

ABSTRACT
Innovations in metazoan development arise from evolutionary modifications of gene regulatory networks (GRNs). We report large cryptic variation in the requirement for two key inputs, SKN-1/Nrf2 and MOM-2/Wnt, into the C. elegans endoderm-determining GRN. Some natural variants show a nearly absolute requirement for SKN-1 and MOM-2, while in others, most of the embryos differentiate endoderm in their absence. GWAS and analysis of recombinant inbred lines reveal multiple genetic regions underlying this broad phenotypic variation. A striking reciprocal relationship is seen in which genomic variants, or debilitation of genes involved in endoderm formation, that result in high SKN-1 requirement show low MOM-2/Wnt requirement and vice-versa. Thus, cryptic variation in the endoderm GRN may be tuned by opposing requirements for these two key regulatory inputs. These findings reveal that while the downstream components in the endoderm GRN are common across metazoans, initiating regulatory inputs are remarkable plastic even within a single species.
INTRODUCTION
While the core regulatory machinery that specifies embryonic germ layers and major organ identity in the ancestor of modern animals has been bequeathed to all extant animals, GRN architecture must be able to accommodate substantial plasticity to allow for evolutionary innovation in developmental strategies, changes in selective pressures, and genetic drift [1,2]. Genetic variation, often with neutral effects on fitness, provides for plasticity in GRN structure and implementation [2]. Although studies of laboratory strains of model organisms with a defined genetic background have been highly informative in identifying the key regulatory nodes in GRNs that specify developmental processes [3–5], these approaches do not reveal the evolutionary basis for plasticity in these networks. Which parameters of GRN architecture provide the greatest opportunity for genetically driven evolutionary change, and which are more rigidly fixed? The variation and incipient changes in GRN function and architecture can be discovered by analyzing phenotypic differences resulting from natural genetic variation present in distinct isolates of a single species [6–8].
The endoderm has been proposed to be the most ancient of the three embryonic germ layers in metazoans [9,10], having appeared prior to the advent of the bilateria about 600 Mya [11]. It follows, therefore, that the GRN for endoderm in extant animals has undergone substantial modifications over the long evolutionary time span since its emergence. However, the core transcriptional machinery for endoderm specification and differentiation appears to share common mechanisms across metazoan phylogeny. For example, cascades of GATA-type transcription factors function to promote endoderm development not only in triploblastic animals but in the most ancient creatures that possess endoderm [12–16]. Among the many observations supporting a common regulatory mechanism for establishing the endoderm, it has been found that the endoderm-determining GATA factor, END-1, in the nematode C. elegans, is sufficient to activate endoderm development in cells that would otherwise become ectoderm in Xenopus [17]. This indicates that the role of GATA factors in endoderm development has been preserved since the nematodes and vertebrates diverged from a common ancestor that lived perhaps 600 Mya.
To assess the genetic basis for evolutionary plasticity and cryptic variation underlying early embryonic germ layer specification, we have analyzed the well-described GRN for endoderm specification in C. elegans. The E cell, which is produced in the very early C. elegans embryo, is the progenitor of the entire endoderm, which subsequently gives rise exclusively to the intestine. The EMS blastomere at the four-cell stage divides to produce the E founder cell and its anterior sister, the MS founder cell, which is the progenitor for much of the mesoderm [18]. Both E and MS fates are determined by maternally provided SKN-1, an orthologue of the vertebrate Nrf2 bZIP transcription factor [19–21]. In the laboratory N2 strain, elimination of maternal SKN-1 function (through either knock down or knockout) results in fully penetrant embryonic lethality as a result of misspecification of EMS cell descendants. In these embryos, the fate of MS is transformed to that of its cousin, the mesectodermal progenitor C cell. E cells similarly adopt a C cell-like fate in a majority, but not all, of these embryos [19]. SKN-1 initiates mesendoderm development via the GRN in E and MS cells in part by activating zygotic expression of the MED-1/2 divergent GATA transcription factors [22,23]. This event mobilizes a cascade of GATA factors in the E cell lineage that ultimately direct intestinal differentiation [21,24,25].
This differential requirement for SKN-1 in endoderm (E) and mesoderm (MS) development is determined by its combinatorial action with triply redundant Wnt, MAPK, and Src signaling systems, which act together to polarize EMS [26–29]. MOM-2/Wnt acts through the MOM-5/Frizzled receptor, mobilizing WRM-1/β-catenin, resulting in its cytoplasmic accumulation in the posterior side of EMS. WRM-1, together with LIT-1/NLK kinase, alters both the nucleocytoplasmic distribution and activity of the Wnt effector POP-1/Tcf [29–31], converting it from a repressor of endoderm in the MS cell lineage to an activator in the E cell lineage [32–37]. Loss of MOM-2 expression in the laboratory N2 strain results in a partial gutless phenotype, while removal of both MOM-2 and SKN-1, through either knockdown or knockout, leads to a completely penetrant loss of gut [29], revealing their genetically redundant roles.
The regulatory relationship between SKN-1 and POP-1, the effector of Wnt signaling, shows striking variation even in relatively closely related species, suggesting substantial evolutionary plasticity in this key node in the endoderm GRN. C. elegans embryos lacking maternal POP-1 always make gut, both in the normal E cell lineage and in the MS cell lineage. However, in embryos lacking both SKN-1 and POP-1, endoderm is virtually never made, implying that these two factors constitute a Boolean “OR” logic gate. In contrast, removal of either SKN-1 or POP-1 alone in C. briggsae causes >90% of embryos to lack gut, indicative of an “AND” logic gate (Fig. 1A, B) [38].

A) Under normal conditions, signaling from the posterior P2 cell (Wnt, MAPK and Src) results in asymmetric cortical localization of Wnt signaling pathway components in EMS leading to POP-1 asymmetry in the descendants of EMS, with high levels of nuclear POP-1 in anterior MS and low levels of nuclear POP-1 in the posterior, E, daughter cell. In the anterior MS cell, high nuclear POP-1 represses the END genes, allowing SKN-1 to activate MS fate. In the posterior E cell, which remains in contact with P2, POP-1 is converted to an activator and, along with SKN-1, activates the END genes, resulting in endoderm fate. Loss of skn-1, either by RNAi or in loss-of-function mutants, causes 100% of the embryos to arrest; in 70% of the arrested embryos, EMS gives rise to two C-like cells, while in the remaining 30% only MS is converted to a C fate; the posterior daughter retains its E fate. Loss of mom-2 leads to embryonic arrest with a partially penetrant E→MS cell fate transformation, resulting in MS-like daughter cells. (B) Regulatory logic of SKN-1 and POP-1 in E specification in C. elegans, C. briggsae and a hypothetical intermediate state. POP-1* denotes the activated state. (C-H) Gut visualization in embryos affected by skn-1 RNAi. (C-E) arrested embryos without endoderm, (F-H) arrested embryos with endoderm. (C, F) DIC images of arrested embryos ∼12 hours after egg laying. (D, G) the same embryos expressing the gut-specific elt-2::GFP reporter, and (E,H) birefringent gut granules under polarized light. All embryos showing gut birefringence also show elt-2::GFP expression. (I, J) Fields of arrested skn-1(RNAi) embryos in wild isolate strains JU1491 (I) and JU440 (J), which reflect the extremes in the spectrum of requirement of SKN-1 in gut development at 0.9% and 60%, respectively.
In this study, we sought to determine whether the aforementioned changes in regulatory logic of the two major inputs into endoderm development are evident within the radiation of a single species. The availability of many naturally inbred variants (isotypes) of C. elegans that show widespread genomic variation [39–41], provides a genetically rich resource for investigating potential quantitative variation in developmental GRNs. We report here that the requirement for activation of the endoderm GRN by SKN-1 or MOM-2, but not POP-1, is profoundly variable between natural C. elegans isolates, and even between very closely related isotypes. Thus, the key regulatory inputs into this major embryonic decision is subject to exceedingly rapid evolutionary modification. Genome-wide association studies in isolates from the natural populations and targeted analysis of recombinant inbred lines (RILs), revealed that a multiplicity of loci and their interactions are responsible for the variation in the developmental requirement for SKN-1 and MOM-2. We identified a striking reciprocal requirement for SKN-1 and MOM-2: loci associated with a high requirement for SKN-1 show a lower requirement for MOM-2 and vice-versa. We further identified several other endoderm regulatory factors, including RICT-1, PLP-1, and MIG-5, that show similar reciprocal relationships between these two GRN inputs. These findings reveal that the activation of the GRN network in specifying a germ layer, one of the most critical and early developmental switches in embryos, is subject to remarkable genetic plasticity during the radiation of a species and that the dynamic and rapid change in network architecture reflects influences distributed across many genetic components that affect both SKN-1 and Wnt pathways.
MATERIALS AND METHODS
C. elegans strains and maintenance
All wild isolates, each with a unique haplotype [40], were obtained from the Caenorhabditis Genetics Center (CGC) (see Supplemental file 1). Worm strains were maintained as described [42] and all experiments were performed at 20°C unless noted otherwise. The following mutant and transgenic strains were used in this study: JJ185 dpy-13(e184) skn-1(zu67) IV; mDp1 (IV;f), JR3666 (elt-2::GFP) X; (ifb-2::GFP) IV, EU384 dpy-11(e1180) mom-2(or42) V/nT1 [let-?(m435)] (IV;V), JJ1057 pop-1(zu189) dpy-5(e61)/hT1 I; him-5(e1490)/hT1 V, KQ1366 (rict-1(ft7) II, SU351 mig-5(rh94)/mIn1 [dpy-10(e128) mIs14] II, and RB1711 plp-1(ok2155) IV.
RNAi
Feeding-based RNAi experiments were performed as described [43]. RNAi clones were obtained from either the Vidal [44] or Ahringer libraries [45]. RNAi bacterial strains were grown at 37°C in LB containing 50 μg/ml ampicillin. The overnight culture was then diluted 1:10. After 4 hours of incubation at 37°C, 1 mM of IPTG was added and 60μl was seeded onto 35mm agar plates containing 1 mM IPTG and 25 μg/ml carbenicillin. Seeded plates were allowed to dry and used within five days. Five to 10 L4 animals were placed on RNAi plate. 24 hours later, they were transferred to another RNAi plate and allowed to lay eggs for four or 12 hours (12 hours for skn-1 RNAi and four hours for the other RNAi). The adults were then removed, leaving the embryos to develop for an extra 7-9 hours. Embryos were quantified and imaged on agar pad using Nikon Ti-E inverted microscope.
Antibody staining
The embryonic gut cells and nuclei of all cells were stained with MH33 (mouse anti-IFB-2, deposited to the DSHB by Waterston, R.H.) and AHP418 (rabbit anti-acetylated histone H4, Serotec Bio-Rad) respectively. Fixation and permeabilization were carried out as described previously [46]. Goat anti-mouse Alexa Fluor® 594 and goat anti-rabbit Alexa Fluor® 488 secondary antibodies were used at 1:1000 dilution.
Quantification of endoderm specification
Gut was scored by presence of birefringent gut granule in arrested embryos [47,48]. For skn-1(RNAi), the laboratory strain N2, which shows invariable ∼30% of embryos with endoderm, was used as a control for all experiments.
Introgression of skn-1(zu67), pop-1(zu189), and mom-2(or42) alleles into wild isolate backgrounds
To introgress skn-1(zu67) into wild isolates (WI), males from the wild isolate strains were crossed to JJ186 dpy-13(e184) skn-1(zu67) IV; mDp1 (IV;f) hermaphrodites. mDp1 is a free duplication that rescues the Dpy and lethal phenotypes of dpy-13(e184) and skn-1(zu67) respectively. Animals that have lost the free duplication will be Dpy and produce dead offspring. Wild type F1 hermaphrodites that have lost the free duplication as determined by presence of a ¼ Dpy progeny in the F2 were selected. 10 single non-Dpy F2 hermaphrodite descendants from F1 animals heterozygous for skn-1(zu67) (2/3 of which would be of the genotype WI dpy-13(+) skn-1(+)/ dpy-13(e184) skn-1(zu67) were backcrossed to their respective parental wild strain. 10 F3 hermaphrodites were picked to individual plates. Half of the F3 cross progeny would be heterozygous for dpy-13(e184) skn-1(zu67), as evidenced by presence of F4 Dpy progeny that produced dead embryos. Non-Dpy siblings were used to continue the introgression as described. This was repeated for at least 5 rounds of introgression. The embryonic gutless phenotype in the progeny of the Dpy animals was quantified.
Similarly, to introgress pop-1(zu189) or mom-2(or42) alleles into wild isolates, JJ1057 pop-1(zu189) dpy-5(e61)/hT1 I; him-5(e1490)/hT1V or EU384 dpy-11(e1180) mom-2(or42) V/nT1 [let-?(m435)] (IV;V) were used, respectively. The mutant strain was crossed to the wild isolates. Non-Dpy F2 animals heterozygous for the chromosomal mutation were selected and backcrossed to their respective parental wild strain for at least four rounds of introgression for pop-1 and seven rounds for mom-2. The embryonic gutless phenotype in the progeny of the Dpy animals was quantified, as above.
Statistical Analyses: GWAS and EMMA
All data were analyzed and plotted using R software v 3.2.3 (https://www.r-project.org/). GWAS for both phenotypes was performed using C. elegans wild isolates and a previously published SNP map containing 4,690 SNPs [40] with the EMMA R package. P-values were calculated using mixed model analysis [49] (emma.REML.t() function) and IBS kinship matrix to account for population structure. For skn-1 and mom-2 RNAi phenotypic data, a genome-wide permutation-based FDR was also calculated for the EMMA results from 10,000 permuted values [50,51]. In addition, a linear model GWAS was performed with the same SNP map (but no kinship matrix) on both mom-2 and skn-1 datasets, with FDR calculations obtained from 10,000 permuted values. However, owing to the skewed nature of the mom-2(RNAi) data (Supplemental Fig. 4), genome-wide permutation-based FDR thresholds did not reveal any significant loci. The p-values for each individual SNP were then adjusted based on 1000 permutations at each locus. Significance thresholds were set at p<0.01 and 0.001.
Correlation Analysis
In order to test for the relationship between mom-2 (RNAi) and skn-1 (RNAi) phenotypic data, the difference between median phenotypic values for each SNP were calculated independently on both a genome-wide level (N = 4690) and at the SNPs most significantly associated with the mom-2 (RNAi) phenotype (N = 45, p < 0.01). Pearson’s correlation test was used to calculate correlation between median phenotypic values for genome-wide analysis using a sliding window (N = 50 SNPs). Spearman’s Rho was used to calculate the correlation using only SNPs most significantly associated with mom-2 GWAS.
RIL construction and Genotype-By-Sequencing (GBS)
Recombinant inbred lines (RILs) were created by crossing an N2 hermaphrodite and an MY16 male. 120 F2 progeny were cloned to individual plates and allowed to self-fertilize for 10 generations. A single worm was isolated from each generation to create inbred lines. A total of 95 lines were successfully created and frozen stocks were immediately created and kept at −80°C (Supplemental File 2), prior to DNA sequencing.
DNA was extracted using Blood and Tissue QIAGEN kit from worms from each of the RILs grown on four large NGM plates (90×15mm) with OP50 E. coli until starved (no more than a day). Samples were submitted in 96-well plate format at 10 ng/µl < n < 30 ng/µl. GBS libraries were constructed using digest products from ApeKI (GWCGC), using a protocol modified from [52]. After digestion, the barcoded adapters were ligated and fragments < 100bp were sequenced as single-end reads using an Illumina HiSeq 2000 lane (100 bp, single-end reads).
SNP calling was performed using the GBSversion3 pipeline in Trait Analysis by aSSociation, Evolution and Linkage (TASSEL) [53]. Briefly, fastq files were aligned to reference genome WS252 using BWA v. 0.7.8-r455 and SNPs were filtered using vcftools [54]. Samples with greater than 90% missing data and SNPs with minor allele frequencies (mAF) of <1% were excluded from analysis, identifying 27,396 variants.
QTL mapping using R/qtl
Variants identified by GBS pipeline were filtered to match the SNPs present in the parental MY16 strain (using vcftools –recode command), and variants were converted to a 012 file (vcftools –012 command). Single-QTL analysis was performed in R/QTL [55] using 1770 variants and 95 RILs. Significant QTL were determined using Standard Interval Mapping (scanone() “em”) and genome-wide significance thresholds were calculated by permuting the phenotype (N =1,000). Change in log-likelihood ratio score of 1.5 was used to calculate 95% confidence intervals and define QTL regions [56].
RESULTS
Extensive natural cryptic variation in the requirement for SKN-1 in endoderm specification within the C. elegans species
The relationship between SKN-1 and Wnt signaling through POP-1 in the endoderm GRN has undergone substantial divergence in the Caenorhabditis genus [38]. While neither input alone is absolutely required for endoderm specification in C. elegans, each is essential in C. briggsae, which has been estimated to have diverged from C. elegans ∼20-40 Mya [57,58]. In contrast to the C. elegans N2 laboratory strain, removal of either SKN-1 or POP-1 alone results in fully penetrant conversion of the E founder cell fate into that of the mesectodermal C blastomere and of E to MS fate, respectively, in C. briggsae [38]. These findings revealed that the earliest inputs into the endoderm GRN are subject to substantial evolutionary differences between these two species (Fig. 1B). We sought to determine whether incipient evolutionary plasticity in this critical node at the earliest stages of endoderm development might be evident even within a single species of the Caenorhabditis genus by assessing their requirement in C. elegans wild isolates and testing whether the quantitative requirements of each input were correlated.
Elimination of detectable maternal SKN-1 from the laboratory N2 strain by either a strong (early nonsense) chromosomal mutation (skn-1(zu67)), or by RNAi knockdown, results in a partially penetrant phenotype: while the E cell adopts the fate of the C cell in the majority of embryos, and gut is not made, ∼30% of arrested embryos undergo strong gut differentiation, as evidenced by the appearance of birefringent, gut-specific rhabditin granules, or expression of elt-2::GFP, a marker of the developing and differentiated intestine (Fig. 1C-H). We found that RNAi of skn-1 in different N2-derived mutant strains gave highly reproducible results: 100% of the embryos derived from skn-1(RNAi)-treated mothers arrest (n>100,000) and 32.0 ± 1.9% of the arrested embryos exhibited birefringent gut granules (Fig. 2A; Supplemental Fig. 1). We found that the LSJ1 laboratory strain, which is derived from the same original source as N2, but experienced very different selective pressures in the laboratory owing to its constant propagation in liquid culture over 40 years [59], gave virtually identical results to that of N2 (31.0% ± s.d 1.2%), implying that SKN-1-independent endoderm formation is a quantitatively stable trait. The low variability in this assay, and high number of embryos that can be readily examined (≥500 embryos per experiment), provides a sensitive and highly reliable system with which to analyze genetic variation in the endoderm GRN between independent C. elegans isolates.

(A) Spectrum of skn-1(RNAi) effects across the C. elegans isolates. The effects of skn-1(RNAi) are quantified as the average percentage of arrested embryos with endoderm (y-axis). All wild isolates treated with skn-1(RNAi) resulted in 100% embryonic arrest (n >500 embryos per replicate per isotype and at least two replicates per isotype). (B) Comparison of skn-1(RNAi) phenotype using two different gut markers (birefringent gut granules and MH33 staining of IFB-2) in five different genetic backgrounds. In all cases, no significant statistical difference was found between the two quantitative methods. Fisher’s exact test (NS p-value>0.05). (C) Comparison of skn-1(RNAi) and skn-1(zu67) effects on endoderm development in six different genetic backgrounds. For each color-coded strain, the first value is of the skn-1(RNAi) results (5 replicates), while the second is the result for the skn-1(zu67) allele introgression (10 replicates). For all strains (with the exception of MY16), no significant statistical difference was found between the RNAi knockdown and corresponding skn-1(zu67) allele effects on endoderm development. Student t-test (NS p-value>0.05, * p-value<0.05).
To assess variation in SKN-1 requirement within the C. elegans species, we analyzed the outcome of knocking down SKN-1 by RNAi in 96 unique C. elegans wild isolates [40]. Owing to their propagation by self-fertilization, each of the isolates (isotypes) is a naturally inbred clonal population that is virtually homozygous and defines a unique haplotype. The reported estimated population mutation rate averages 8.3×10−4 per bp [40], and we found that a substantial fraction (29/97) of isotypes were quantitatively indistinguishable in phenotype between the N2 and LSJ1 laboratory strains (Fig. 2A). We found that all strains, with the exception of the RNAi-resistant Hawaiian CB4856 strain, invariably gave 100% embryonic lethality with skn-1(RNAi), showing that on the basis of that criterion all strains are fully sensitive to RNAi. However, we observed dramatic variation in the fraction of embryos with differentiated gut across the complete set of strains, ranging from 0.9% to 60% (Fig. 2A). Repeated measurements with >500 embryos per replicate per strain revealed very high reproducibility (Supplemental Fig. 1), indicating that even small differences in the fraction of embryos generating endoderm could be reproducibly measured. Further, we found that some wild isolates that were subsequently found to have identical genome sequences also gave identical results. We note that these results contrast with those of Paaby et al. [60], who found that RNAi in liquid culture of a set of 55 wild isolates resulted in much weaker effects, both on lethality and on gut differentiation. This difference is likely attributable to variability in RNAi efficacy in the latter study [61,62].
Although birefringent and autofluorescent rhabditin granules have been used as a marker of gut specification and differentiation in many studies [47,48], it is conceivable that the variation in fraction of embryos containing this marker that we observed might reflect variations in gut granule formation rather than in gut differentiation per se. We note that embryos from all strains showed a decisive “all-or-none” phenotype: i.e., they were either strongly positive for gut differentiation or completely lacked gut granules, with virtually no intermediate or ambiguous phenotypes. A threshold of gene activity in the GRN has been shown to account for such an all-or-none switch in gut specification [22,63,64]. This observation is inconsistent with possible variation in gut granule production: if SKN-1-depleted embryos were defective in formation of the many granules present in each gut cell, one might expect to observe gradations in numbers or signal intensity of these granules between gut cells or across a set of embryos. Nonetheless, we extended our findings by analyzing expression of the gut-specific intermediate filament IFB-2, a marker of late gut differentiation, in selected strains representing the spectrum of phenotypes observed (Fig. 2B). As with gut granules, we found that embryos showed all-or-none expression of IFB-2. In all cases, we found that the fraction of embryos containing immunoreactive IFB-2 was not significantly different (Fisher’s exact test, p-values > 0.05) from the fraction containing gut granules, strongly suggesting that the strains vary in endoderm specification per se and consistent with earlier studies of SKN-1 function [19,22].
Although we found that skn-1(RNAi) was 100% effective at inducing embryonic lethality in all strains (with the exception of the RNAi-defective Hawaiian strain, CB4856), it is conceivable that, at least for the strains that showed a weaker phenotype than for N2 (i.e., higher number of embryos specifying endoderm), the variation observed between strains might be attributable to differences in RNAi efficacy rather than in the endoderm GRN. To address this possibility, we introgressed the strong loss-of-function skn-1(zu67) chromosomal mutation into five wild isolates whose phenotypes spanned the spectrum observed (ranging from 2% of embryos with differentiated gut for MY16 to 50% for MY1) (Fig. 2C). In all cases, we found that introgression of the allele through five rounds of backcrosses resulted in a quantitative phenotype that was similar or identical to that of the effect observed with skn-1(RNAi). The phenotypes of the introgressed allele were significantly different (p-values <0.01) from that of the parental N2 skn-1(zu67) strain, except for DL238, whose skn-1(RNAi) phenotype was indistinguishable from that of N2. The results obtained by introgression from four of the isotypes (CX11262, DL238, EG4724 and MY1), were not statistically different (Student t-test, p-values >0.05) from the corresponding RNAi knock down results (Fig. 2C) (i.e., the phenotype was suppressed or enhanced relative to N2 in these genetic backgrounds to the same extent as with skn-1(RNAi)). However, while the MY16 skn-1(zu67) strain shifted in the predicted direction (i.e., became stronger) when compared to the N2 strain, it was a weaker effect than was evident by RNAi knockdown, even following eight rounds of introgression. Nonetheless, diminished RNAi efficacy in MY16 cannot explain the large difference in skn-1(RNAi) phenotype between N2 and MY16, as the latter phenotype is much stronger, not weaker, than the former. As described below, we identified a modifier locus in the MY16 strain that is closely linked to the skn-1 gene; it seems likely that the N2 chromosomal segment containing this modifier was carried with the skn-1(zu67) mutation through the introgression crosses, thereby explaining the somewhat weaker phenotype of the introgressed allele compared to the RNAi effect in MY16. The results of introgression of the skn-1(zu67) chromosomal mutation confirm that the extreme variation in skn-1(RNAi) phenotype between the wild isolates results from bona fide cryptic variation in the endoderm GRN, rather than differences in RNAi efficacy.
We note that the strength of skn-1(RNAi) phenotype does not correlate with phylogenetic relatedness between the strains (Mantel test r = 0.21, NS). To illustrate, while some closely related strains (e.g., MY16 and MY23) showed a similar gut developmental phenotype, some very closely related strains (e.g., JU1491 and JU778) had phenotypes on the opposite ends of the phenotypic spectrum (Fig. 3A). We also did not observe any significant correlation between geographical distribution and skn-1 (RNAi) phenotype (Fig. 3B). These findings suggest that the endoderm GRN may be subject to rapid intraspecies evolutionary divergence and suggests that a small number of loci may underlie variation in the trait.

(A) skn-1(RNAi) phenotype of 97 isolates arranged with respect to the neighbor-joining tree constructed using 4,690 SNPs and pseudo-rooted to QX1211. Red asterisk indicates an example of closely related strains (MY23 and MY16) with similar phenotype, while black asterisks indicate example sister strains (JU778 and JU1491; JU561 and JU1652) with distinct phenotype. Phylogenetic relatedness and phenotype (measured as Euclidean distance) are not significantly correlated (Mantel test, r = 0.21, NS). (B) Worldwide distribution of skn-1(RNAi) phenotype across 97 wild isolates. Each circle represents a single isotype.

(A) Spectrum of mom-2(RNAi) effects across the C. elegans isolates. The effects of mom-2(RNAi) are quantified as the average percentage of arrested embryos with endoderm (y-axis). Each column represents the mean for each wild isolate (n >500 embryos were scored for each experiment with at least two replicates per isotype). (B) Comparison of mom-2(RNAi) phenotype using two different gut markers (birefringent gut granules and MH33 staining of IFB-2) in three different genetic backgrounds. In all cases, no significant statistical difference was found between the two quantitative methods. Fisher’s exact test (NS p-value>0.05). (C) Comparison of the effect of mom-2(or42) on endoderm development after introgression into four different genetic backgrounds. At least three independent introgressed lines were studied for each wild isotype. The results were compared to N2; mom-2(or42) shown by dashed line. Student t-test (*** p-value<0.001).
Cryptic variation in the quantitative requirement for MOM-2 Wnt, but not POP-1, in endoderm development
The switch in the relationship of the SKN-1 and Wnt inputs between C. elegans (“OR” operator) and C. briggsae (“AND” operator) [38], and the extensive variation in the requirement for SKN-1 seen across C. elegans isolates, raised the possibility that the quantitative requirement for Wnt components might vary between unique isolates of C. elegans. It has been shown that signaling from Ras pathway varies in different C. elegans wild isolates and hyperactive Wnt signaling can compensate for reduced Ras activity in the vulva signaling network [6,65]. Given that removal of the maternal Wnt input also results in a partially penetrant gut defect (through either knock-out or knockdown of Wnt signaling components), it is conceivable that a compensatory relationship may exist between the SKN-1 and Wnt inputs. We investigated this possibility by examining the requirement for the MOM-2/Wnt ligand in the same wild isolates that were tested for the SKN-1 gut developmental requirement. Indeed, we observed broad variation in the requirement for MOM-2/Wnt in activation of the endoderm GRN between isotypes. mom-2(RNAi) of 94 isotypes resulted in embryonic arrest, indicating that, as with skn-1(RNAi), mom-2(RNAi) was effective at least by the criterion of lethality. Two isotypes, CB4853 and EG4349, did not exhibit mom-2(RNAi)-induced lethality and were omitted from further analyses. In the affected strains, the fraction of mom-2(RNAi) embryos with differentiated gut varied from ∼40% to ∼99% (Fig. 4A). As with skn-1(RNAi), the mom-2(RNAi) phenotype of isotypes N2, JU440, and JU1213 was further confirmed by immunostaining with IFB-2 (Fig 4B), again demonstrating that birefringence of gut granules is a reliable proxy for endoderm formation for this analysis.
To assess whether the observed variation in the mom-2(RNAi) phenotype reflected differences in the GRN or RNAi efficacy, the mom-2(or42) allele was introgressed into three different genetic backgrounds chosen from the extreme ends of the phenotypic spectrum. mom-2(RNAi) of the laboratory N2 strain resulted in the developmental arrest of embryos. Of those, ∼90% contained differentiated endoderm, a result that was highly reproducible. In contrast, the introgression of an apparent loss-of-function allele, mom-2(or42), into the N2 strain results in a more extreme phenotype: only ∼28% of embryos show endoderm differentiation (Fig. 4C) [29]. While this discrepancy can partly be explained by incomplete RNAi efficacy, it is notable that the penetrance of mom-2 alleles vary widely [29]. We observed strain-specific variation in embryonic lethality response to RNAi of mom-2 between the different isotypes. However, we found that the mom-2(or42) introgressed strains show qualitatively similar effects to those observed with mom-2 RNAi. For example, the mom-2(or42) allele introgressed into the isotype JU1213 background resulted in a severe gutless phenotype (5.7% ± s.d 2.4%; n=2292) a similar but more extreme effect than was seen with RNAi (34.0% ± s.d 1.5%; n=1876). This is the strongest phenotype that has been reported for any known mom-2 allele. On the other hand, introgression of the mom-2 mutation gave rise to a significantly higher fraction of embryos with endoderm in isotypes DL226 (55.2% ± s.d 1.2%, n=1377) and PB303 (65.5% ± s.d 4.9%, n=2726), relative to the laboratory strain N2 (29.1% ± s.d 3.1%; n=1693), consistent with the RNAi phenotypes (Fig. 4C). These findings indicate that the differential requirement for MOM-2 is, at least in part, attributable to genetic modifiers in these strains.
As the MOM-2/Wnt signal is mediated through the POP-1 transcription factor, we sought to determine whether the requirement for POP-1 might also vary between isolates. We found that, while pop-1(RNAi) resulted in 100% embryonic lethality across all 96 RNAi-sensitive isolates, 100% of the arrested embryos contained a differentiated gut (n>500 for each isolate scored) (results not shown). Thus, all isolates behave similarly to the N2 strain with respect to the requirement for POP-1. These results were confirmed by introgressing a strong loss-of-function pop-1(zu189) allele into four wild isolates (N2, MY16, JU440, and KR314) (Supplemental Fig. 2). The lack of variation in endoderm specification after loss of POP-1 is not entirely unexpected. As has been observed in a pop-1(-) mutant strain, elimination of the endoderm-repressive role of POP-1 in the MS lineage (which is not influenced by the P2 signal) supersedes its endoderm activating role in the presence of SKN-1. Indeed, the original observation that all pop-1(-) embryos in an N2 background contain gut masked the activating function for POP-1, which is apparently only in the absence of SKN-1 [32,34,36]. It is likely that, as with the N2 strain, gut arises from both E and MS cells in all of these strains; however, as we have scored only for presence or absence of gut, it is conceivable that the E lineage is not properly specified in some strains, a possibility that cannot be ruled out without higher resolution analysis.
Genome-wide association studies (GWAS) and analysis of RILs identify multiple genomic regions underlying variation in the two major endoderm GRN inputs
We sought to examine the genetic basis for the wide variation in SKN-1 and Wnt requirements across C. elegans isolates and to evaluate possible relationships in the variation seen with the SKN-1 and Wnt inputs by performing linear-model GWAS using the available SNP makers and map [40]. This analysis identified two highly significantly associated regions on chromosome IV and V (FDR < 1.5) that underlie the variation in SKN-1 requirement (Supplemental Fig. 3A). To ensure that these two QTLs were not an artifact of genetic relatedness between sets of strains, we applied the more stringent EMMA (Efficient Mixed-Model Analysis) algorithm, which adjusts for population structure (Fig. 5A) [49,66]. This approach also identified the same significant location on chromosome IV. The two mapping approaches show a moderate linear correlation (Spearman correlation coefficient = 0.43) (Supplemental Fig. 3B). In each case, the most statistically significant SNPs within each of the two identified QTLs are highly associated with the observed variance in SKN-1 requirement (Fig 5C; Supplemental Fig. 3C, D).

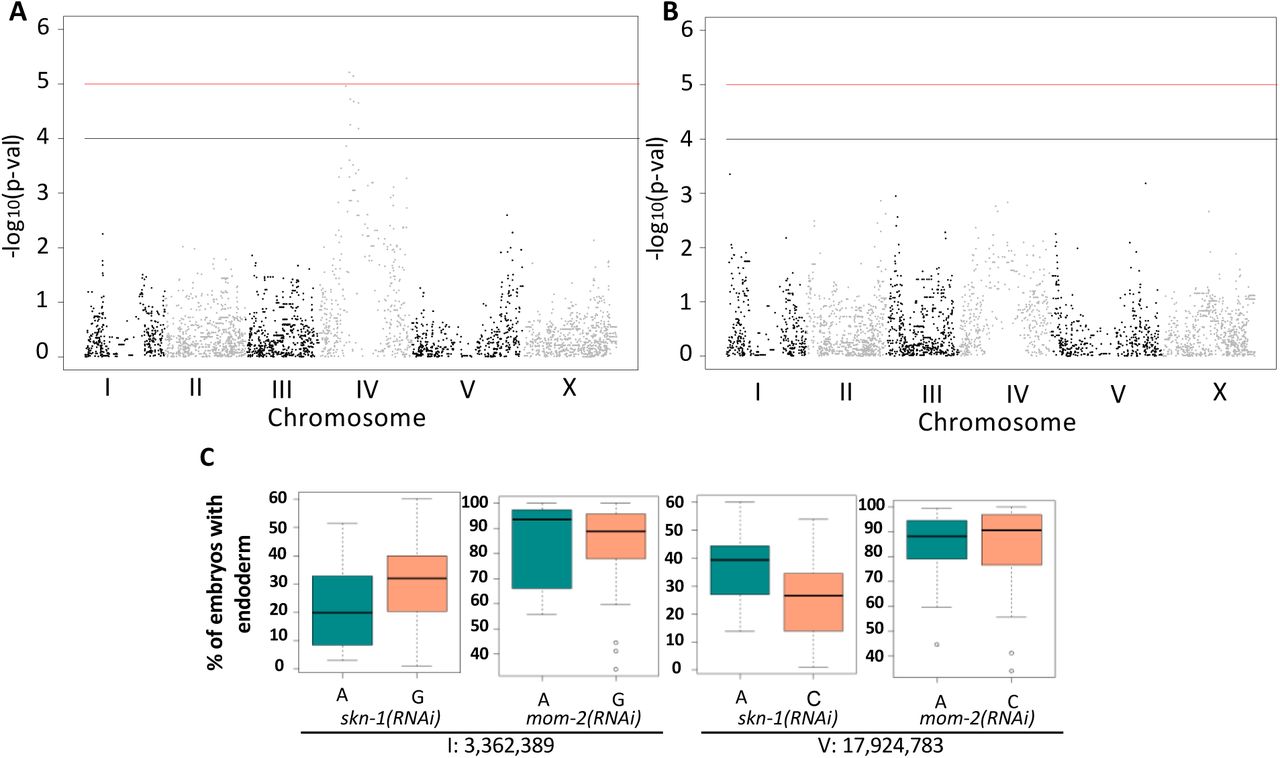
(A) Manhattan plot of skn-1(RNAi) EMMA. The red line indicates a genome-wide 1.5% FDR (permutation-based FDR, from 10,000 permutated results). Black line represents 3.0% FDR. The chromosomes are color-coded. The y axis is the –log10 of p-value. (B) Manhattan plot of mom-2 (RNAi) EMMA. The y axis is the – log10 of p-value. Genomic regions are shown on the x-axis. (C) Effect plots of the significant SNPs from skn-1(RNAi) GWAS at position 3,362,389 bp on chromosome I and position 17,924,783 bp on chromosome V (see Supplemental Fig. 3). Horizontal lines within each box represent the median, and the boxes represent 25th–75th percentile.
GWAS analysis on the mom-2(RNAi) phenotypic variation proved more challenging because this phenotype showed a highly skewed distribution (Shapiro-Wilk’ test W =0.8682, p-value = 1.207X10-7) (Supplemental Fig. 4). Nevertheless, by applying a linear model GWAS and adjusting the individual p-values using a permutation-based approach (see Materials and Methods) and EMMA (Fig. 5B, Supplemental Fig. 5A), both GWAS and EMMA revealed highly correlated results (Pearson’s R = 0.95, p-value < 2.2e-16) (Supplemental Fig. 5B). Although GWAS identified 45 significant SNPs distributed across the genome (GWAS adjusted p-values < 0.01), EMMA did not reveal any significant genomic regions for mom-2(RNAi) variation, suggesting that the MOM-2 requirement is a highly complex trait influenced by many loci.
However, when we compared the p-values of individual SNPs from skn-1(RNAi) and mom-2(RNAi) EMMA, a substantial overlap in the central region of chromosome of chromosome IV was observed (Supplemental Fig. 6). This genomic region showed striking reciprocality in phenotype compared to the SKN-1 results, as described below.

(A) mom-2(RNAi) (left) and skn-1(RNAi) (right) phenotype of RILs. The phenotype of the parental strains, MY16 and N2 are shown by red and blue lines, respectively. (B) QTL analyses (interval mapping) of skn-1(RNAi) (blue line) and mom-2(RNAi) (red line) phenotype shown in (A). Genomic regions are shown on the x-axis and LOD score is shown on the y-axis. (C-F) Effect plots of significant SNPs from mom-2(RNAi) (C, D) and skn-1(RNAi) (E, F) QTL analyses of RILs. Each dot represents a RIL. The parental alleles are shown on the x-axis, and skn-1(RNAi) or mom-2(RNAi) phenotypes on the y-axis. Confidence intervals for the average phenotype in each genotype group are shown.
In an effort to narrow in on causal loci underlying the skn-1(-) and mom-2(-) phenotypic variation, and to assess possible relationships between these two GRN inputs, we prepared and analyzed 95 recombinant inbred lines (RILs) between two C. elegans isotypes, N2 and MY16. These strains were chosen for their widely varying differences in requirement for both SKN-1 and MOM-2 (see Materials and Methods). In contrast to the very low variation seen between multiple trials of each parental strain, analysis of the RNAi treated RIL strains (>500 embryos/RIL) revealed a very broad distribution of phenotypes. We found that, while some RILs gave phenotypes similar to that of the two parents, many showed intermediate phenotypes and some were reproducibly more extreme than either parent, indicative of transgressive segregation [67]. For skn-1(RNAi), the phenotype varied widely across the RILs, with 1 to 47% of embryos containing gut (Fig. 6A). This effect was even more striking with mom-2(RNAi), for which virtually the entire possible phenotypic spectrum was observed across a selection of 31 RILs representing the span of skn-1(RNAi) phenotypes. The mom-2(RNAi) phenotypes ranged from RILs showing 3% of embryos with gut to those showing 92% (Fig. 6A). It is noteworthy that one RIL (JR3572, Supplemental File 2) showed a nearly completely penetrant gutless phenotype, an effect that is much stronger than has been previously observed for mom-2(-) [29]. These results indicate that a combination of natural variants can nearly eliminate a requirement for MOM-2 altogether, while others make it virtually essential for endoderm development. Collectively, these analyses reveal that multiple quantitative trait loci (QTL) underlie SKN-1- and MOM-2-dependent endoderm specification.
To identify QTLs from the recombinant population, we performed linkage mapping for both phenotypes using both interval mapping and marker regression. For skn-1(RNAi), two major peaks were revealed on chromosomes II and IV (above 1% FDR estimated from 1,000 permutations). Two minor loci were found on chromosomes I and X (suggestive linkage, above 20% FDR) (Fig 6B). For mom-2(RNAi), two major independent QTL peaks were found on Chromosomes I and II (above the 5% FDR estimated from 1,000 permutations). Although the candidate peaks observed on Chromosome IV for skn-1(RNAi) (Fig. 6B) did not appear to overlap with those for mom-2(RNAi), overlap was observed between the Chromosomes I and II candidate regions for these two phenotypes. Moreover, many N2 and MY16 alleles in the QTL showed a reciprocal effect on SKN-1 and MOM-2 dependence (Fig. 6C-F).
A cryptic compensatory relationship between the SKN-1 and Wnt regulatory inputs
As with skn-1(RNAi) findings, we found no correlation between the mom-2(RNAi) phenotype and phylogenetic relatedness or geographical distribution (Supplementary Fig. 7A, B), suggesting rapid intraspecies developmental system drift. This, together with the preceding findings, unveiled wide cryptic variation in the requirements for both SKN-1 and MOM-2/Wnt in the endoderm GRN and raised the possibility of a functional overlap in this variation. Comparisons of the GWAS and QTL mapping results for skn-1 and mom-2 showed an overlap in candidate QTL regions on chromosome I, II and IV (Fig. 5, Fig 6, Supplemental Fig. 6), suggesting a possible relationship between the genetic basis underlying these two traits. It is conceivable that some genetic backgrounds are generally more sensitive to loss of either input (e.g, the threshold for activating the GRN is higher) and others more robust to single-input loss. Alternatively, a higher requirement for one input might be associated with a relaxed requirement for the other, i.e., a reciprocal relationship.
As an initial assessment of these alternatives, we examined whether the requirements for SKN-1 and MOM-2 across all strains were significantly correlated. This analysis revealed no strong relationship between the cryptic variation in the requirement for these inputs seen across all the strains (Spearman correlation R=0.18, p-value=0.07) (Fig. 7A). This apparent lack of correlation at the level of strains is not unexpected, as many factors likely contribute to the cryptic variation and the comparison reflects the collective effect of all causal loci in the genome of each strain.

(A) Comparison of skn-1(RNAi) and mom-2(RNAi) phenotype in 94 strains tested. No correlation was found (Spearman correlation R=0.1844, p-value=0.07). Each dot corresponds to a wild isolate. Y-axis, skn-1(RNAi) phenotype, x-axis, mom-2(RNAi) phenotype. (B) Negative correlation of skn-1(RNAi) and mom-2(RNAi) allelic differences at the top SNPs from the mom-2(RNAi) GWAS as calculated by subtracting the median (of the skn-1(RNAi) and mom-2(RNAi) phenotypes) of one allele to the median of the second. Pearson’s correlation R=−0.6099, p-value=0.0001. (C) Genome-wide negative correlation in allelic effects. Each dot represents a SNP, and 4,690 SNPs in total were analyzed. The chromosomes and their corresponding regression lines are color-coded. The R-value of each chromosome is indicated. Correlation sliding window of (D) chromosome I and (E) chromosome IV. Windows of 50 SNPs were used to calculate the correlation coefficient and p-value. Black circles represent the correlation coefficient (R value) for each window (scale on the x-axis). Black line indicates the 0 threshold. Grey line represents the –log10 of the p-values for the corresponding correlation windows (scale on the y-axis). Grey horizontal line is the significance threshold set at p-value=0.01. Blue dotted lines divide chromosomal region into left, middle and right arms.
We next sought to examine possible relationships between the two GRN inputs at higher resolution by comparing association of specific genetic regions with the quantitative requirement for each input. We took advantage of the available sequence data for all the isotypes tested [40] and examined the impact of each allele on the skn-1(RNAi) and mom-2(RNAi) phenotypes for the SNPs that were most highly associated with the variation in requirement for SKN-1 by calculating the difference between the phenotypic medians for each allele at each SNP. Comparisons of the GWAS analyses for variation in the requirement for SKN-1 and MOM-2 showed a particularly strong overlap in candidate QTL regions for the two phenotypes on chromosome IV (Fig. 5A, B, Supplemental Fig. 3A, 5, 6). To assess the relationship between these and other significant SNPs, we analyzed the top 45 SNPs from the mom-2 GWAS data and found a strong negative correlation between the allelic effects for SKN-1 and MOM-2 dependence. With very few exceptions, SNPs associated with a milder skn-1(RNAi) phenotype (higher % with endoderm) showed a stronger mom-2(RNAi) phenotype (low %) and vice versa (Fig. 7B), with an overall highly significant negative correlation (Pearson’s correlation R=−0.6099, p-value=0.0001).
The strong negative correlation we observed between the strength of the skn-1(RNAi) and mom-2(RNAi) phenotypes for the SNPs that are most significantly associated with the variation might be explained in part by the large blocks of linkage disequilibrium observed in C. elegans [40]. Thus, in principle, relatively few genomic regions might, by chance, show the reciprocal relationship, in which case all linked high-significance SNPs would similarly show the negative correlation. It was therefore important to assess how widespread and consistent this effect is across the entire genome. We dissected the relationship of the SKN-1 and MOM-2 requirements across all chromosomes by analyzing the phenotypic strength in sliding windows of 50 SNPs each across each chromosome, using all 4,690 SNPs. This analysis revealed a striking overall trend: for all six chromosomes, regions associated with high SKN-1 requirement showed a tendency toward lower MOM-2/Wnt requirement and vice-versa. This effect was most pronounced on chromosome I, which showed a very strong negative correlation (R=−0.69). The effect was also clearly evident on chromosomes IV (R=−0.55), III (R=−0.45), and II (R=−0.43). Though weaker for chromosomes V and X (R=−0.23 for both), the correlation was nonetheless negative for these chromosomes as well (Fig. 7C-E; Supplemental Fig. 8A-E). Thus, the inverse relationship between the MOM-2 and SKN-1 requirement appears to be distributed across the entire genome. The sequences underlying the cryptic variation we observed might not be expected to be uniformly distributed throughout the genome and, indeed, we found that strength of the correlation varied widely between and even within chromosomes (Fig. 7C-E, Supplemental Fig. 8B-E).

(A, B) Loss of RICT-1 or PLP-1 enhances the mom-2(or42) loss-of-endoderm phenotype and suppresses skn-1(zu67) and skn-1(RNAi) phenotype. (C) Loss of MIG-5 enhances the skn-1(zu67) and skn-1(RNAi) phenotype and suppresses mom-2(or42) phenotype. At least three replicates were performed per experiment. Student t-test (*** p-value<0.001). Data represented with Standard Deviations.
Multiple factors reciprocally regulate the requirement for SKN-1 and MOM-2/Wnt
We further explored this relationship between the requirement for SKN-1 and MOM-2 by testing other candidate genes implicated in endoderm development [68–70]. We found that loss of RICT-1, the C. elegans orthologue of the human RICTOR (Rapamycin-insensitive companion of mTOR; [71]), a component of the TORC2 complex, which has been shown to antagonize SKN-1 function [68], results in opposite effects on skn-1(-) and mom-2(-) mutants (Fig. 8A). Specifically, while rict-1(RNAi) suppresses the absence of gut in skn-1(zu67) embryos (skn-1(zu67): 34.3% ± s.d 4.1% with gut vs. skn-1(zu67); rict-1(RNAi): 48.3% ± s.d 4.9%; p=<0.001), we found that it enhances this phenotype in mom-2(or42) mutants (mom-2(or42): 23.8% ± s.d 2.0%; vs. mom-2(or42); rict-1(RNAi): 11.2% ± s.d 3.2%; p<0.001). Confirming this effect, a similar outcome was observed when SKN-1 was depleted by RNAi in rict-1(ft7) chromosomal mutants (skn-1(RNAi): 31.6% ± s.d 4.3% with gut vs. rict-1(ft7); skn-1(RNAi): 45.9% ± s.d 6.3%; p<0.05) (Fig. 8A). Similarly, RNAi depletion of PLP-1, the C. elegans homologue of the Pur alpha transcription factor that has been shown to bind to and regulate the end-1 promoter [69], reciprocally affects the outcome of removing these two inputs in the same direction: loss of PLP-1 function suppresses the skn-1(-) phenotype (to 48.0% ± s.d 6.6%), and strongly enhances the mom-2 phenotype (to 6.9% ± s.d 1.6%). Again, this result was confirmed by RNAi of skn-1 in a plp-1(ok2156) chromosomal mutant (Fig. 8B). Thus, as observed with the effect across the genome with natural variants, we observed a striking reciprocal effect of both of these genes on loss of SKN-1 and MOM-2.
We also observed a reciprocal effect on the SKN-1 and Wnt inputs with MIG-5/dishevelled, a component of the Wnt pathway that acts downstream of the Wnt receptor [70]; however, in this case the effect was in the opposite direction as seen for RICT-1 and PLP-1. Loss of MIG-5 as a result of chromosomal mutation or RNAi leads to enhancement of the skn-1(-) phenotype (mig-5(rh94); skn-1(RNAi): 6.6% ± s.d 2.3%; skn-1(zu67); mig-5(RNAi): 9.4% ± s.d 1.4%) and suppression of the mom-2(-) phenotype (88.6% ± s.d 4.0%) (Fig 8C).
Together, these findings reveal that, as observed with the natural variant alleles (Fig.5C, Fig. 6C-F; Fig. 7B, C), RICT-1, PLP-1, and MIG-5 show opposite effects on the phenotype of removing SKN-1 and MOM-2, suggesting a prevalence of genetic influences that reciprocally influence the outcome in the absence of these two inputs.
DISCUSSION
The remarkable variety of forms associated with the ∼36 animal phyla [72] that emerged from a common metazoan ancestor >600 Mya is the product of numerous incremental changes in GRNs underlying the formation of the body plan and cell types. Here, we describe an unexpectedly broad divergence in the deployment of SKN-1/Nrf and MOM-2/Wnt signaling in generating the most ancient germ layer, the endoderm, within wild isolates of a single animal species, C. elegans. In this study, we report five major findings: 1) while the quantitative requirement for two distinct regulatory inputs that initiate expression of the endoderm GRN (SKN-1 and MOM-2) are highly reproducible in individual C. elegans isolates, there is wide cryptic variation between isolates. 2) Cryptic variation in the requirement for these regulatory factors shows substantial differences even between closely related strains, suggesting that these traits are subject to rapid evolutionary change in this species. 3) Quantitative genetic analyses of natural and recombinant populations revealed multiple loci underlying the variation in the requirement for SKN-1 and MOM-2 in endoderm specification. 4) The quantitative requirements for SKN-1 and MOM-2 in endoderm specification are negatively correlated across the genome, as shown by allelic effect analysis, implying a reciprocal requirement for the two inputs. 5), rict-1, plp-1, and mig-5 reciprocally influence the outcome of skn-1(-) and mom-2(-), substantiating the reciprocal influences on the two GRN inputs. These findings reveal substantial plasticity and complexity underlying SKN-1 and MOM-2/Wnt regulatory inputs in mobilizing a conserved system for endoderm specification.
Together, these findings indicate that, while the core genetic toolkit for the development of the endoderm, the most ancient of the three germ layers, appears to have been preserved for well over half a billion years, the molecular regulatory inputs that initiate its expression in C. elegans vary extremely rapidly over short evolutionary time scales within the species.
Evolutionary plasticity in maternal regulators of embryonic GRNs
The finding that the key regulatory inputs that initiate the endoderm GRN show dramatic plasticity is in accordance with the “hourglass” concept of embryonic development [73–75], in which divergent developmental mechanisms converge on a more constant state (i.e., a “phylotypic stage” at the molecular regulatory level). Indeed, it appears that a downstream GATA factor cascade that directs endoderm specification and differentiation is a highly conserved feature not only across Caenorhabitis species [38,76,77] but, in fact, across the broad spectrum of animal phyla [12–17]. These observations are also consistent with the notion that, while the late stages in organ differentiation involve activation of a very large number of target differentiation genes by a limited set of transcription factors, thereby restricting evolutionary divergence at that stage in the regulatory circuitry, the early stages involve the action of transcription factors on far fewer target genes, hence allowing for much greater evolutionary plasticity [21].
In Drosophila, early maternally acting genes show more rapid evolution than those expressed zygotically [78]. Moreover, maternal patterning systems that spatially regulate conserved patterning gene networks between broadly divergent insect species are highly divergent [79,80]. Further comparisons of early embryonic transcripts across many Drosophila species and Aedes aegypti revealed that maternal transcript pools that, like those of C. elegans skn-1, are present only transiently during early embryogenesis, and expression levels are highly variable across these species, spanning ∼60 My of evolution [81]. What is particularly striking about our findings is that the varying requirement for key maternal regulatory components is seen within the relatively recent radiation of a single species with low genetic diversity [40]. Variation in gene expression predicts phenotypic severity of mutations in different genetic backgrounds [82]. As quantitative transcriptional profiling of C. elegans isotypes advances, it will be of interest to assess whether the highly evolvable requirement for maternal regulatory inputs into the endoderm GRN similarly correlates with rapid divergence in quantitative levels of maternal transcripts that are transiently deployed in early embryos of this species.
Multigenic variation in the requirement for SKN-1 and MOM-2
GWAS and EMMA revealed several major candidate QTLs (Fig. 5, Supplemental Fig. 3, 5), implying that multigenic factors are causally responsible for the differences in requirement for SKN-1 and MOM-2 between isotypes. This multigenic influence was also apparent from analysis of RILs derived from N2 and MY16 parental strains, which identified several loci associated with both traits. In addition, we found substantial epistasis between the different genomic regions underlying this variation. Transgressive segregation of the requirement for both SKN-1 and MOM-2 was seen in the RIL sets (Fig. 6A). For example, the MY16 strain which shows an almost fully penetrant requirement for SKN-1 for gut development, appear to harbor cryptic variants that suppress the requirement for SKN-1, allowing enhanced gut development when combined with genetic factors in the N2 strain.
We observed substantial overlap on chromosome IV in the GWAS/EMMA analyses of the skn-1 and mom-2 requirements in wild isotypes (Fig. 5, Supplemental Fig. 6) and on chromosome II from analyses using RILs (Fig. 6B). This finding raises the possibility that some QTLs may influence requirement for both inputs into the endoderm specification pathway: as SKN-1 and Wnt converge to regulate expression of the end-1/3 genes, it is conceivable that common genetic variants might modulate the relative strength or outcome of both maternal inputs. However, our findings do not resolve whether these genetic variants act independently to influence the maternal regulatory inputs.
Genetic interactions are often neglected in large-scale genetic association studies [83] owing in part to the difficulty in confirming them [84]. Many studies [85–88], including ours here, showed that epistasis can strongly influence the behavior of certain variants upon genetic perturbation. In addition, selection on pleiotropically acting loci facilitates rapid developmental system drift [87,89,90]. Together, epistasis and selection on pleiotropic loci play important roles in the evolution of natural populations [89–92].
Potential compensatory relationships between SKN-1 and MOM-2/Wnt
Although we did not observe a direct correlation between the skn-1(-) and mom-2(-) phenotypes across the isotypes studied here, a clear inverse correlation was observed when testing associated individual SNPs in significantly linked loci (Fig. 7). This reciprocal effect seen across large portions of the genome may be attributable in part to the large LD blocks present on all chromosomes (Fig. 7, Supplemental Fig. 8) [40]. However, our finding that this effect is seen across the entire genome raises the possibility that the SKN-1 and MOM-2/Wnt inputs might compensate for each other and that genetic variants that enhance the requirement for one of the inputs relaxes the requirement for the other. This reciprocality might reflect cross-regulatory interactions between these two maternal inputs or may be the result of evolutionary constraints imposed by selection on these genes, which act pleiotropically in a variety of processes.
We identified two genes, rict-1 and plp-1, that show similar inverse effects on the requirements from skn-1 and mom-2: debilitation of either gene enhances the phenotype of mom-2(-) and suppresses that of skn-1(-). RICT-1 function extends lifespan in C. elegans through the action of SKN-1 [68], and loss of RICT-1 rescues the misspecification of the MS and E blastomeres and lethality of skn-1(-) embryos [68], consistent with our finding. However, the mechanism by which loss of rict-1 synergizes with a defect in the Wnt pathway is not clear. We previously showed that PLP-1, a homologue of the vertebrate transcription factor pur alpha, binds to the end-1 promoter and acts in parallel to the Wnt pathway and downstream of the MAPK signal [69], thereby promoting gut formation. PLP-1 shows a similar reciprocal relationship with SKN-1 and MOM-2 as with RICT-1 (Fig. 8). Given that PLP-1 binds at a cis regulatory site in end-1 near a putative POP-1 binding site [69], and that SKN-1 also binds to the end-1 regulatory region [64], it is conceivable that this reciprocality reflects integration of information at the level of transcription factor binding sites. As the architecture of the GRN is shaped by changes in cis-regulatory sequences [1,3], analyzing alterations in SKN-1 and Wnt/POP-1 targets among C. elegans wild isolates may provide insights into how genetic changes are accommodated without compromising the developmental output at microevolutionary time scale.
MIG-5, a dishevelled orthologue, functions in the Wnt pathway in parallel to Src signaling to regulate asymmetric cell division and endoderm induction [28,70]. We found that the loss of mig-5 function enhances the gut defect of skn-1(-) and suppresses that of the mom-2(-), the opposite reciprocal relationship to that of rict-1 and plp-1, and consistent with a previous report (Fig. 8) [28]. These effects were not observed in embryos lacking function of dsh-2, the orthologue of mig-5 (data not shown), supporting a previous study that showed overlapping but non-redundant roles of MIG-5 and DSH-2 in EMS spindle orientation and gut specification [70]. Recent studies showed that Dishevelled can play both positive and negative roles during axon guidance [93,94]. Dishevelled, upon Wnt-activation, promotes hyperphosphorylation and inactivation of Frizzled receptor to fine-tune Wnt activity. It is tempting to speculate that MIG-5 may perform similar function in EMS by downregulating activating signals (Src or MAPK), in the absence of MOM-2.
We hypothesize that compensatory mechanisms may evolve to fine-tune the level of gut-activating regulatory inputs. Successful developmental events depend on tight spatial and temporal regulation of gene expression. For example, anterior-posterior patterning in the Drosophila embryo is determined by the local concentrations of the Bicoid, Hunchback, and Caudal transcription factors [95]. We postulate that SKN-1 and Wnt signaling is modulated so that the downstream genes, end-1/3, which control specification and later differentiation of endoderm progenitors, are expressed at optimal levels that ensure normal gut development. Suboptimal END activity leads to poorly differentiated gut and both hypo- and hyperplasia in the gut lineage [96–98]. Hyper- or hypo-activation of Wnt signaling has been implicated in cancer development [99], bone diseases [100,101], and metabolic diseases [102,103], demonstrating the importance of regulating the timing and dynamics of such developmental signals within a quantitatively restricted window.
Cryptic variation and evolvability of GRNs
This study revealed substantial cryptic genetic modifications that alter the relative importance of two partially redundant inputs into the C. elegans endoderm GRN, leading to rapid change in the developmental network architecture (Fig. 9). Such modifications may occur through transitional states that are apparent even within the radiation of this single species. For example, the finding that POP-1 is not required for gut development even in a wild isolate (e.g., MY16) that, like C. briggsae, shows a near-absolute requirement for SKN-1 may reflect a transitional state between the two species: i.e., a nearly essential requirement for SKN-1 but non-essential requirement for POP-1, an effect not previously seen in either species. In addition, duplicated GATA factors (the MEDs, ENDs, and ELTs) and partially redundant activating inputs (SKN-1, Wnt, Src, and MAPK) in endoderm GRN, provide an opportunity for genetic variation to accumulate and “experimentation” of new regulatory relationships without diminishing fitness [2,104,105].

Accumulation of cryptic genetic modifications drives rapid rewiring of the GRN, causing broad variation of SKN-1 and MOM-2/Wnt dependence in endoderm (E) specification among C. elegans isotypes. Wnt-signaled POP-1 (indicated by *) acts as an E activator, while unmodified POP-1 in the MS blastomere acts as a repressor of E fate in all C. elegans isotypes. The relative strength of the inputs is indicated by the thickness of the arrow.
Redundancy in the system may act to ‘rescue’ an initial mutation and allow for secondary mutations that might eventually lead to rewiring of the network. For example, loss of either MyoD or Myf5, two key regulators of muscle differentiation in metazoans, produces minimal defects in myogenesis as a result of compensatory relationship between the myogenic factors [106]. In vertebrates, gene duplication events have resulted in an expansion of Hox genes to a total of >200, resulting in prevalent redundancy [107–109]. This proliferation of redundant genes provides opportunities for evolutionary experimentation and subsequent specialization of new functions [109]. In C. elegans, loss of GAP-1 (a Ras inhibitor) or SLI-1 (a negative regulator of EGFR signaling) alone does not produce obvious defects, while double mutations lead to a multivulva phenotype [110]. Many other similar redundant relationships between redundant partners exist in the animal. Notably, the relative importance of Ras, Notch, and Wnt signals in vulva induction differ in various genetic backgrounds [6,65] and physiological conditions [111,112], resulting in flexibility in the system. While vulval development in C. elegans, when grown under standard laboratory conditions, predominantly favors utilization of the EGF/Ras signaling pathway [111], Wnt is the predominant signaling pathway in the related Pristionchus pacificus, which is ∼250 MY divergent [113]. In addition, while Cel-lin-17 functions positively to transduce the Wnt signal, Ppa-lin-17/Fz antagonizes Wnt signaling and instead the Wnt signal is transmitted by Ppa-lin-18/Ryk, which has acquired a novel SH3 domain not present in the C. elegans ortholog [114].
Thus, extensive rewiring of signaling networks and modularity of signaling motifs contribute to developmental systems drift.
The broad cryptic variation we have observed in this study may drive developmental system drift, giving rise to GRN architectures that differ in the relative strength of the network components. In the developmental hourglass model of evolvability of animal development, the early stages of embryonic development showed the least constraint in gene expression compared to either the phylotypic stage or post phylotypic stage. This is likely attributable either to positive selection during early embryonic and later larval stages or to developmental constraints. Analysis of developmental gene expression in mutation accumulation lines, which have evolved in the absence of any positive selection, showed similarity to the developmental hourglass model of evolvability, consistent with strong developmental constraints on the phylotypic stage [115]. However, they do not rule out the possibility that early and late stages of development might be more adaptive and therefore subject to positive selection. It will be of interest to learn the degree to which the divergence in network architecture might arise as a result of differences in the environment and selective pressures on different C. elegans isotypes.
COMPETING INTERESTS
The authors declare no competing or financial interests.
Supplementary figures

A minimum of two replicates were obtained, with >500 embryos per replicate. Box-plot represents median with range bars showing upper and lower quartiles.

Strains are shown on the x-axis and fraction of arrested embryos with endoderm are shown on the y-axis. Four introgressed lines were studied for each mutant strain. >200 embryos were scored per experiment.

(A) Manhattan plot of skn-1(RNAi) GWAS. Red line represents 1.5% FDR (obtained from 10,000 permutated results) and black line represents 3.0% FDR. The chromosomes are color-coded. The y axis is the –log10 of p-value. (B) Correlation between skn-1(RNAi) GWAS and EMMA (see Fig. 5A). y-axis is the –log10 of the p-values from EMMA, x-axis is –log10 of the p-values from GWAS. A modest linear relationship is found (Spearman correlation coefficient = 0.43) as shown by the blue line. (C) Effect plot of the top SNP revealed by skn-1(RNAi) EMMA (see Fig. 5A). (D) Effect plot of the top SNP on Chromosome V revealed by skn-1(RNAi) GWAS. The variant position and genotype are shown on the x-axis, while the phenotype of strains carrying the alleles after skn-1(RNAi) treatment is shown on the y-axis. Confidence intervals for the average phenotype in each genotype group are shown.

A beta-distribution is observed (skewed to the right). Shapiro-Wilk normality test (W=0.8682, p-value=1.207X10-7).
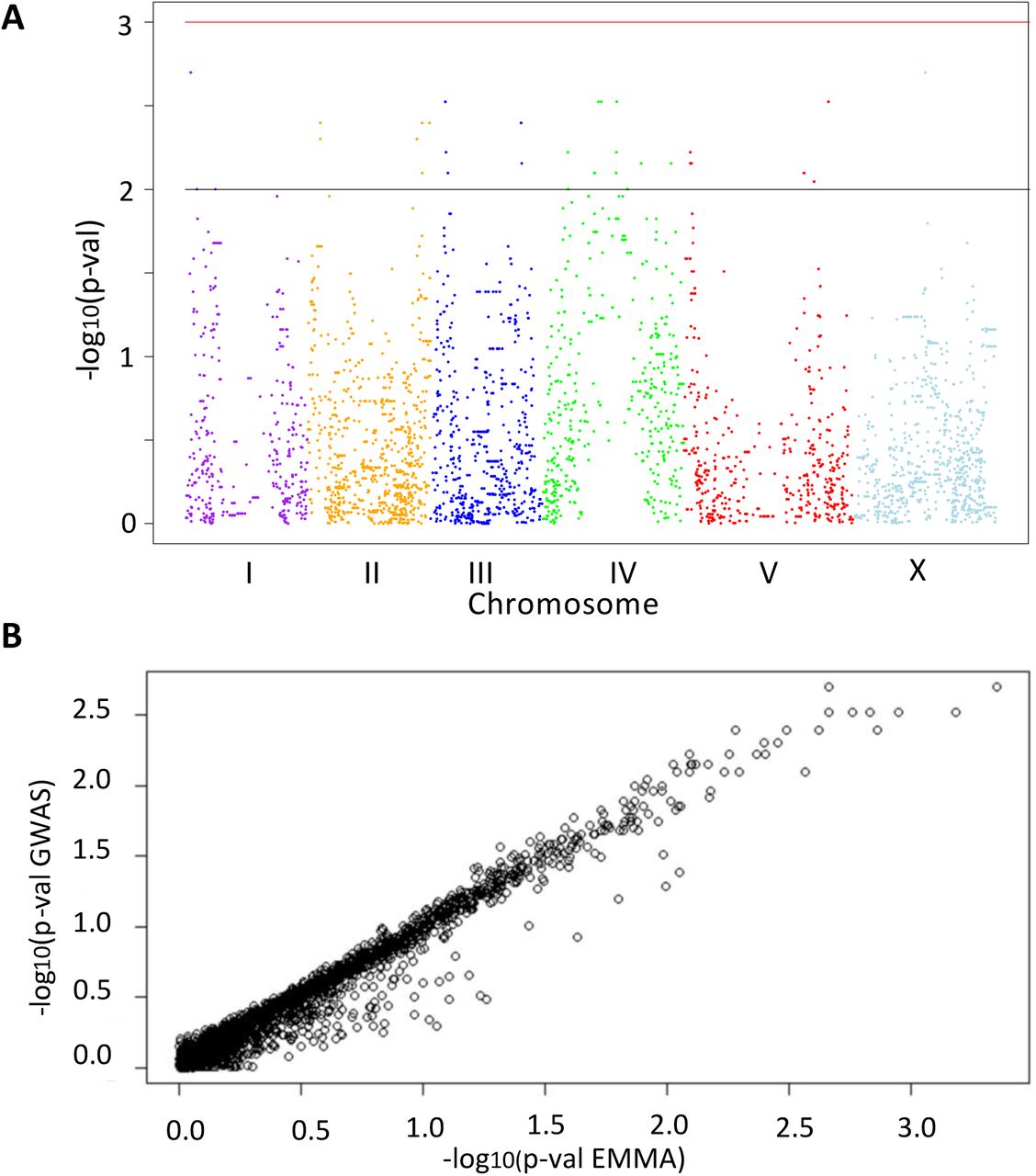
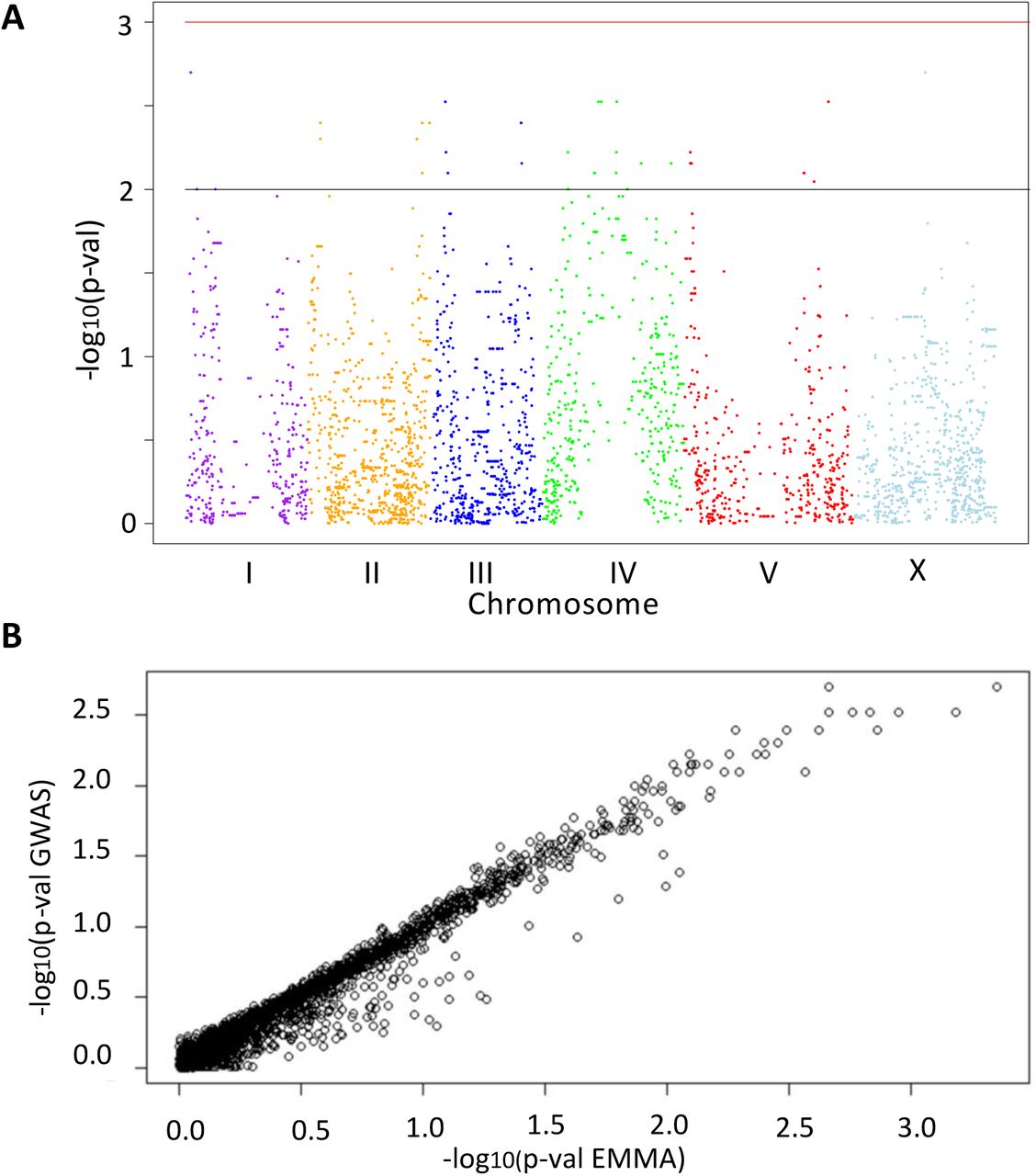
(A) Manhattan plot of mom-2(RNAi) GWAS (permutation-adjusted p-values). Black line represents p-value<0.01, while red line represents p-value<0.001. Genomic regions are shown on the x-axis. The chromosomes are color-coded. The y axis is the –log10 of p-value in the linear model. (B) Correlation between mom-2(RNAi) GWAS and EMMA. The y-axis represents the −log10 of the p-values from the GWAS approach, while the x-axis represents –log10 of the p-values from the EMMA approach. A strong linear relationship is found (Pearson’s correlation R = 0.95, p-value < 2.2e-16).

Heatmap of p-values for mom-2(RNAi) (left) and skn-1(RNAi) (right) as calculated in the EMMA analyses (see Fig. 5A, B). Strength of association between genotype and endoderm formation phenotypes is represented as –log10(p-value), here depicted as a heatmap (lighter colors – weaker association, darker colors – stronger association). An overlap (indicated by arrow head) is found in a small region of chromosome IV, but no further correlations are observed.

(A) mom-2 (RNAi) phenotype of 94 isolates arranged with respect to the neighbor-joining tree constructed using 4,690 SNPs and pseudo-rooted to QX1211. Red asterisk indicates an example of closely related strains (JU394 and CB4851) with similar phenotypes, while black asterisks indicate examples sister strains (JU792 and JU1242; JU1440 and JT11398) with distinct phenotypes. (B) Worldwide distribution of mom-2(RNAi) phenotype across 94 isolates. Each circle represents a single isolate.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Heat map of allelic differences per SNP for skn-1(RNAi) and mom-2(RNAi), as calculated by the phenotypic median differences per allele at each SNP. Each line represents a color-coded result of a single locus, covering the entire genome. Correlation sliding window of (B) chromosome II, (C) chromosome III, (D) chromosome V and (E) chromosome X. Windows of 50 SNPs were used to calculate the correlation coefficient and p-value. Black circles represent the correlation coefficient (R value) for each window (scale on the x-axis). Black line indicates the 0 threshold. Grey line represents the –log10 of the p-values for the corresponding correlation windows (scale on the y-axis). Grey horizontal line is the significance threshold set at p-value=0.01. Dotted blue lines divide chromosomal region into left, middle and right arms.
ACKNOWLEDGMENTS
We thank members of the Rothman, especially Sagen Flowers and Kristoffer C. Mellingen for experimental assistance, and Snell labs, particularly Dr. Kien Ly, for helpful advice and feedback. We thank Dr. Kathy Ruggiero (University of Auckland, New Zealand) for helpful advice on GWAS methodology and Dr. James McGhee (University of Calgary, Canada) for providing the MH33 antibody. Nematode strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health - Office of Research Infrastructure Programs (P40 OD010440). Y.N.T.C was supported during part of this work by a University of Auckland Doctoral Scholarship. This work was supported by grants from the NIH (#1R01HD082347 and # 1R01HD081266) to J.H.R.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.
- 5.↵
- 6.↵
- 7.
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.
- 14.
- 15.
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.
- 28.↵
- 29.↵
- 30.
- 31.↵
- 32.↵
- 33.
- 34.↵
- 35.
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵




