RISC) loading. Most importantly, its expression is positively correlated with the expression of target RNAs that contain cognate MREs for the miRNA it sponges.
Transcripts containing oppositely oriented Alus in close vicinity tend to form double-stranded RNA by intramolecular pairing and are recognized by ADAR1, undergo A-to-I hyperediting (Bazak et al. 2014) and usually retained within nuclear paraspeckles (Chen and Carmichael 2009; Goldstone et al. 2010). Since our transcript of interest fulfils all these criteria, we checked its level using RT-qPCR in both nuclear and cytosolic fractions and found that it is predominantly localized to the cytosol (Figure 4a). Even using a stringent cut-off for MRE prediction (binding energy≤ −25kcal/mol), we observed miRNAs with as many as 43 MREs and binding energy as low as −47kcal/mol. Out of the 994 miRNAs, 140 have ≥10 MREs spread across the length of the UTR. We checked for the presence of bulge within the MREs for the 23 prioritized miRNAs (listed in Table 2) using miRanda on default parameters. To screen for MREs with efficient miRNA docking without leading to cleavage of CYP20A1 RNA, two major criteria were followed – complete match in (2-7)6mer seed site and presence of mismatch or insertion at 9-12 position. 6mer sites with wobble base pairing were also retained as 2 wobble-pairs were maximally present in some of the MREs. We found five such sites for miR-6724-5p, 2 each for miR-1254, miR-4767 and miR-3620-5p and one each for miR-941, miR-4446-3p, miR-296-3p, miR-619-5p, miR-6842-3p and miR-1226-5p (Table 3). In all these sites, insertion was present in CYP20A1 UTR suggesting bulge formation in the sponge RNA itself.
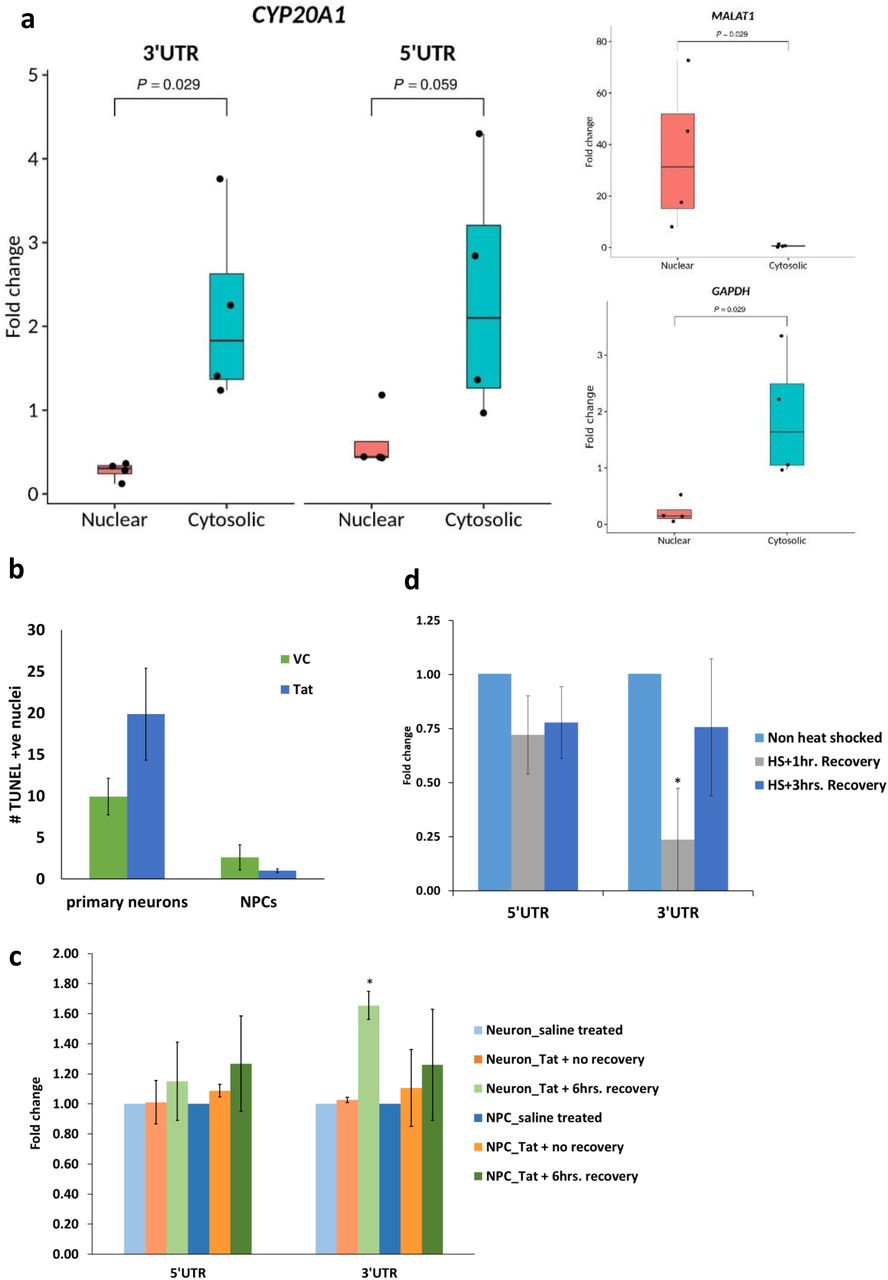
a. Cytosolic localisation of CYP20A1 isoform 1 confirmed by RT-qPCR. Fold change was calculated with respect to total RNA, after internal normalization using the primers against spiked-in control. The error bars represent the SD of four independent experiments and the average of two technical replicates were used for each experiment. Quality controls for assessing the purity of nuclear (MALAT1) and cytosolic (GAPDH) fractions are shown on the right.
The RT-qPCR data were analyzed in accordance with the MIQE guidelines (Bustin et al. 2009) (see supplementary information for details).
b. Late apoptotic cells in primary neurons and NPCs in response to HIV1-Tat treatment were scored by the number of TUNEL positive nuclei. Tat is neurotoxic and kills ∼10% more neurons compared to the vehicle control (VC i.e., saline), whereas the difference is not statistically significant for NPCs (p-values 0.04 and 0.21 for primary neurons and NPCs, respectively, for Student’s t-test assuming equal variance). The data represent the mean and SD of three independent experiments and >1000 nuclei were scored per condition for each experiment.
c and d. Expression of CYP20A1 isoform 1 in response to HIV1-Tat [c] and heat shock [d] treatment was assessed by RT-qPCR using both 5’ and 3’UTR primers. The 3’UTR was found to be upregulated following 6hrs recovery after Tat treatment in neurons (p value = 0.035 *p value <0.05, Student’s t-test), but not in NPCs (p value = 0.348) [c]. It was also strongly downregulated in neurons (p value = 0.031) immediately after heat shock (HS+1hr recovery) but lost statistical significance as the cells continued to recover (p value = 0.310; HS+3hrs recovery) [d]. In both these cases, the 5’UTR primer exhibits the same trend as the 3’UTR but does not qualify the statistical significance cut-off of p< 0.05 Fold change was calculated with respect to saline (vehicle) treatment, after internal normalization with the geomean of GAPDH, ACTB and 18S rRNA in [c] and with respect to non heat shock treated cells, after internal normalization with the geomean of GAPDH and ACTB in [d]. The error bars represent the SD of three independent experiments and the average of 2-3 technical replicates was taken for each experiment.
Features of MREs with seed site match and presence of bulge
In order to probe if the alteration in CYP20A1 long-3’UTR isoform level would be consequential to the expression of transcripts containing cognate MREs, we looked for conditions where it is likely to be altered. Since CYP20A1 has been identified as a candidate from a set of Alu exonized genes that map to apoptosis, we asked if this would respond to triggers that induce cell death. HIV1-Tat is a potent neurotoxin, killing ∼50% more neurons compared to vehicle (Figure 4b). Upon treating primary human neurons with HIV1 full length Tat protein, followed by 6hrs recovery, isoform 1 was found to be significantly upregulated (1.65 fold). However, progenitor cells, which are immune to Tat (Figure 4b), did not show any such trend (Figure 4c).
CYP20A1 3’UTR also carries 17 high confidence binding sites for HSF1, 14 of them within Alus, which show positional conservation in agreement with previous studies (Pandey et al. 2011). This suggests that CYP20A1 may be amenable to antisense-mediated downregulation during heat shock response as demonstrated by an earlier work from our lab(Pandey et al. 2011). We found isoform 1 to be significantly downregulated in primary neurons upon heat shock, followed by 1hr recovery (Figure 4d).
In order to query the expression of other cognate targets of the 9 prioritized miRNAs in these two conditions, we performed stranded RNA sequencing in primary neurons. In accordance with our RT-qPCR results, we found isoform 1 to be significantly downregulated 2.68 folds (log2FC = −1.42) upon heat shock recovery (HS) and 1.21 folds upregulated (log2FC = 0.28) during Tat response. The latter, however, did not cross our stringent statistical significance threshold. Out of the 3876 genes with significant differential expression in HS or Tat, 380 genes exhibit positively correlated expression pattern as CYP20A1 (Figure 5, Supplementary Table S6). All of these 380 genes contain at least one MRE for one or more of the 9 prioritized miRNAs and majority of their MREs are canonical and not Alu-derived. Whereas most genes contain a few MREs for one or two of these miRNAs, CYP20A1 is at the 2nd position with a total of 116 total MREs (Supplementary Table S6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Log2FC values of 380 genes upregulated in response to Tat treatment (red) and downregulated during heat shock recovery (green) in primary neurons, mirroring the exact trend exhibited by CYP20A1 isoform 1. All of them contain one or more MREs for the 9 miRNAs that can potentially be sponged by isoform 1 in neurons and represent potential cognate targets whose expression can be regulated by CYP20A1 isoform 1 perturbation. Genes are plotted in order as supplementary table S6.
Next, we performed enrichment analysis for this gene set using Toppfun and found enrichment of blood coagulation pathways and neuronal development as major hubs in biological processes. The top five processes were hemostasis (28 genes), axon guidance (25 genes), neutrophil degranulation (23 genes), platelet signalling, activation and aggregation (18 genes) and ECM organisation (18 genes). mRNA processing and mitochondria translation, metabolism, amino acid and nucleotide synthesis and antigen presentation were amongst others (Supplementary Table S7). Blood coagulation factors have been reported to affect pathophysiology of CNS via coagulation protein mediated signal transduction (De Luca et al. 2017). Although the exact biological role of CYP20A1 remains be explored, yet enrichment of coagulation pathways in gene set showing correlated expression with CYP20A1 suggests that it may have some role in neuro-coagulopathy, which remains to be tested.
Discussion
Long non-coding RNAs, as competing endogenous RNAs (ceRNAs), are being implicated in rewiring miRNA gene regulatory networks. These could contribute to lineage specific novelties. For example, lncND has been shown to drive brain expansion in the primate lineage by sequestering miR-143-3p that regulates the expression of Notch-1 and Notch-2 (Rani et al. 2016). ceRNA-mediated regulation has been demonstrated in neurodevelopment, myoblast differentiation, pluripotency maintenance and in various cancers (Wang et al. 2010; Cesana et al. 2011; Fan et al. 2013; Wang et al. 2013) and their expression is spatio-temporally regulated. Circular RNAs, produced by back-splicing, have been previously reported as potent miRNA sponges (Hansen et al. 2013; Barrett and Salzman 2016). However, to the best of our knowledge, TE-derived endogenous miRNA sponge has not been characterized so far. We present an interesting case of Alu exonization that lengthens the 3’UTR of an orphan protein coding gene CYP20A1 and creates a novel lncRNA isoform. The initial characterization of this isoform (Isoform 1) reveals exaptation of 23 Alus from different subfamilies in its 3’UTR. These have provided the substrate for evolution of multiple miRNA binding sites and resulted in its neo-functionalization to a potential multi-miRNA sponge. The event seems to be specific to the human lineage.
CYP20A1 is an orphan member of the cytochrome P450 superfamily of oxidoreductases that metabolize and detoxify drugs (Guengerich et al. 2005). These genes are also involved in steroidogenesis. It contains unusual heme-binding motifs and is highly expressed in the liver. In copepods it has been shown to be responsive to stressors like bisphenol A(28) and UV-B (Puthumana et al. 2017). Elevated expression of CYP20A1 RNA has been reported in the gastric mucosa of splenasthenic syndrome (chronic superficial gastritis) patients as well as in the leukocytes of blunt trauma patients (Brandfellner et al. 2013). Besides, high levels of CYP20A1 mRNA in both unfertilized zebrafish eggs (Goldstone et al. 2010) and developing mouse embryos (Choudhary et al. 2003) suggest its involvement in early developmental stages. However, these observations are correlational and it is still contentious whether the gene could function as an RNA or a protein.
We report both coding as well as non-coding transcripts from CYP20A1 locus. While the protein is highly conserved, we observe a novel transcript with a skipped exon that results in an out-of-frame CDS. Further, the novel RNA isoform has a substantially longer UTR with an increased scope of involvement in regulatory events post-transcription. This we confirm and validate experimentally in multiple cell lines. This isoform is also human specific. It is not only expressed in 75% of single nuclei derived from different layers of the human cerebral cortex but also in human specific rosehip neurons. Its specific expression to human lineage is also strengthened by the fact that the 3’UTR genomic region is neither conserved at the DNA level in high order primates nor is present in transcripts derived from RNA seq of closely related primates. The protein coding isoform exhibits a significantly greater conservation across the evolutionary lineages.
miRNA sponges so far have been reported to contain MREs that are targets for a single or a few related miRNA species (Franco-Zorrilla et al. 2007; Poliseno et al. 2010; Hu et al. 2018). Artificially created sponges with MREs >=4 have been effectively used in a few studies(Ebert et al. 2007; Ebert and Sharp 2010) but the CYP20A1 isoform 1 having >=10 MREs for as many as 140 miRNAs clearly stands out. The presence of MREs for multiple miRNAs raises the exciting possibility that it can exert a systemic effect through titrating the cellular levels of many miRNAs simultaneously. A modulated expression of this transcript could alter spatio-temporal dynamics and stoichiometry of the miRNA pools. In a previous study from our lab, we have reported the functional effects of MREs within 3’UTR-resident exonized Alus in fine tuning the p53 regulatory network in heat-shock response (Pandey et al. 2016). In CYP20A1, out of 4742 total MREs more that 80% are contained within Alu which could potentially participate in miRNA regulatory network and are not just repetitive sequence artifacts. Further, all the 23 Alus in this 3’UTR have been shown harbour antisense transcripts and are substrate for A-to-I RNA editing (Mandal et al. 2013). These events though dynamic could further disrupt or create new miRNA binding sites from existing MREs thereby increasing the regulatory possibilities. For example, miR-513 and miR-769 targets 3’UTR of DNA fragmentation factor alpha gene in an adenosine deamination dependent manner (Borchert et al. 2009). Since A-to-I editing events are preponderant in brain (Paz-Yaacov et al. 2010; Rosenthal and Seeburg 2012), these editing events could further contribute to phylogenetic novelties.
We observe that expression of isoform 1 in human primary neurons is inducible in response to HIV1-Tat and decreased during heat shock response. Expression of ceRNAs is also tightly regulated and often, specific to tissue, development stage or some stress conditions (Cesana et al. 2011; Tay et al. 2014). High levels of sponge RNA could yield high expression of the cognate targets as miRISC complexes are sequestered by the sponge (Ebert et al. 2007; Ebert and Sharp 2010), whereas reduced expression of sponge could relieve these miRNA for the latter to bind, inhibit and/or degrade their cognate targets (Figure 3c). We make similar observations in a set of 380 genes which correlate with the expression pattern of CYP20A1 i.e., downregulated during heat-shock and upregulated upon Tat treatment. These genes mapped to processes that are majorly involved in blood coagulation and neuronal pathways. Neurocoagulopathy has been implicated in the pathophysiology of CNS via coagulation protein mediated signal transduction. Besides coagulation, these proteins interfere with synaptic homeostasis. They also affect neurite outgrowth and morphological changes in neurons, blood brain barrier integrity, ECM stability, ROS generation from astrocytes, secretion of inflammatory cytokines, as well as impair excitability, increase inflammation, mitosis of astrocytes and/or microglia and ultimately, affect neuronal viability. This process is being linked to several neurodegenerative diseases including multiple sclerosis, cancer of the CNS, addiction and mental health (De Luca et al. 2017). Exposure to HIV1-Tat is known to cause axonal damage, loss of blood brain barrier integrity, changes in neurite outgrowth, etc. These are mediated by astrocyte activation, inflammatory cytokine expression, inducing mitochondrial injury and rearrangement of microtubules. The set of 380 genes were also found to be enriched in similar pathways like axon guidance, hemostasis, platelet activation and aggregation, ECM organization, regulation of actin cytoskeleton, antigen presentation, Golgi to ER transport and mitochondrial translation. In the light of our observations, it is possible that the changes observed upon Tat exposure could partly be mediated and synergised by the sponging effect of CYP20A1. Upon activation by Tat, the sponge could titrate out the miRNA that target the 380 genes and hence modulates all the pathways simultaneously. Validation of this lncRNA in the context of neuronal damage might shed further light on this plausible involvement. It could also play a role in normal neuronal functions through fine tuning expression NPCs during neurogenesis, neuronal migration during differentiation, etc.
Multiple exonized Alus in CYP20A1 3’UTR can potentially make it highly structured and lead to altered bioavailability for MREs. Future studies on secondary structure simulations would allow us to assess the availability and accessibility of these MREs. Closely spaced sites of same or different miRNAs can cooperatively sequester multiple miRNAs thus making the process fast and leading to robust outcomes (Denzler et al. 2016). This would be an exciting possibility to explore. With multiple miRNA that target members of a co-regulated network it could exhibit a systemic response. Also, this could work as an effective regulatory switch for a faster response or return to baseline. We have not yet looked at its turnover rates, however multiple possibilities exist such that it could be transiently over-expressed induced in response to specific cues, regulated through a negative feedback, cleared via transcriptional shutdown by certain miRNAs or have an inherently shorter half-life due to a rapid turnover. Other possibilities such as independent transcription events from this UTR or additional polyadenylation sites, cannot however be ruled out. During the course of this study, we noticed reads mapping beyond the longest 3’UTR annotated in UCSC (Supplementary Figure 5). Beyond its role as miRNA sponge, this UTR can also be involved in sequestering RBPs and deplete the cellular reserves, thereby indirectly affecting other genes (Kelley et al. 2014).
Conclusion
In conclusion, the present study further substantiates the fact that non-random distribution of Alu in human genome can alter conserved regulatory networks and reshape them in lineage specific manner. The contribution of repeats in evolving regulatory networks has been undermined and overlooked until recently. Extensive Alu mediated 3’UTR lengthening in case of CYP20A1 provides a platform for creation of multiple MREs which makes it capable of functioning as multi-miRNA sponge. CYP20A1 provides an interesting model for studying lncRNA derived ceRNAs that could co-regulate multiple genes and participate in complex networks that govern intermediate phenotypes such as neuro-coagulation. These could play important roles as regulatory switches in response to biological cues. This could result in differential cellular outcomes due to rapid release or sequestering of miRNAs.
Materials and Methods
Genome-wide 3’UTR length distribution
The coordinates for all human NCBI RefSeq transcripts were downloaded from RefSeq version 74 (hg38). Both NM and XM ID containing transcripts were included in the analysis. The 3’UTR length was computed from its annotated translation end site through the transcription end site and for every gene, only the longest 3’UTR was considered. The summary statistics for the distribution were calculated using R scripts.
Conservation Analyses
DNA
DNA sequence conservation across different species was checked using UCSC genome browser. We have used multiple alignment across 20 species generated by multiz (Blanchette 2004) which uses a combination of two algorithms (phastCons and phyloP) to estimate evolutionary conservation, with the null hypothesis of neutral evolution. Both gaps as well as unaligned sequences were treated as ‘missing’ data.
For calculating Jukes-Cantor (JC) divergence, DNA sequences with 1kb upstream and downstream were downloaded from UCSC for human CYP20A1 5’UTR, 3’UTR and 10kb promoter proximal region (chr2:203,228,449-203,238,448; hg38). The corresponding regions were fetched for pair-wise alignment between human-chimp (panTro4), human-gorilla (gorGor4), human-bonobo (panPan1) and human-macaque (rheMac8), which were subsequently used to calculate the JC-score.
Protein
CYP20A1 protein sequences (from different species) were taken from the top hits obtained in NCBI pBLAST by using the human protein as a reference. Multiple sequence alignment was performed using Clustal Omega (O 1.2.2). It uses seeded guide trees and hidden Markov model (HMM) to estimate the similarity among three or more sequences.
Ka/Ks ratio was calculated using the method detailed in a previous publication by our group (Gautam et al. 2015). Protein sequence IDs of CYP20A1 ortholog for each species was identified as the first hit of NCBI pBLAST using human NP-ID as the query term. The NM-IDs of corresponding mRNAs were also obtained from NCBI and sequences were fetched for these NP and NM-IDs. For a pair of orthologous genes, the protein-coding DNA alignment between human and each of the other species was constructed using ParaAT (a Parallel Alignment and back-Translation tool) (Zhang et al. 2012). The protein sequence information was used to match each triplet codon with its respective amino acid. Aligned coding DNA between a pair (human vs. another species) was provided as an input to KaKs_Calculator (Zhang et al. 2006). Model averaging (MA), which takes an average of substitution rates of seven models, was used as a maximum likelihood method to calculate the Ka/Ks value.
Cell culture
MCF-7 cell line was procured from National Centre for Cell Sciences (Pune, India) and cultured in GlutaMax-DMEM high glucose (4.5gm/l) (Gibco, 10569044) supplemented with 10% heat inactivated FBS (Gibco, US origin, 10082147), HEPES (Gibco, 11560496) and 1X antibiotic-antimycotic (Gibco, 15240096). The culture was maintained at 70-80% confluency at 37°C, 5% CO2. We had authenticated the cell line identity by STR profiling (supplementary information). The cells were routinely screened for mycoplasma using MycoAlert Mycoplasma detection kit (Lonza, LT07-218.) as per manufacturer’s protocol and confirmed to be free from contamination. Additionally, 16S rRNA PCR was performed using both genomic DNA from cells and cell-free DNA from spent media to confirm the absence of bacterial contamination.
Primary human neuron and astrocyte cultures comply with the guidelines approved by the Institutional Human Ethics Committee of NBRC as well as the Stem Cell and Research Committee of the Indian Council of Medical Research (ICMR) (Fatima et al. 2017). Briefly, neural progenitor cells (NPCs) derived from the telencephalon region of a 10-15 week old aborted foetus were isolated, suspended into single cells and plated on poly-D-lysine (Sigma, P7886) coated flasks. The cells were maintained in neurobasal media (Gibco, 21103049) containing N2 supplement (Gibco, 17502048), Neural Survival Factor 1 (Lonza, CC-4323), 25ng/ml bovine fibroblast growth factor (bFGF) (Sigma, F0291) and 20ng/ml human epidermal growth factor (hEGF) (Sigma, E9644) and allowed to proliferate over one or two passages. The stemness of NPCs was functionally assayed by i.) formation of neurospheres, and ii.) ability to differentiate into neurons or astrocytes. Additionally, NPCs were also checked for the presence of specific markers like Nestin. For commitment to the neuronal lineage, NPCs were starved of bFGF and EGF; 10ng/ml each of PDGF(Sigma, P3326) and BDNF(Sigma, B3795) were instead added to the media cocktail. Differentiation of NPCs to astrocytes required Minimum Essential Medium (MEM) (Sigma, M0268-10x) supplemented with 10% FBS. The process of neuronal differentiation completes in exactly 21 days; our experiments were completed within a week post-differentiation. Differentiated cultures of primary neurons and astrocytes were also checked for specific markers by immunostaining to determine the efficiency of the differentiation process (Supplementary Figure S6).
RNA isolation, cDNA synthesis and RT-qPCR
Total RNA was isolated using TRIzol (Ambion, 15596-026) as per manufacturer’s protocol and its integrity was checked on 1% agarose gel followed by Nanodrop quantification (ND1000, Nanodrop technologies, USA). cDNA was prepared from oligo(dT)-primed DNase-treated RNA [3 units of TURBO DNase (Invitrogen, AM1907) was used for 5µg RNA; incubated at 37°C for 20’] using 300 units SuperScript III RT (Invitrogen, 18080-044) (50°C for 90’, 70°C for 15’). RNA template was digested from the cDNA using 2 units of E. coli RNaseH (37°C for 20’) (Invitrogen, 18021071). Primers were designed using Primer3 (version 4.0.0) and were synthesized by Sigma (supplementary information). To insure against spurious amplification, we had designed two pairs of overlapping primers both on the 5’ as well as 3’ends of our transcript of interest and run ‘minus-RT’ controls in every reaction. Additionally, we had sequenced three amplicons (1, 5 and 10) with both forward and reverse primers to check the specificity of amplification. BLASTN (NCBI; 2.4.0+) against the corresponding in silico predicted amplicons had revealed >95% sequence identity with an average query cover of 90%; BLAT against the whole genome (hg38) gave CYP20A1 as the top hit in every case.
RT-qPCR was performed in 10µl volume using 5µl 2X SYBR Green I master mix (Kappa; KK3605), 0.2µl each of 10µM forward and reverse primers and cDNA (stock: ∼80ng/µl RNA equivalent, diluted 1:40 in the final reaction). The reaction was carried out in Roche Light Cycler 480 (USA) and the cycling conditions were: 95°C for 3’, 45 cycles of (95°C for 30”, 58°C for 30”, 72°C for 40”), 72°C for 3’; followed by melting: 95°C for 5”, 65°C for 1’. Melting curves were confirmed to contain a single peak and the fold change was calculated by ΔΔCt method. MIQE guidelines were followed for data analysis.
3’RACE
cDNA for 3’RACE was prepared using RLM-RACE kit (Ambion, AM1700) with 1µg MCF-7 total RNA as per the manufacturer’s recommendation. Nested PCR was performed with FP10 and inner primer using the amplicon produced by FP9 and outer primer. The product of this nested PCR was run on 2% agarose gel and four major bands were observed, which were gel-eluted (As per manufacturer’s protocol, Qiaquick gel extraction kit, Qiagen, 28704) and subsequently sequenced. BLASTN (NCBI; 2.6.0+) against the 861bp in silico amplicon revealed 98% and 95% identity for the ∼800bp and 700 bp amplicons, respectively; BLAT on hg38 showed that these sequences (including a part of AluSc) map to CYP20A1 3’UTR with 97% identity.
Sanger sequencing
The sequencing reaction was carried out using Big dye Terminator v3.1 cycle sequencing kit (ABI, 4337454) in 10µl volume (containing 2.5µl purified DNA, 0.8µl sequencing reaction mix, 2µl 5X dilution buffer and 0.6µl forward/ reverse primer) with the following cycling conditions - 3 mins at 95°C, 40 cycles of (10 sec at 95°C, 10 sec at 55°C, 4 mins at 60°C) and 10 mins at 4°C. Subsequently, the PCR product was purified by mixing with 12µl of 125mM EDTA (pH 8.0) and incubating at RT for 5 mins. 50µl of absolute ethanol and 2µl of 3M NaOAc (pH 4.8) were then added, incubated at RT for 10 mins and centrifuged at 3800rpm for 30 mins, followed by invert spin at <300rpm to discard the supernatant. The pellet was washed twice with 100µl of 70% ethanol at 4000rpm for 15 mins and supernatant was discarded by invert spin. The pellet was air dried, dissolved in 12µl of Hi-Di formamide (Thermo fisher, 4311320), denatured at 95°C for 5 mins followed by snapchill, and linked to ABI 3130xl sequencer. Base calling was carried out using sequencing analysis software (v5.3.1) (ABI, US) and sequence was analyzed using Chromas v2.6.5 (Technelysium, Australia).
CYP20A1 expression in non-human primates
In order to query if the long-3’UTR isoform is expressed in other primate species, we checked for reads mapping to the 3’UTR in publicly available chimp and macaque RNA-seq datasets [GSM1432846, 55, 65 (SRR1510158, 167, 177); GSM2265102, 4, 6 (SRR4012405, 08, 09, 13)]. Reads were mapped to both human and chimp/macaque 3’UTR to increase fidelity and mapping on housekeeping genes like ACTB, GAPDH and EIF4A2 was also checked to control for data quality and mapping parameters. To query more expression datasets, we took advantage of the skipped exon 6 in our isoform of interest. In CYP20A1 long-3’UTR isoform, exon 5 and exon 7 are contiguous and if we have reads mapping to the region spanning the junction of these two exons, we can reliably say that this isoform is expressed. Towards this, we performed BLAST against human datasets in SRA using a 289bp sequence reconstructed by joining exon 5 and 7 and further confirmed the hits by checking for read mapping to the 3’UTR.
miRNA target prediction
miRNA target sites (MREs) on CYP20A1 3’UTR were predicted using miRanda (version 3.3a) (Enright et al. 2003), with the parameters set as follows: score threshold(-sc): 100, gap opening penalty(-go): −8, gap extension penalty(-ge): −2, binding energy(-en): −25kcal/mol., ‘strict’ (i.e., G:U pairs and gaps were not tolerated in the seed region). miRanda uses miRBase (which contains ∼2500 miRNAs) for annotation.
For bulge analysis, target prediction for 23 miRNAs on CYP20A1 3’UTR was performed using miRanda offline version 3.2a with default parameters (gap opening penalty= −8, gap extension= −2, score threshold= 50, energy threshold= −20kcal/mol, scaling parameter= 4).
Small RNA sequencing and analyses
cDNA libraries were prepared as per manufacturer’s protocol using TruSeq small RNA library prep kit (Illumina, RS-122-2202) from 1µg total RNA (RIN ≥8.5). Briefly, 3’ and 5’ adapters were ligated, followed by enrichment of RNA fragments with adapter molecules on both ends. Reverse transcription followed by amplification using primers that anneal to the adapter ends created the cDNA libraries which was then purified and size selected (∼147nt) to enrich for small RNAs by running on 6% native PAGE. Subsequently, the libraries were concentrated by ethanol precipitation and quality checked on Agilent technologies 2100 Bioanalyzer (USA) using DNA high sensitivity chips (Agilent, 5067-4626). Using 10mM Tris-HCl (pH 8.5) libraries were normalized to 2nM, denatured and applied to cBot for cluster generation (Illumina, PE-401-3001), which were then sequenced on HiSeq2000 (Illumina, California, USA) using TruSeq SBS kit (Illumina, FC-401-2202) for 50 cycles.
The data were quality checked using FastQC (version 0.11.2), followed by adapter trimming by cutadapt (version 1.18); reads were not discarded. As expected, around 95% of the of adapter trimming events happened on the 3’end of the reads. Length filtering and quality filtering were carried out by cutadapt; Q30 reads with sequence length >15 but < 35nt. were retained for mapping. ∼80% of the reads were retained after these filtering steps. Size distribution of the reads and kmer position were (21-25, 28-32) and 26-28 respectively. Subsequently, these reads were mapped onto hg38 using Bowtie2. On an average, 61% of the reads were uniquely mapped. miRDeep2 was run to obtain the read counts as TPM.
Nuclear cytosolic RNA isolation
Nuclear and cytosolic RNA were isolated using PARIS kit (Ambion, AM1921) as per manufacturer’s protocol. Briefly, ∼10 million cells were resuspended in fractionation buffer, incubated on ice and centrifuged at 4°C to separate the nuclear and cytosolic fractions. The nuclear pellet was additionally treated with cell disruption buffer before mixing with the 2X lysis/binding solution and absolute ethanol and passing through a column. The RNA was subsequently eluted in hot elution buffer, quantified using Nanodrop and its integrity was checked on 1% agarose gel. Nuclear RNA contains an additional hnRNA band above the 28S rRNA band and is usually of lower yield than cytosolic RNA.
Cell lysate preparation and Western Blotting
Cells were harvested with 0.25% trypsin-EDTA (Gibco, 25200-072) and pelleted. 30% of this pellet (2-3 million cells) was lysed in 100µl RIPA (Sigma, R0278) (reconstituted with 0.1M DTT and 1µl protease inhibitor cocktail (Sigma, P8340) and incubated on ice for 30’ with intermittent vortexing. The total cell lysate was purified by centrifugation at 14000rpm for 45’ (at 4°C) and the supernatant was stored at −20°C.
For nuclear cytosolic fractionation, ∼2 million cells were gently lysed with 200µl cell lysis buffer (10mM Tris-HCl pH 7.5, 0.15% NP-40 and 150mM NaCl) and incubated on ice for 20’. The lysate was then layered on top of 2.5 volumes of chilled 24% sucrose cushion (prepared in lysis buffer) and centrifuged at 14000rpm for 10’ at 4°C. The supernatant (cytosolic fraction) was collected and stored at −20°C. The nuclear fraction was obtained by re-suspending the pellet in 200µl pre-chilled glycerol buffer (20mM Tris-HCl pH 7.5, 75mM NaCl, 0.5mM EDTA, 50% glycerol and 0.85mM DTT) and an equal volume of nuclear lysis buffer (10mM Tris-HCl pH 7.5, 1mM DTT, 75mM MgCl2, 0.2mM EDTA, 0.3M NaCl, 1M urea and 1% NP-40) with brief vortexing.
The protein concentration was determined by Pierce BCA Protein Assay Kit (Thermo Scientific, PI23225). 25µg of denatured protein (90°C for 10’) was loaded on 15% SDS PAGE and was resolved at 60V, followed by overnight transfer (30V at 4°C) onto 0.2µm PVDF membrane (MDI, SVFX8301XXXX101). The transfer was confirmed by Ponceau staining (USB, 32819). Blocking was done in 5% skimmed milk (Himedia, GRM1254) for 1hr, followed by incubation with primary and secondary Ab. The blot was developed by ECL (Abcam, AB133406) staining and analyzed using ImageJ (NIH, US) (antibody details provided in supplementary information).
Immunostaining
Cells were seeded on coverslips and cultured in 4-well plates for this experiment. Spent media was discarded and cells were briefly washed with 1X PBS (to remove residual media completely). The cells were then fixed with 4% PFA (freshly prepared in 1X PBS) for 20 minutes, followed by 3 washes in 1X PBS (5 minutes each) to completely remove traces of PFA. Blocking was done with 4% BSA containing 0.5% Triton-X (to permeabilize the cells) for 1hr with gentle rocking. Incubation in primary antibody (MAP2, Tuj1, NeuN, Nestin, GFAP; all diluted 1:200 in 0.1% BSA) was done overnight at 4°C on a rocker, except GFAP (1hr at RT), followed by 3 washes in 1X PBS (5 minutes each). Incubation with fluorescently labeled secondary antibody (diluted 1:1000 in 0.1% BSA) was performed for 1hr in the dark with shaking. This was followed by five washes in 1X PBS (5 minutes per wash) on a rocker. The coverslips were mounted in DAPI hardset (Vectashield, USA) on clean glass slides. Images were captured from 7-10 random fields using AxioImager.Z1 microscope (Carl Zeiss, Germany).
Stress treatment
Cells were gently washed once with 1X PBS and fresh media was replenished before treatment for accurate quantification of stress response. Heat shock was given at 45°C (±0.2) for 30’ in a water bath. Subsequent to the treatment, cells were transferred to 37°C/5% CO2 for recovery and harvested after 1hr, 3hrs and 24hrs. For Tat treatment, full-length lyophilized recombinant HIV1 Tat protein was purchased from ImmunoDX, LLC (Woburn, Massachusetts, USA) and reconstituted in saline. The dosage for treatment was determined by drawing a ‘kill curve’ using graded dose of Tat on neurons (Supplementary Figure S7). Finally, treatment was performed for 6hrs with 100ng/ml Tat and cells were either harvested just after the treatment or allowed to recover at 37°C/5% CO2 for another 6hrs prior to harvesting.
TUNEL Assay
The assay was performed with in situ Cell Death Detection kit, TMR red (Roche, Germany) which uses a proprietary terminal deoxynucleotidyl transferase (TdT) to incorporate a fluorescently tagged nucleotide at the free 3’-OH ends of damaged DNA molecules, a hallmark of late apoptotic cells.
∼20,000 cells were seeded per well (on coverslips) in 12-well plates. Post Tat treatment, cells were washed once with 1X PBS and instantly fixed with 4% PFA, followed by three brief washes in 1X PBS, permeabilization and blocking with 4% BSA containing 0.5% Triton-X 100, incubation with TdT for 1hr in the dark and three thorough washes with 1X PBS. Coverslips were then mounted on clean glass slides using hardset mounting media containing DAPI (Vectashield, USA). 6-8 random fields were imaged for each experimental group using AxioImager.Z1 microscope (Carl Zeiss, Germany). Fixed cells treated with 2 units of DNaseI (for 10 minutes at RT, followed by the addition of EDTA to stop the reaction) were used as a positive control in this experiment. TUNEL positive nuclei were scored using ImageJ software (NIH, USA). At least 1000 cells were scored for each replicate.
Library preparation, RNA sequencing and data analyses
Libraries were prepared following Illumina’s TruSeq stranded total RNA protocol as per manufacturer’s recommendations, using three biological replicates per experimental condition. Integrity of the RNA samples was checked on Agilent 2100 Bioanalyzer (Agilent RNA 6000 Nano Kit); RIN values ranged from 8.6-9.6 for our samples. 500ng of total RNA was taken in a volume of 10μl and ribosomal RNA was removed using Ribozero depletion kit. Briefly, total RNA was denatured at 68°C for 5’, followed by removal of rRNA using magnetic beads, subsequent clean up with RNAClean XP beads (Beckman Coulter, A63987), two consecutive washes in freshly prepared 70% ethanol and then elution (by heating at 94°C for 8’; to fragment and prime the RNA for cDNA synthesis). First strand synthesis was carried out with random hexamers using the following program (25°C for 10’, 42°C and 70°C for 15’ in each). This was followed by second strand cDNA synthesis at 16°C for 1hr for elimination of the template RNA, replacement of dUTP by dTTP and to finally produce blunt-ended cDNA, which was then cleaned up using AMPure XP beads (Beckman Coulter, A63881) and two washes in 80% ethanol. Next, the 3’ends of these blunt cDNA molecules were adenylated to create a single ‘A’ overhang (incubation at 37°C for 30’, followed by 70°C for 5’) such that these can form complementary base pairs with the ‘T’s on the adapter ends and also do not concatenate with each other to form chimera during adapter ligation. Then, adapters were ligated (30°C for 10’) at both the ends of these double-stranded cDNA molecules so that these can hybridize to the probes on the sequencing flow-cell. The fragments with adapter ligated on both ends (libraries) were enriched and amplified using a 15-cycle PCR (the PCR primer cocktail anneal to both ends of the adapters; PCR conditions: 98°C for 30”, 15 cycles of (98°C for 10”, 60°C for 30”, 72°C for 30”), final extension 72°C for 5’). This was followed by their clean up using AMPure XP beads and two washes in 80% ethanol. The libraries were quantified using Qubit fluorometer (Invitrogen) and subsequently validated on Bioanalyzer (Agilent DNA 1000 kit) to confirm the insert size. The size of the libraries was in the range of 260-280bp. This was followed by normalization, dilution of the libraries to 10nM and pooling 3 samples together. The 10nM libraries were denatured using NaOH, diluted to 8pM and clusters were generated on a paired end flowcell (1 pool/lane) using TruSeq PE cluster kit v3-cBOT-HS on cBot system. Paired end sequencing was carried out on HiSeq 2000 using TruSeq SBS kit v3-HS (200 cycles).
Sample-wise data obtained in the RNAseq has been summarized in the supplementary. Briefly, .fastq files were checked using FASTQC and overall Q score was found to be >20. There was no adapter contamination. Overrepresented sequences were not removed. Reads were mapped on hg38 using Tophat, followed by isoform quantification (Cufflinks) and collation (Cuffmerge). Overall read mapping rate was between 59-86.3% and concordant pair alignment ranged between 53.1 and 81.1%. Cuffdiff was used to calculate the differential expression (D.E.; calculated for each experimental condition against untreated).
MTT assay
∼10,000 cells were seeded per well in 96 well plate. Post treatment, freshly prepared MTT (HiMedia, TC191) solution (in 1X PBS) was added to the cells at a final concentration of 0.5mg/ml. Cells were incubated for 3hrs at 37°C in dark and then checked for formazan crystals under the microscope. After removing the solution, the formazan crystals were dissolved in 100µl DMSO (Sigma, D2650) at RT for 5-10 minutes and then absorbance was acquired at 560nm on Tecan.
The raw data for small RNA and mRNA sequencing generated in this study have been submitted to GEO.
dbGap link for the human temporal cortex (MTG) raw sequence reads https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001790.v1.p1
Conflict of interest
None to disclose
Authors contribution
MM and AB designed the study and co-wrote the manuscript along with KS. AB performed conservation analyses, miRNA target prediction, cell culture and molecular biology experiments and helped in RNA seq data analysis. VJ analyzed mRNA and small RNA seq data, ran miRNA target prediction on miRanda and helped in data visualization. KS performed molecular biology experiments, ran miRNA bulge analysis with DD and seminally contributed to improving data visualization along with RK. MF carried out primary human neuron and NPC culture under the supervision of PS. DS performed some of the cellular assays. GC prepared NGS libraries and carried out sequencing, assisted by AB and KS. TEB analyzed the sn-RNA seq data. BP contributed reagents, helped in troubleshooting experiments and provided critical inputs. MM carried out data analysis and supervised the overall study.
All authors have read and approved the final version of the manuscript.
Acknowledgements
Financial support from CSIR in the form of fellowships to AB and KS and through MLP-901 project to MM are acknowledged. VJ was supported by Persistent Systems LTD. GC and DD were supported by fellowships from the University Grants Commission (UGC) and Department of Biotechnology (DBT), respectively. MF and PS acknowledge the support of the facilities provided through Distributed Information Centre at NBRC, Manesar, under the Biotechnology Information System Network (BTISNET) grant, DBT, India. MF was supported by a fellowship from CSIR, New Delhi and PS was partially supported by research grants from DBT and NBRC core funds.
The authors acknowledge Chitra Mohindar Singh Singal, NBRC for her help with the TUNEL assay, Parashar Dhapola for his help with data visualization in the initial phase of this work, Dr. Amit Chaurasia for Jukes Cantor divergence analysis and Drs. Debojyoti Chakraborty and Rakesh Dey for providing reagents and many fruitful discussions.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵




